

Abstract
BiP, an essential and ubiquitous Hsp70 chaperone in the endoplasmic reticulum, plays a key role in protein folding and quality control. BiP contains two functional domains: a nucleotide binding domain (NBD) and a substrate-binding domain (SBD). NBD binds and hydrolyzes ATP; the substrates for SBD are extended polypeptides. ATP binding allosterically accelerates polypeptide binding and release. Although crucial to the chaperone activity, the molecular mechanisms of polypeptide binding and allosteric coupling of BiP are poorly understood. Here we present crystal structures of an intact human BiP in the ATP-bound state, the first intact eukaryotic Hsp70 structure, and isolated BiP SBD with a peptide substrate bound representing the ADP-bound state. These structures and our biochemical analysis demonstrate that BiP has a unique NBD-SBD interface that is highly conserved only in eukaryotic Hsp70s found in the cytosol and ER to fortify its ATP-bound state to promote the opening of its polypeptide-binding pocket.
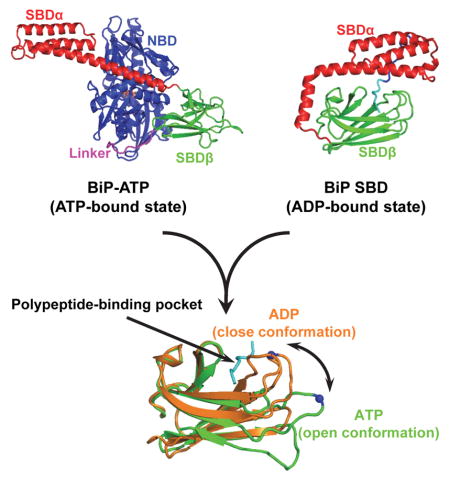
Graphical abstract
INTRODUCTION
The key functions of the endoplasmic reticulum (ER) are folding, assembly, and quality control for secreted and membrane proteins(Hammond and Helenius, 1995). Binding immunoglobulin protein (BiP), an essential and ubiquitous Hsp70 molecular chaperone resident in the lumen of ER, plays a crucial role in all of these ER functions(Dudek et al., 2009; Hendershot, 2004).
Hsp70s are a class of conserved and abundant molecular chaperones that play multiple essential roles in maintaining cellular protein homeostasis by assisting protein folding, assembly, translocation into organelles, and degradation(Bukau et al., 2000; Hartl and Hayer-Hartl, 2009; Mayer and Bukau, 2005; Young, 2010). Hsp70s have been found in the cytosol of both prokaryotes and eukaryotes, and in all the cellular compartments of eukaryotes including the ER and mitochondria. All Hsp70s including BiP have two conserved functional domains: a nucleotide binding domain (NBD) at the N-terminus and a substrate binding domain (SBD) at the C-terminus(Bukau and Horwich, 1998; Mayer and Bukau, 2005). NBD binds ATP and hydrolyzes it to ADP. SBD binds hydrophobic polypeptides in an extended conformation as substrates(Blond-Elguindi et al., 1993; Rudiger et al., 1997; Zhu et al., 1996). Extensive structural efforts for the past three decades have yielded a number of isolated domain structures from both prokaryotic and eukaryotic Hsp70s. These structures have shown the conserved structural basis of each domain in binding its substrates. NBD is composed of two large lobes, between which is the nucleotide-binding site(Flaherty et al., 1990; Mayer and Bukau, 2005). SBD is divided into two subdomains: SBDβ and SBDα (Chang et al., 2008; Leu et al., 2014; Liebscher and Roujeinikova, 2009; Zhu et al., 1996). The polypeptide-binding pocket is formed between two loops on SBDβ while SBDα functions as a lid covering the pocket.
The chaperone activity of Hsp70s is powered by ATP through allosteric coupling of the two functional domains(Buchberger et al., 1995; Mayer and Bukau, 2005). In the ADP-bound and nucleotide-free (apo) states, the two domains have little interaction(Bertelsen et al., 2009; Buchberger et al., 1995; Chang et al., 2008; Swain et al., 2007). The polypeptide substrate binding properties of this state are like those of the isolated SBD, high affinity with both slow binding and release rates(Flynn et al., 1989; Schmid et al., 1994). In contrast, in the ATP-bound state, the two domains are tightly coupled, which results in drastically accelerated kinetics in both binding and release of polypeptide substrates, although the resulting affinity is 2 to 3 orders of magnitude lower(Schmid et al., 1994). This ATP-induced allosteric coupling is crucial for efficient chaperone activity(Mayer and Bukau, 2005). Thus, the molecular mechanism of allostery had been highly sought through obtaining crystal structures of intact Hsp70s in the ATP-bound state. However, due to the transient nature of the ATP-bound state, only recently, the captured DnaK-ATP structures first revealed the molecular mechanism of this allosteric coupling in E. coli(Kityk et al., 2012; Qi et al., 2013).
However, DnaK shares only 40–50% sequence identity to various human Hsp70s, and more importantly, DnaK’s cellular functions differ from those of human Hsp70s especially BiP(Dudek et al., 2009; Ma and Hendershot, 2004). Moreover, previous studies have shown that the biochemical properties of eukaryotic Hsp70s are significantly different from those of DnaK including the kinetics for peptide substrate binding, the molecular radius of the ATP-bound state and ATP-induced allosteric coupling(Mapa et al., 2010; Marcinowski et al., 2013; Shi et al., 1996; Wilbanks et al., 1995), suggesting there may be unknown important mechanistic differences between DnaK and eukaryotic Hsp70s. Thus, the exact molecular mechanism of the ATP-driven allosteric coupling in human Hsp70s is ill-defined. To directly answer this question, we have solved a crystal structure of an intact human BiP in the ATP-bound state, the first eukaryotic Hsp70 structure in the ATP-bound state. Moreover, to understand the structural basis of peptide substrate binding in the ADP-bound and apo states, we solved structures of the isolated SBD of BiP. Together with our biochemical analysis, these structures support the hypothesis that the opening of the polypeptide-binding pocket upon ATP binding is conserved in BiP and most likely in other eukaryotic Hsp70s.
RESULTS
Human Hsp70 BiP constructs for crystallization studies
To understand the molecular mechanism of BiP peptide substrate binding and allosteric regulation by ATP, we aimed to solve crystal structures of human BiP in two states: the isolated SBD, which represents the ADP-bound and apo states, and a functionally complete BiP in the ATP-bound state. First, we tested whether purified human BiP binds to the NR peptide (sequence NRLLLTG), a well-characterized peptide substrate for DnaK(Zhu et al., 1996). As shown in Fig. 1a, BiP binds NR peptide with good affinity: the Dissociation Constant (Kd) is about 2 times lower than that of DnaK (Fig. 1b). However, the binding kinetics, both on and off rates, are much slower than those of DnaK (Fig. S1a,b), which is consistent with previous reports with other peptide substrates(Marcinowski et al., 2011; Marcinowski et al., 2013). Moreover, addition of ATP drastically reduced the affinity of BiP for NR, supporting allosteric regulation of peptide substrate binding by ATP (Fig. 1a). As expected, the isolated SBD of BiP binds NR with affinity comparable to full-length BiP (Fig. 1a,b).
Figure 1. Constructs for crystallization of a full-length BiP and its isolated SBD.

a, BiP binds NR peptide through its SBD and in an ATP-sensitive manner. Fluorescence polarization assay with serial dilutions of BiP proteins was used to measure NR binding. DnaK was used as a positive control.
b, Peptide NR binding affinities for DnaK and BiP proteins. Dissociation constants (Kd) were calculated based on the results in a. Standard errors were calculated from at least 6 assays on more than two protein purifications.
c, Schematics of BiP domain structure and the construct for crystallization of a full-length BiP. The coloring of domains is: NBD (blue), Linker (purple), SBDβ (green), and SBDα (red). The signal sequence (first 24 residues) and the last 20 residues are not colored. The residue numbers marking domains are labeled on the top.
d, Schematics of the BiP-SBD constructs for crystallization. Domain coloring is the same as in c. The Tev linker and linked NR peptide are shown as an orange line and in cyan, respectively.
e, Neither BiP SBD-Tev–NR nor SBD-L3,4′-Tev-NR showed appreciable binding to NR peptide. WT BiP SBD was used as a positive control. Binding assay was carried out as in a.
f, BiP-T229A mutant binds NR peptide in an ATP-sensitive manner like that of WT BiP, and L3,4′ modification drastically compromised the NR peptide binding. WT BiP was used as a positive control. Binding assay was carried out as in a.
To solve a crystal structure of the isolated SBD of BiP, we linked the NR peptide to the C-terminus of BiP SBD through a linker containing a Tev protease digest site (sequence: SENLYFQGS; Fig. 1c–d). Linking the NR peptide to the C-terminus of BiP completely blocks the binding of free NR peptide in solution (Fig. 1e), suggesting that the linked NR is bound to the isolated SBD as substrate. We named this construct SBD-Tev-NR, and solved its crystal structure at 2.57 Å resolution (Table 1).
Table 1.
Data collection and refinement statistics
| BiP-T229A L3,4′ (native) | SBD-Tev-NR (native) | SBD-L3,4′-Tev-NR (Native) | SBD-L3,4′-Tev-NR (Se-SAD) | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P3221 | C2221 | C2221 | C2221 |
| Cell dimensions | ||||
| a, b, c (Å) | 222.468, 222.468, 209.460 | 36.378, 91.661, 141.797 | 34.536, 82.593, 150.776 | 34.553, 82.661, 150.840 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Wavelength | 0.979 | 0.979 | 0.979 | 0.979 |
| Resolution (Å) | 50–3.00 (3.05–3.00) | 50–2.57 (2.61–2.57) | 50–2.68 (2.73–2.68) | 50–2.69 (2.74–2.69) |
| Rsym or Rmerge | 0.118 (0.402) | 0.044 (0.120) | 0.041 (0.092) | 0.036 (0.097) |
| I/σI | 25.4 (4.1) | 56.6 (16.2) | 38.6 (9.6) | 70.1 (23.0) |
| Completeness (%) | 97.8 (98.6) | 99.8 (98.0) | 99.8 (95.8) | 100 (100) |
| Redundancy | 7.3 (7.3) | 6.6 (4.2) | 4.6 (3.0) | 8.6 (6.2) |
| Refinement | ||||
| Resolution (Å) | 40.76–2.99 | 38.49–2.57 | 41.30–2.68 | |
| No. reflections | 111753 | 7908 | 6391 | |
| Rwork/Rfree | 24.3%/28.7% | 21.9%/26.4% | 21.60%/25.55% | |
| No. atoms | 28436 | 1891 | 1803 | |
| Protein | 28147 | 1838 | 1787 | |
| ATP/Zn/Mg | 186/24/17 | - | - | |
| Water | 32 | 48 | 16 | |
| B-factors | 76.68 | 30.56 | 56.25 | |
| Protein | 76.98 | 30.67 | 56.41 | |
| ATP/Zn/Mg | 49.61/79.98/55.03 | - | - | |
| Water | 48.51 | 26.34 | 38.46 | |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.011 | 0.003 | 0.003 | |
| Bond angles (°) | 1.495 | 0.746 | 0.709 | |
One crystal was used for each structure or dataset.
Values in parentheses are for the highest-resolution shell.
To obtain a functional intact BiP in the ATP-bound state, we took advantage of two mutations analogous to those used in our recently reported DnaK-ATP structure(Qi et al., 2013): T229A and loop L3,4 alternative L3,4′ (Fig. 1c). The BiP T229A mutation significantly compromised the ATPase rate (Fig. S1c) but maintains allosteric coupling as shown by the significantly reduced affinity for NR peptide in the presence of ATP (Fig. 1f). These properties of T229A are consistent with a previously characterized BiP T229G mutant(Wei et al., 1995). The L3,4′ mutation has its L3,4 replaced with a shortened L1,2 sequence (TASDNQP→VGG). This BiP-L3,4′ protein has a drastically reduced affinity for NR peptide (Fig. 1e,f). Thus, this BiP-T229A L3,4′ construct helped stabilize BiP in the ATP-bound state and solved the self-association problem that complicates crystallization. Moreover, since the first 24 residues are a signal sequence and the last 20 residues are largely disordered based on sequence alignments, both were removed to facilitate crystallization (Fig. 1c). We obtained crystals of BiP-T229A L3,4′ grown only in the presence of ATP. We solved this BiP-ATP structure by molecular replacement, and the final model was refined to 3.0 Å resolution (Table 1).
To test the structural impact of the L3,4′ modification in BiP, we introduced it into the SBD-Tev-NR construct and solved the crystal structure of the resulting SBD-L3,4′-Tev-NR construct at 2.68 Å resolution (Table 1).
Structures of isolated BiP-SBD with peptide substrate bound
As expected, the isolated BiP-SBD structure contains both SBDβ and SBDα subdomains (Fig. 2a). The linked NR peptide is bound to the polypeptide-binding site formed between L1,2 and L3,4 on SBDβ. SBDα covers the polypeptide-binding site as a lid with the signature kink between helices αA and αB. The Tev linker that connects the NR peptide to the C-terminus of BiP-SBD packs against helices αB and αD/E of SBDα, and facilitates crystal contacts. Overall, the BiP-SBD structure is highly similar to that of the isolated DnaK SBD structure in complex with NR peptide, the first isolated SBD structure(Zhu et al., 1996), except for the Tev linker (Fig. 2b, S2a). The linked NR peptide binds to BiP in a fashion similar to that of DnaK with almost identical main-chain conformation (Fig. 2b, S2a,b). However, the register of amino acids is shifted one residue toward the N-terminus (Fig. 2c, S2b): instead of Leu4 in DnaK, Leu5 is in the center of the polypeptide-binding pocket, which could due to the constraints from the covalent linkage of the NR peptide to the C-terminus of SBD. A similar shift is observed in the isolated DnaK SBD structure with the L3,4′ modification, DnaK SBD- L3,4′, in which the inter-domain Linker from a symmetry mate binds to the polypeptide-binding site like a peptide substrate(Qi et al., 2013) (Fig. S2d–e). These shifts suggest the flexibility of the polypeptide-binding site in recognition of substrates.
Figure 2. Structural analysis of isolated BiP-SBD structures.

a, Ribbon diagrams of the BiP SBD-Tev–NR structure. Domain coloring is the same as Fig. 1c with the TEV linker and NR peptide shown in blue and cyan, respectively.
b, Comparison of the BiP SBD-Tev–NR structure with the DnaK SBD structure (PDB code: 1DKZ). The coloring of BiP SBD-Tev–NR is the same as in a. The DnaK SBD structure is shown in orange with the bound NR peptide in purple. The structures were superimposed based on Cα atoms of SBDβ.
c, Comparison of the bound NR peptide in the BiP SBD-Tev–NR (top) and DnaK SBD (bottom; PDB code:1DKZ) structures. The two structures were superimposed as in b. The side-chains of NR peptides are highlighted in stick presentation, and the carbon atoms of the NR peptide are colored in grey and orange for the BiP SBD-Tev–NR and DnaK SBD structures, respectively.
d, Y570 and R492 form novel hydrophobic contacts with L4 of bound NR peptide in the BiP-SBD structure. The BiP-SBD structure is shown in ribbon representation and colored as in a. L4 (NR peptide), V429, R492 and Y570 are highlighted as sticks.
e, Ribbon diagram of the BiP SBD-L3,4′-Tev-NR structure. Domain coloring is the same as in Fig. 1c with the Tev linker and NR peptide in blue and cyan, respectively.
f, Superposition of BiP SBD-L3,4′-Tev-NR (purple) with BiP SBD-Tev–NR (domain coloring is the same as in a) based on Cα atoms of SBDβ.
g, Superposition of the NR peptides in BiP SBD-L3,4′-Tev-NR (purple) and BiP SBD-Tev–NR (cyan). The two structures were superimposed as in f. The side-chains are shown in stick representation with carbon atoms shown in orange for SBD-L3,4′-Tev-NR and grey for SBD-Tev–NR.
h, Comparison of NR-contacting residues (in stick representation) between BiP SBD-Tev–NR (orange) and SBD-L3,4′-Tev-NR (green). The two structures were superimposed based on Cα atoms of SBDβ.
In the polypeptide-binding pocket, except for V429 in BiP and M404 in DnaK, the residues that form hydrophobic contacts with NR peptide in BiP are identical to those in DnaK (Fig. S2c), and more importantly, the side chains of these residues assume almost identical conformations, suggesting high conservation of the polypeptide-binding site among Hsp70s. Mutating V429 in hamster BiP to methionine has been shown to change the kinetics of peptide substrate binding similar to those of DnaK(Marcinowski et al., 2013), confirming that the difference between V429 in BiP and M404 in DnaK is the structural basis for their different peptide binding kinetics.
Interestingly, unlike M404 forming hydrophobic contacts with SBDα, V429 is too small to form any contact with SBDα. Surprisingly, Y570 from SBDα and R492 in L5,6 form direct hydrophobic contacts with Leu4 of the NR peptide (Fig. 2d), suggesting the direct involvement of SBDα and L5,6 in binding peptide substrates, which was not observed in any previous SBD structures. These novel contacts may stabilize the BiP-peptide complex and thus explain the much slower peptide substrate binding kinetics of BiP than those of DnaK. The smaller side chain of V429 in BiP may allow enough space for Y570 and R492 to contact Leu4 of NR, which was not possible for the larger side chain of M404 in DnaK. Mutating V429 to methionine in BiP may disrupt the contacts from Y570 and R492 to NR peptide, resulting in increased kinetics similar to DnaK. Thus, Y570 and R492 may contribute to the special substrate binding properties of BiP. Consistent with this observation, the SBDα of BiP has been indicated in direct binding of polypeptide substrates(Marcinowski et al., 2011). Interestingly, a previous study identified two arginine residues in SBDβ of hamster BiP, R470 and R492 (R470 and R492 in human BiP), which were modified by ADP, and then destabilized peptide substrate binding(Chambers et al., 2012). Based on the DnaK SBD structure, R470 (in L4,5) and R492 form contacts with SBDα and thus stabilize the covering of the SBDα lid over the polypeptide- binding pocket. This is also true for the BiP SBD structure. Consistent with this feature, R470E mutant has a peptide binding defect similar to the deletion of SBDα characterized by increased dissociation rate. However, R492E mutant has a surprisingly much stronger peptide binding defect than the deletion of SBDα, which can not be explained by the DnaK SBD structure. The direct hydrophobic contacts with bound NR peptide from R492 observed in the isolated BiP-SBD structure may provide an explanation. Moreover, R467C mutant in DnaK (R492 in BiP) has a similar defect in peptide substrate binding as BiP R470E instead of R492E (data not shown), supporting the contacts between R492 and NR peptide may be unique for BiP.
The structure of BiP SBD with the L3,4′ alternation, SBD-L3,4′-Tev-NR, is almost identical to that of BiP SBD-Tev-NR except for the L3,4′ modification (Fig. 2e,f, S2f). Although the BiP protein carrying the L3,4′ alteration has very low affinity for peptide substrates (Fig. 1f), the linked NR peptide binds to the polypeptide-binding site almost identically to that of SBD-Tev-NR, with the same amino acid register and virtually identical main-chain conformation, except that the last glycine residue of the NR peptide is missing(Fig. 2g). Except for the shortening of L3,4, the polypeptide-binding site of SBD-L3,4′-Tev-NR is essentially identical to that of SBD-Tev-NR (Fig. 2h, S2g). Thus, the L3,4′ modification has no appreciable structural impact on BiP SBD.
The human BiP-ATP structure
There are six BiP molecules per asymmetric unit in the BiP-ATP structure and the six molecules are almost identical (Fig. S3a–d). As expected, each protomer contains NBD, linker, SBDβ and SBDα domains, and ATP (Fig. 3a). Thus, this human BiP-ATP structure is the first intact eukaryotic Hsp70 structure in the ATP-bound state. Except for small changes in several loops, the NBD and SBDβ are virtually identical for the six protomers (Fig. S3b–d). Whereas, the SBDα regions showed more difference although the overall differences are still small. We mainly used protomer A for the following structural analysis and comparison.
Figure 3. Overall structure of BiP-ATP.

a, Ribbon diagram of human BiP-ATP structure. Domain coloring is the same as in Fig. 1c. The bound ATP is in stick presentation, and its associating Zn ion is shown as a grey ball. Left, the classic front-face view of NBD; right, view orthogonal to that of the left panel.
b, Comparison of human BiP-ATP structure with our previously published DnaK-ATP (PDB code: 4JNE) structure. The domain coloring of BiP-ATP is the same as a. The domain coloring for DnaK-ATP: NBD (cyan), Linker (orange), SBDβ (yellow) and SBDα (grey). The superposition was based on Cα atoms. The view point is the same as the right panel of a.
c, Superposition of SBDβ domains from the BiP-ATP (green) and DnaK-ATP (yellow) structures. The loops are labeled. The superposition was based on Cα atoms.
d, A unique hydrogen bond was formed between D483 and D529 in the BiP-ATP structure. D483, D529, T527, N528 and Q530 are shown in stick presentation. Hydrogen bonds are shown as dotted lines.
One interesting feature about the crystal packing of the BiP-ATP structure is, among the six protomers, two pack as a dimer similar to our previously published DnaK-ATP structure(Qi et al., 2013) (Fig. S3e). We have recently shown this dimer to be essential for Hsp40 interaction(Sarbeng et al., 2015). Thus, BiP, like DnaK, may also form a dimer in solution in the ATP-bound state, facilitating Hsp40 interaction.
Consistent with various biochemical studies on a number of Hsp70s including BiP(Buchberger et al., 1995; Mapa et al., 2010; Marcinowski et al., 2011; Swain et al., 2007; Wei et al., 1995), the domains form extensive contacts in the BiP-ATP structure. SBDβ binds on the back of NBD contacting both lobes while SBDα docks on the side of lobe I with the first two helices fused into one long helix. The highly conserved inter-domain linker is fitted in the bottom groove between the two lobes of NBD. Overall, the BiP-ATP structure is similar to the two recently published DnaK-ATP structures(Kityk et al., 2012; Qi et al., 2013) although the sequence identity is only 48%, suggesting an overall conserved molecular mechanism of ATP-elicited allosteric coupling among Hsp70 members despite substantial evolutionary distances. For comparison we used our DnaK-ATP structure, which is almost identical to the other available structure. The relative orientation of the domains in BiP-ATP is similar to that of DnaK-ATP with a rotation in SBDα (Fig. 3b), consistent with the high conservation of the NBD-SBDβ interface and relatively low conservation of the NBD-SBDα interface (see below for detailed comparison). The NBD shares the most resemblance with virtually identical conformation (Fig. S4a), suggesting high conservation in ATP binding. The bound ATP molecules are superimposable although Zn2+ replaced Mg2+ in BiP-ATP due to the high concentration of Zn2+ in crystallization solution (Fig. S4b). Moreover, BiP proteins have lower ATPase activity in the presence of Zn2+ than that of Mg2+(Fig. S1c), consistent with the presence of ATP in the BiP-ATP structure.
Despite the above similarities, there are notable differences between the BiP-ATP and DnaK-ATP structures.
NBD: There are three insertion/deletion segments in the NBD between BiP and DnaK: 1) β8-β9; 2) β13-β14; and 3) β15-β16 (Fig. S4a), whose functions had largely been a mystery. BiP has a longer β8-β9, but shorter β13-β14 and β15-β16. Based on sequence alignment of these segments, Hsp70s can be divided into two groups: eukaryotic cytosolic/ER Hsp70s (BiP-like) and prokaryotic/mitochondrial Hsp70s (DnaK-like) (Fig. S4c–e). The sequences are conserved in each group. In the BiP-ATP structure, the β8-β9 insertion is involved in the NBD-SBDα interface, and discussed in more details below. β13-β14 has been proposed to be involved in Hsp40 interaction(Ahmad et al., 2011); thus, it may contribute to the different properties in Hsp40 interaction. β15-β16 in DnaK contributes to the interaction with the nucleotide-exchange factor GrpE(Harrison et al., 1997), a cochaperone only existed in prokaryotes and eukaryotic mitochondria, which may explain the conservation only in these Hsp70s.
SBD: there are apparent differences in both SBDβ and SBDα although the overall conformations are similar to those of our DnaK-ATP structure (Fig. 3b,c, S4f). Notably, there are significant differences in the loops on the L3,4 side of the polypeptide-binding site (including L3,4, L5,6, and L7,8), consistent with the high flexibility of these loops(Kityk et al., 2012), which is further supported by the larger difference among the six promoters in the BiP-ATP structure (Fig. S3c–d). Intriguingly, L7,8 is significantly longer in the BiP-ATP structure (Fig. 3c) while it is virtually identical in the isolated SBD structures (Fig. 2b). It seems that β8 slides towards L7,8 against the rest of the structure. This could partially be due to the insertion of the highly conserved R532 in the Lα,β in BiP (see below for details). Furthermore, D529 on β8 forms a short strong hydrogen bond with D483 on β5 in BiP-ATP (Fig. 3d), which could stabilize the position of β8 relative to β5 in addition to the main-chain hydrogen bonds between these two β strands. Supporting the conformation of D529, two salt bridges are formed between N527 and Q530. All these residues, D483, D529, N527 and Q530, are only conserved in the eukaryotic cytosolic/ER Hsp70s (Fig. S4g), suggesting these features of the BiP-ATP structure may be conserved among these Hsp70s, but not in prokaryotic/mitochondrial Hsp70s.
In SBDα, reflecting the relatively low sequence conservation, the loops connecting the helices have different conformations from those of DnaK-ATP (Fig. S4f). Interestingly, the C-terminus of αA/B seems less bent than that of DnaK-ATP, which correlates with the NBD-SBDα interface difference between BiP and DnaK as described below. Moreover, among the six almost identical protomers in the BiP-ATP structure, SBDα has the biggest difference (Fig. S3b–c), further supporting the intrinsic flexibility of SBDα.
Taken together, the differences and similarities described above support an overall conserved but differently regulated molecular mechanism of allostery among Hsp70s.
The unique role of NBD-SBDα interface and Lα,β in eukaryotic Hsp70 allosteric coupling
The distinct conformation of the BiP-ATP structure is due to the extensive contacts between domains upon ATP binding. Like the DnaK-ATP and Hsp110-ATP structures(Kityk et al., 2012; Liu and Hendrickson, 2007; Qi et al., 2013), there are also three major inter-domain contacts in BiP-ATP: NBD-linker, NBD-SBDβ, and NBD-SBDα (Fig. 3a). Hsp110s are distant homologs of Hsp70s and our previously solved Hsp110-ATP structure suggests that Hsp110-ATP is an evolutionary vestige of Hsp70-ATP(Liu and Hendrickson, 2007). Notably, the NBD-SBDα interface is divergent, which is consistent with the low sequence conservation in SBDα. Two clusters of hydrophobic contacts are featured at this interface in BiP-ATP (Fig. 4a). The first is mediated by L533 and M541 at the N-terminus of helix αA/B. This cluster is highly conserved in DnaK-ATP (L507 and M515) (Fig. 4b), and similar contacts are observed in Hsp110-ATP (L542 and L550) (Fig. 4c). For the second cluster, F548 in the middle of helix αA/B forms extensive van der Waals contacts with 6 residues from NBD: F68, R74, I132, K138, F140 and M148 (Fig. 4a). This cluster is similar to that in Hsp110-ATP; whereas, this interaction is replaced by a salt-bridge between N522 and E118 in DnaK-ATP. Moreover, all the residues in this second cluster are highly conserved in the eukaryotic cytosolic/ER Hsp70s, but not in the prokaryotic/mitochondrial Hsp70s (Fig. 4d, S5a–b). Interestingly, three residues in NBD that form hydrophobic contacts with F548 are in the segment of β8-β9 (Fig. S5b–c), one of the three insertions/deletions in NBD between the eukaryotic cytosolic/ER and prokaryotic/mitochondrial Hsp70s (Fig. S4a,c). Thus, this NBD-SBDα contact may be unique to eukaryotic cytosolic/ER Hsp70s. Moreover, our previous mutational work has shown the importance of F548 in the chaperone activity of Ssa1, the major cytosolic Hsp70 in yeast(Liu and Hendrickson, 2007), supporting the importance of this NBD-SBDα contact.
Figure 4. The unique NBD-SBDα interface and NBD-Lα,β contact in BiP-ATP.

a–c, Ribbon diagrams of NBD-SBDα interfaces in the BiP-ATP (a), DnaK-ATP (PDB code: 4JNE) (b), and Sse1-ATP (PDB code: 2QXL) (c) structures. NBDs are in blue and SBDαs are in red. Residues forming the two clusters of contacts are shown in stick presentation. Residues labeled in green are highly conserved between BiP-ATP and DnaK-ATP; residues labeled in orange are conserved between BiP-ATP and Sse1-ATP.
d, Sequence alignment among Hsp70s. Secondary structure assignments are labeled on the top with cylinder for helix and arrow for strand. R532 and F548 are highlighted in red and green, respectively. h, human; d, Drosophila melanogaster; b, bovine; v, Virgibacillus halodenitrificans. DnaK is from E.coli. Kar2, Ssa1 and Ssc1 are from saccharomyces cerevisiae.
e, The unique contact of NBD-Lα,β in the BiP-ATP structure. R532 forms two hydrogen bonds with D178 on NBD (blue). SBDβ and SBDα are in green and red, respectively.
f, Fluorescence anisotropy assay of NR peptide binding affinity for BiP R532E mutant. WT BiP was used as a control. Assays were carried out as in Fig. 1 in the presence of ATP (+ATP) or ADP (+ADP).
g, BiP R532E has a defect in releasing NR peptide upon addition of ATP. BiP proteins were incubated with F-NR peptide in the presence of ADP. After binding reached equilibrium, ATP was added (indicated by an arrow), and the release of F-NR was monitored over time.
In BiP, Lα,β, the small linker between SBDα and SBDβ, is one residue longer than in DnaK, with an extra arginine, Arg532 (Fig. 4d). This Arg residue is highly conserved among Hsp70s from eukaryotic cytosol and ER, but missing in prokaryotic and mitochondrial Hsp70s (Fig. 4d), which had been a mystery until our BiP-ATP structure. Interestingly, Arg532 forms two hydrogen bonds with D178 from NBD in the BiP-ATP structure (Fig. 4e) while it is on the surface of the isolated SBD structure with no obvious function (Fig. S2h), suggesting its importance in stabilizing the ATP-bound state. Thus, we hypothesized that it may play a role in the ATP-induced allosteric coupling of Hsp70s from eukaryotic cytosol and ER. To test this idea, we changed it to glutamate in BiP. This R532E mutant has normal affinity for NR peptide in the presence of ADP (Fig. 1b,4f), consistent with its location on the surface of isolated SBD with no apparent role in binding peptide substrate. In contrast, in the presence of ATP, the affinity of R532E is higher than that of WT BiP (Fig. 4f), suggesting the ATP-bound state is compromised by R532E. To further confirm the ATP-induced allosteric coupling is compromised in R532E, we assayed bound-peptide release triggered by ATP binding. Upon ATP addition, WT BiP released the majority of its bound NR peptide (Fig. 4g). In contrast, R532E mutant demonstrated significantly reduced release of bound NR peptide. Thus, we conclude that this conserved arginine is important for the ATP-induced allosteric coupling in BiP, supporting its role in stabilizing the ATP-bound state as observed in the BiP-ATP structure.
Consistent with the high conservation of the NBD-linker and NBD-SBDβ interfaces, these interfaces in BiP-ATP are highly similar to those in DnaK-ATP.
Conserved opening of the polypeptide-binding pocket
Both NBD and SBD of the BiP-ATP structure undergo a number of radical conformational changes relative to the isolated domain structures, which represent the ADP-bound and apo states. There are several isolated NBD structures of BiP in complex with different nucleotides including ADP and ATP(Macias et al., 2011; Wisniewska et al., 2010). All assume virtually the same conformation even when ATP is bound. The conformation of these structures is basically identical to that of the isolated bovine Hsc70 NBD structure with ADP bound, the first NBD structure(Flaherty et al., 1990). Thus, this conformation has been believed to be the ADP-bound conformation. In contrast, the NBD of BiP-ATP adopts a drastically different conformation with a rotation of more than 20 degrees between the two lobes (Fig. 5a, S6), suggesting that the SBD and its interaction with NBD is required to stabilize the ATP-bound conformation of NBD. Thus, ATP binding mainly induces a large rotation of the two lobes against each other to form a more closed conformation of the nucleotide-binding site. This is consistent with DnaK-ATP and Hsp110-ATP(Kityk et al., 2012; Liu and Hendrickson, 2007; Qi et al., 2013). This more closed conformation of NBD provides suitable surface for SBD subdomains and the inter-domain linker to form extensive contacts, and then propagates to SBD to cause striking conformational changes in both subdomains of SBD.
Figure 5. Comparisons of the BiP-ATP structure with the isolated domain structures.

a, Comparison of the NBD from the BiP-ATP structure with the isolated BiP NBD structure in complex with ADP (3IUC). Subdomain coloring for NBD of BiP-ATP is: Lobe I (blue), and Lobe II (red); for isolated BiP NBD structure: Lobe I (brown), and Lobe II (green). Left panel, the classic front-face view; right panel, top view of the left panel. NBDs are in backbone worm representation and superimposed on the basis of Lobe I Cα positions.
b, Superposition of the BiP-ATP structure to the BiP SBD-Tev–NR structure based on the Cα positions of SBDβ. Domain coloring for BiP-ATP is the same as Fig. 3a. BiP SBD-Tev–NR is colored in orange with the NR peptide highlighted in cyan.
c, Superposition of the BiP-ATP structure to the BiP SBD-L3,4′-Tev-NR structure based on the Cα positions of SBDβ. Domain coloring for BiP-ATP is the same as Fig. 3a. BiP SBD-L3,4′-Tev-NR is colored in purple with the NR peptide highlighted in cyan.
d,e, Close-up view of b and c, respectively. Only SBDβ domains are shown. The Cα atoms of R492 are shown as blue spheres.
f–h, Comparisons of polypeptide-binding site conformations. The polypeptide-binding site for BiP-ATP (f), superposition of f with BiP SBD-Tev–NR structure (g), and superposition of f with BiP SBD-L3,4′-Tev-NR structure (h) are shown in backbone worms representations. The superposition is based on Cαs in L1,2 and L4,5. Residues that form van der Waals contacts with NR peptide in BiP SBD-Tev–NR and SBD-L3,4′-Tev-NR are highlighted in stick representation.
It is well-established that the isolated SBD structures represent the ADP-bound and apo states(Bertelsen et al., 2009). With NR peptide bound, the polypeptide-binding site adopts a closed conformation in both the BiP-SBD and DnaK-SBD structures (Fig. 2a,b). The peptide-binding loops, L1,2 and L3,4, close on the bound NR peptide. Moreover, the polypeptide-binding site in SBDβ is covered up by SBDα. In contrast, the SBDα of the BiP-ATP structure is peeled away from covering the polypeptide-binding site (Fig. 3a,5b,c). The L1,2 side of the polypeptide-binding site including L1,2 and L4,5 assumes a nearly identical conformation as that of BiP-SBD; in contrast, the L3,4 side of the polypeptide-binding site is wide open: both L3,4 and L5,6 are flipped out and away, and the L3,4 side of β3 and β4 is shifted downward (Fig. 5b–e). L5,6 shifted as much as 16.1 Å at the Cα of R492. Thus, the van der Waals contacts with the NR peptide from this side of the polypeptide-binding site were abolished (Fig. 5f–h). The open conformation of the polypeptide-binding site is consistent with the low affinity and fast kinetics of peptide substrate binding in the ATP-bound state. We observed a similar open conformation in our DnaK-ATP structure(Qi et al., 2013), suggesting an overall conserved opening of the polypeptide-binding site elicited by ATP binding among both prokaryotic and eukaryotic Hsp70s. Consistent with the role of L3,4 and L5,6 in both peptide substrate binding and ATP-induced allosteric coupling, a previous study isolated two mutations in yeast BiP (Kar2), T473L (T453 in L3,4 of BiP) and P515L (P495 in L5,6 of BiP), which showed defects in both peptide substrate binding and allosteric coupling(Kabani et al., 2003).
Different Hsp40 interaction between BiP and DnaK when the polypeptide-binding pocket is open
Comparing the open conformation of the polypeptide-binding pocket in BiP-ATP with the close conformation in the isolated SBD structure, two glycine residues on L5,6, G486 and G493, changed their backbone conformations drastically (Table S1). We observed similar changes in DnaK-ATP with analogous G461 and G468 (Table S1). Previously, we have shown that these glycine residues were crucial for the ATP-induced opening of the polypeptide-binding pocket in DnaK(Qi et al., 2013). Thus, these two glycine residues in BiP most likely play the same crucial role for the opening of the polypeptide-binding site upon ATP binding. To test this hypothesis, we mutated both G486 and G493 to proline as for the DnaK-PP mutant (DnaK-G461P/G468P). We named this mutant BiP-PP. Like the DnaK-PP mutant, BiP-PP has reduced affinity for NR peptide and fast binding kinetics in the presence of ADP, as happens for WT BiP in the presence of ATP (Fig. 6a,b). This is different from the WT BiP in the presence of ADP where binding affinity is high and binding kinetics are slow. Thus, like the DnaK-PP mutant, the BiP-PP mutant locks BiP’s SBDβ into the ATP-bound conformation regardless of the ATP or ADP status of the NBD. In summary, these two glycine residues have a conserved essential role in the allosteric opening of the polypeptide-binding pocket in BiP as in DnaK.
Figure 6. The importance of two conserved glycine residues on L5,6 of Hsp70s.

a, Peptide binding affinity determined by fluorescence polarization assay. Assays were carried out in the presence of ADP (+ADP) or ATP (+ATP).
b, Fluorescence anisotropy assay of peptide substrate binding kinetics. The binding reactions of F-NR peptide were carried out in the presence of either ADP (+ADP) or ATP (+ATP), and the measurements were started right after mixing F-NR with the indicated protein.
c–g, NR peptide and Hsp40 stimulation of BiP and DnaK in a single turn-over ATPase assay. Fold of stimulation was calculated by setting the intrinsic ATPase activity as 1.
c, NR peptide failed to stimulate the ATPase activity of the BiP-PP mutant.
d, f, Neither ERdj3 (d) nor DnaJ (f) showed appreciable stimulation on the ATPase activity of the BiP-PP mutant.
e, g, The DnaK-PP protein manifested significant stimulation by both DnaJ (e) and ERdj3 (g).
It is well-established that peptide substrate binding stimulates the ATPase activity of Hsp70s(Flynn et al., 1989; Mayer and Bukau, 2005), the other half of the allosteric coupling. As expected, NR peptide stimulates the ATPase activity of BiP close to 15 fold at 400 μM in our hands (Fig. 6c). In contrast, BiP-PP showed little appreciable stimulation by NR peptide whereas the intrinsic ATPase rate was similar to that of the WT protein (Fig. 6c). At the same time, we observed similar results for the DnaK-PP mutant protein (data not shown). Therefore, peptide substrate stimulation of the ATPase activity requires the closure of the polypeptide-binding site as in the isolated SBD structures for both BiP and DnaK.
The chaperone activity of Hsp70s is further regulated by two classes of co-chaperones: Hsp40s and nucleotide-exchange factors (NEF)(Hartl and Hayer-Hartl, 2009; Hendrickson and Liu, 2008; Kampinga and Craig, 2010; Mayer and Bukau, 2005; Young, 2010). While NEFs facilitate the exchange of ADP for ATP, Hsp40s have been shown to specifically recognize the ATP-bound state of Hsp70s and stimulate the ATP hydrolysis step by Hsp70s. Since these two conserved glycine residues on L5,6 are crucial for the opening of the polypeptide-binding pocket in the ATP-bound state, we tested whether the open conformation of the polypeptide-binding pocket is essential for Hsp40 interaction. ERdj3, an abundant class I Hsp40 in ER, has been proposed to be an Hsp40 for BiP and shown to stimulate the ATPase activity of BiP(Jin et al., 2009; Otero et al., 2010; Tan et al., 2014). In our hands, ERdj3 stimulated the ATPase activity of BiP more than 20 fold at 4 μM (Fig. 6d). Intriguingly, ERdj3 failed to show any appreciable stimulation of the ATPase rate of BiP-PP (Fig. 6d), suggesting that the open conformation of the polypeptide-binding pocket of the ATP-bound state is not sufficient for ERdj3 stimulation. DnaJ, an Hsp40 co-chaperone for DnaK and founding representative of the class I Hsp40s, stimulates the ATPase rate of DnaK robustly, close to 60 fold at 0.4 μM in our hands (Fig. 6e). In contrast to ERdj3, DnaJ stimulates the ATPase rate of DnaK-PP drastically, about two thirds of that of WT, suggesting the open conformation of the polypeptide-binding pocket is sufficient for DnaJ stimulation of DnaK. Then we tested whether this different stimulation is due to different Hsp40s or Hsp70s. DnaJ stimulated BiP although to less extend as ERdj3; but failed to stimulate BiP-PP appreciably (Fig. 6f), consistent with the ERdj3 stimulation on BiP proteins. At the same time, ERdj3 stimulated DnaK’s ATPase activity robustly, and more interestingly, it also showed significant stimulation on DnaK-PP although to less extend than that of DnaJ (Fig. 6g). Thus, this difference in Hsp40 stimulation is most likely due to an intrinsic difference between BiP, a eukaryotic Hsp70, and DnaK, a prokaryotic Hsp70.
DISCUSSION
In this study, we present crystal structures of human BiP that represent its two functional states: BiP-ATP (ATP-bound state) and isolated BiP-SBD (ADP-bound state). The BiP-ATP structure is the first intact eukaryotic Hsp70 structure in the ATP-bound state, the allosterically active state. The overall similarity of BiP-ATP and DnaK-ATP structures suggests an overall conserved molecular mechanism of the ATP-induced allosteric coupling among Hsp70s in spite of sequence divergence, and different cellular locations and functions. At the same time, the isolated SBD of BiP assumes an overall structure almost identical to that of DnaK, suggesting an overall high conservation on peptide substrate binding among Hsp70s. The unique contacts with bound NR peptide from SBDα and L5,6 due to the smaller side chain of V429 provide a mechanistic explanation for the much slower peptide binding kinetics of BiP than those of DnaK. The most striking conservation between BiP and DnaK is the open conformation of the polypeptide-binding pocket in the ATP-bound structures and closed conformation in the isolated SBD structures (representing the ADP-bound state). Thus, opening of the polypeptide-binding site in the ATP-bound state and closing in the ADP-bound state during the chaperone cycle are conserved among Hsp70s.
Although the BiP-ATP structure shares overall similarity to the DnaK-ATP structure, there are significant differences on the conformation of SBDβ and the NBD-SBDα interface. With more hydrophobic contacts, the NBD-SBDα interface in BiP-ATP seems stronger than that in DnaK-ATP. Sequence alignment suggests this interface is more conserved among eukaryotic cytosolic/ER Hsp70s but is different from prokaryotic/mitochondrial Hsp70s. Moreover, R532 on Lα,β of BiP is highly conserved in eukaryotic cytosolic/ER Hsp70s but is missing in prokaryotic/mitochondrial Hsp70s. This study suggests that R532 forms contacts with NBD and plays a role in stabilizing the ATP-bound state in BiP. Thus, this arginine further strengthens the NBD-SBDα interface in the ATP-bound state. It is possible that the NBD-SBDα interface together with Lα,β plays a more important role in eukaryotic cytosolic/ER Hsp70s than in prokaryotic/mitochondrial Hsp70s in the ATP-induced allosteric coupling. The functional meaning of a stronger NBD-SBDα interface for eukaryotic Hsp70s in the cytosol and ER needs further exploration. It is interesting that sequence alignments on the regions with differences between BiP-ATP and DnaK-ATP suggest that Hsp70s can be divided into two subgroups: eukaryotic cytosolic/ER and prokaryotic/mitochondrial Hsp70s. It appears that BiP-ATP represents the eukaryotic cytosolic/ER Hsp70s while DnaK-ATP represents the prokaryotic/mitochondrial Hsp70s. Interestingly, the BiP-ATP structure is compatible with available data on its interaction with Ire1 in unfolded protein response (Fig. S4h).
Our ATPase assay suggests that closing of the polypeptide-binding pocket is required for the stimulation of the ATPase activity of Hsp70s by peptide substrates. Closing the polypeptide-binding pocket could lead to the dissociation of SBD from NBD, and freed NBD has a faster ATPase rate(Swain et al., 2007; Vogel et al., 2006), which could be the basis of peptide substrate stimulation of the ATPase activity(Kityk et al., 2012; Zhuravleva et al., 2012). In contrast, Hsp40 stimulation presents a more complicated picture. On the one hand, neither ERdj3 nor DnaJ showed appreciable stimulation on the ATPase activity of BiP-PP. On the other hand, both DnaJ and ERdj3 stimulated DnaK-PP significantly. It seems that there is significant difference between BiP and DnaK in interacting with Hsp40s. Consistent with this observation, ERdj3 had been reported to show significant interaction with BiP in the presence of ADP(Marcinowski et al., 2011); in contrast, several previous studies demonstrated that DnaK has little interaction with DnaJ in the presence of ADP(Mayer et al., 1999; Sarbeng et al., 2015; Suh et al., 1999). It is well-established that Hsp40s specifically recognize the ATP-bound state of Hsp70s. It has been proposed that Hsp40 has two binding sites on Hsp70: one is on the NBD, and the other is on SBD, at or near the polypeptide-binding site(Davis et al., 1999; Suh et al., 1998; Suh et al., 1999). For BiP, either the open conformation of the polypeptide-binding pocket is not enough for Hsp40s to bind or closing of the polypeptide-binding pocket is required for the stimulation. Since ERdj3 showed significant interaction with BiP in the ADP-bound state where the polypeptide-binding pocket is closed (Marcinowski et al., 2011), it is possible closing the polypeptide-binding pocket is required for ERdj3 stimulation on BiP. This is different from the DnaK-DnaJ interaction, for which interaction was detected only in the presence ATP. The ADP-bound state of DnaK with closed polypeptide-binding pocket may not be part of the requirement for this interaction. Thus, the open conformation of the polypeptide-binding pocket in the ATP-bound state may be sufficient. The functional implication of this difference in Hsp40 interaction waits for further studies.
METHODS
Protein expression and purification
The BiP-T229A-L3,4′ (residue 25–633) construct used for crystallization was cloned into a pSMT3 vector (a generous gift from Dr. Chris Lima, Sloan-Kettering Institute). After expressing as a Smt3 fusion protein in BL21(DE3)Gold, the fusion protein was first purified on a HisTrap column using 2XPBS buffer. The Smt3 tag was removed by Ulp1 protease. The BiP protein was separated from the Smt3 tag and Ulp1 protease on a second HisTrap column. After further purification using a HiTrap Q and Superdex 200 16/60 columns, the BiP protein was concentrated to ~ 40 mg/ml in a buffer containing 5 mM Hepes-KOH, pH7.5 and 10 mM KCl, and flash frozen in liquid nitrogen.
The two BiP SBD constructs (residue 418–636) used for crystallization, and BiP wild-type and mutant proteins used in biochemical assays were cloned, expressed and purified essentially the same way as that of the BiP-T229A-L3,4′.
DnaK, DnaJ, GrpE, and ERdj3 proteins were expressed and purified as described previously (Jin et al., 2009; Kumar et al., 2011; Sarbeng et al., 2015) (Details are included in the Supplementary Methods). The ERdj3 expression plasmid was a generous gift from Dr. Linda Hendershot.
Crystallization, data collection, and model building
All crystals were obtained with hanging-drop vapor diffusion method. The BiP-T229A-L3,4′ protein was diluted to 10 mg/ml with buffer A (5 mM Hepes-KOH, pH7.5, 10 mM KCl, 5 mM Mg(OAc)2 and 2 mM ATP), and crystals were obtained at 4°C w ith a mother liquor containing 8%–12% PEG 3000, 0.1 M acetate acid, pH 4.5, and 0.2 M zinc acetate. Before flash frozen in liquid nitrogen, crystals were treated with 0.25% glutaraldehyde for at least 12 hours and cryoprotected by 15% MPD (2-Methyl-2, 4-pentanediol) in the mother liquor. Crystals for SBD-Tev-NR and SBD-L3,4′-Tev-NR were grown at 20°C in similar conditions: 2.0 M ammonium sulfate, 0.1 M acetate acid, pH 4.5, and 2.67% acetonitrile for SBD-Tev-NR; and 2.5 M ammonium sulfate, 0.1 M acetate acid, pH 5.0, and 66.7 mM sodium malonate, pH 7.0 for SBD-L3,4′-Tev-NR. Both crystals were cryoprotected with 18–20% glycerol before flash frozen in liquid nitrogen. Selenomethionyl (SeMet) protein was prepared for SBD-L3,4-Tev-NR, and crystals were obtained in the same condition as the native protein.
All diffraction data sets were collected at Beamline X4C of NSLS, Brookhaven National Laboratory at 100 K with cryostream. Indexing, integration, and scaling of the diffraction data were performed using HKL2000. Model building was carried out in COOT.
A 3.0 Å resolution native dataset for BiP-T229A-L3,4′ was collected at wavelength 0.979 Å. Structure solution was obtained by molecular replacement with Phaser using our previously solved DnaK-ATP structure (PDB code: 4JNE) as search model. Refinement was carried out with Refmac. For SBD-L3,4′-Tev-NR, a single-wavelength anomalous diffraction (SAD) dataset was collected at Se peak with a SeMet crystal. Phases were evaluated with hkl2map, and structure was developed and refined with Phenix using a 2.68 Å native dataset. A native dataset at 2.57 Å resolution was collected from a SBD-Tev-NR crystal. Structure was solved by molecular replacement using Phaser with SBD-L3,4′-Tev-NR as search model. The refinement was carried out with Phenix.
Fluorescence anisotropy assays for peptide substrate binding affinity and kinetics
The assay was performed as described previously (Kumar et al., 2011; Xu et al., 2012). Details are in the Supplementary Methods.
Single-turnover ATPase assay
We used single-turnover ATPase assay to determine the ATP hydrolysis rates of BiP and DnaK. The assay was carried out as described before for DnaK(Kumar et al., 2011). Details are in the Supplementary Methods.
Supplementary Material
HIGHLIGHTS.
Crystal structure of an intact human BiP in the ATP-bound state
Crystal structure of isolated BiP SBD with a peptide substrate bound
The structures provided a structural explanation for allosteric coupling in Hsp70s
BiP has a unique NBD-SBD interface that is highly conserved only in eukaryotic Hsp70s
Acknowledgments
We thank Drs. Wayne Hendrickson, Elizabeth Craig, Wei Yang, Lois Greene, Diomedes Logothetis, Tricia Serio, Young-Jai You, and Leon Avery for critically reading the manuscript and providing insightful suggestions. We are grateful to staff at BNL Beamline X4C for their assistance in collecting diffraction data. We thank Dr. Linda Hendershot for the ERdj3 expression plasmid. This work was supported by NIH (1R01GM098592 to Q.L.) and Blick Scholar Award from VCU (to Q.L.). L.Z. is partially supported by 1RO1GM109193 from NIH.
Footnotes
Accession codes: Atomic coordinates and structure factors have been deposited in the RSCB Protein Data Bank under the accession number 5E84, 5E85, and 5E86.
AUTHOR CONTRIBUTIONS
J.Y., L.Z., and Q.L. designed the study. J.Y. carried out most experiments. All authors wrote or edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad A, Bhattacharya A, McDonald RA, Cordes M, Ellington B, Bertelsen EB, Zuiderweg ER. Heat shock protein 70 kDa chaperone/DnaJ cochaperone complex employs an unusual dynamic interface. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18966–18971. doi: 10.1073/pnas.1111220108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci U S A. 2009;106:8471–8476. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blond-Elguindi S, Cwirla SE, Dower WJ, Lipshutz RJ, Sprang SR, Sambrook JF, Gething MJ. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell. 1993;75:717–728. doi: 10.1016/0092-8674(93)90492-9. [DOI] [PubMed] [Google Scholar]
- Buchberger A, Theyssen H, Schroder H, McCarty JS, Virgallita G, Milkereit P, Reinstein J, Bukau B. Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. The Journal of biological chemistry. 1995;270:16903–16910. doi: 10.1074/jbc.270.28.16903. [DOI] [PubMed] [Google Scholar]
- Bukau B, Deuerling E, Pfund C, Craig EA. Getting newly synthesized proteins into shape. Cell. 2000;101:119–122. doi: 10.1016/S0092-8674(00)80806-5. [DOI] [PubMed] [Google Scholar]
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- Chambers JE, Petrova K, Tomba G, Vendruscolo M, Ron D. ADP ribosylation adapts an ER chaperone response to short-term fluctuations in unfolded protein load. The Journal of cell biology. 2012;198:371–385. doi: 10.1083/jcb.201202005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YW, Sun YJ, Wang C, Hsiao CD. Crystal structures of the 70-kDa heat shock proteins in domain disjoining conformation. The Journal of biological chemistry. 2008;283:15502–15511. doi: 10.1074/jbc.M708992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JE, Voisine C, Craig EA. Intragenic suppressors of Hsp70 mutants: interplay between the ATPase- and peptide-binding domains. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:9269–9276. doi: 10.1073/pnas.96.16.9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek J, Benedix J, Cappel S, Greiner M, Jalal C, Muller L, Zimmermann R. Functions and pathologies of BiP and its interaction partners. Cellular and molecular life sciences : CMLS. 2009;66:1556–1569. doi: 10.1007/s00018-009-8745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KM, DeLuca-Flaherty C, McKay DB. Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature. 1990;346:623–628. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- Flynn GC, Chappell TG, Rothman JE. Peptide binding and release by proteins implicated as catalysts of protein assembly. Science. 1989;245:385–390. doi: 10.1126/science.2756425. [DOI] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Quality control in the secretory pathway. Current opinion in cell biology. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science. 1997;276:431–435. doi: 10.1126/science.276.5311.431. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nature structural & molecular biology. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- Hendershot LM. The ER function BiP is a master regulator of ER function. The Mount Sinai journal of medicine, New York. 2004;71:289–297. [PubMed] [Google Scholar]
- Hendrickson WA, Liu Q. Exchange we can believe in. Structure. 2008;16:1153–1155. doi: 10.1016/j.str.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Jin Y, Zhuang M, Hendershot LM. ERdj3, a luminal ER DnaJ homologue, binds directly to unfolded proteins in the mammalian ER: identification of critical residues. Biochemistry. 2009;48:41–49. doi: 10.1021/bi8015923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabani M, Kelley SS, Morrow MW, Montgomery DL, Sivendran R, Rose MD, Gierasch LM, Brodsky JL. Dependence of endoplasmic reticulum-associated degradation on the peptide binding domain and concentration of BiP. Molecular biology of the cell. 2003;14:3437–3448. doi: 10.1091/mbc.E02-12-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nature reviews Molecular cell biology. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kityk R, Kopp J, Sinning I, Mayer MP. Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Molecular cell. 2012;48:863–874. doi: 10.1016/j.molcel.2012.09.023. [DOI] [PubMed] [Google Scholar]
- Kumar DP, Vorvis C, Sarbeng EB, Cabra Ledesma VC, Willis JE, Liu Q. The four hydrophobic residues on the Hsp70 inter-domain linker have two distinct roles. Journal of molecular biology. 2011;411:1099–1113. doi: 10.1016/j.jmb.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Zhang P, Murphy ME, Marmorstein R, George DL. Structural basis for the inhibition of HSP70 and DnaK chaperones by small-molecule targeting of a C-terminal allosteric pocket. ACS chemical biology. 2014;9:2508–2516. doi: 10.1021/cb500236y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebscher M, Roujeinikova A. Allosteric coupling between the lid and interdomain linker in DnaK revealed by inhibitor binding studies. Journal of bacteriology. 2009;191:1456–1462. doi: 10.1128/JB.01131-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Hendrickson WA. Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell. 2007;131:106–120. doi: 10.1016/j.cell.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. ER chaperone functions during normal and stress conditions. Journal of chemical neuroanatomy. 2004;28:51–65. doi: 10.1016/j.jchemneu.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Macias AT, Williamson DS, Allen N, Borgognoni J, Clay A, Daniels Z, Dokurno P, Drysdale MJ, Francis GL, Graham CJ, et al. Adenosine-derived inhibitors of 78 kDa glucose regulated protein (Grp78) ATPase: insights into isoform selectivity. Journal of medicinal chemistry. 2011;54:4034–4041. doi: 10.1021/jm101625x. [DOI] [PubMed] [Google Scholar]
- Mapa K, Sikor M, Kudryavtsev V, Waegemann K, Kalinin S, Seidel CA, Neupert W, Lamb DC, Mokranjac D. The conformational dynamics of the mitochondrial Hsp70 chaperone. Molecular cell. 2010;38:89–100. doi: 10.1016/j.molcel.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Marcinowski M, Holler M, Feige MJ, Baerend D, Lamb DC, Buchner J. Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nature structural & molecular biology. 2011;18:150–158. doi: 10.1038/nsmb.1970. [DOI] [PubMed] [Google Scholar]
- Marcinowski M, Rosam M, Seitz C, Elferich J, Behnke J, Bello C, Feige MJ, Becker CF, Antes I, Buchner J. Conformational selection in substrate recognition by Hsp70 chaperones. Journal of molecular biology. 2013;425:466–474. doi: 10.1016/j.jmb.2012.11.030. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cellular and molecular life sciences : CMLS. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Laufen T, Paal K, McCarty JS, Bukau B. Investigation of the interaction between DnaK and DnaJ by surface plasmon resonance spectroscopy. Journal of molecular biology. 1999;289:1131–1144. doi: 10.1006/jmbi.1999.2844. [DOI] [PubMed] [Google Scholar]
- Otero JH, Lizak B, Hendershot LM. Life and death of a BiP substrate. Seminars in cell & developmental biology. 2010;21:472–478. doi: 10.1016/j.semcdb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi R, Sarbeng EB, Liu Q, Le KQ, Xu X, Xu H, Yang J, Wong JL, Vorvis C, Hendrickson WA, et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nature structural & molecular biology. 2013;20:900–907. doi: 10.1038/nsmb.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudiger S, Buchberger A, Bukau B. Interaction of Hsp70 chaperones with substrates. Nature structural biology. 1997;4:342–349. doi: 10.1038/nsb0597-342. [DOI] [PubMed] [Google Scholar]
- Sarbeng EB, Liu Q, Tian X, Yang J, Li H, Wong JL, Zhou L, Liu Q. A functional DnaK dimer is essential for the efficient interaction with Hsp40 heat shock protein. The Journal of biological chemistry. 2015;290:8849–8862. doi: 10.1074/jbc.M114.596288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid D, Baici A, Gehring H, Christen P. Kinetics of molecular chaperone action. Science. 1994;263:971–973. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- Shi L, Kataoka M, Fink AL. Conformational characterization of DnaK and its complexes by small-angle X-ray scattering. Biochemistry. 1996;35:3297–3308. doi: 10.1021/bi951984l. [DOI] [PubMed] [Google Scholar]
- Suh WC, Burkholder WF, Lu CZ, Zhao X, Gottesman ME, Gross CA. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15223–15228. doi: 10.1073/pnas.95.26.15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh WC, Lu CZ, Gross CA. Structural features required for the interaction of the Hsp70 molecular chaperone DnaK with its cochaperone DnaJ. The Journal of biological chemistry. 1999;274:30534–30539. doi: 10.1074/jbc.274.43.30534. [DOI] [PubMed] [Google Scholar]
- Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Molecular cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YL, Genereux JC, Pankow S, Aerts JM, Yates JR, 3rd, Kelly JW. ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher’s disease. Chemistry & biology. 2014;21:967–976. doi: 10.1016/j.chembiol.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel M, Mayer MP, Bukau B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. The Journal of biological chemistry. 2006;281:38705–38711. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- Wei J, Gaut JR, Hendershot LM. In vitro dissociation of BiP-peptide complexes requires a conformational change in BiP after ATP binding but does not require ATP hydrolysis. The Journal of biological chemistry. 1995;270:26677–26682. doi: 10.1074/jbc.270.44.26677. [DOI] [PubMed] [Google Scholar]
- Wilbanks SM, Chen L, Tsuruta H, Hodgson KO, McKay DB. Solution small-angle X-ray scattering study of the molecular chaperone Hsc70 and its subfragments. Biochemistry. 1995;34:12095–12106. doi: 10.1021/bi00038a002. [DOI] [PubMed] [Google Scholar]
- Wisniewska M, Karlberg T, Lehtio L, Johansson I, Kotenyova T, Moche M, Schuler H. Crystal structures of the ATPase domains of four human Hsp70 isoforms: HSPA1L/Hsp70-hom, HSPA2/Hsp70–2, HSPA6/Hsp70B’, and HSPA5/BiP/GRP78. PloS one. 2010;5:e8625. doi: 10.1371/journal.pone.0008625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Sarbeng EB, Vorvis C, Kumar DP, Zhou L, Liu Q. Unique peptide substrate binding properties of 110-kDa heat-shock protein (Hsp110) determine its distinct chaperone activity. The Journal of biological chemistry. 2012;287:5661–5672. doi: 10.1074/jbc.M111.275057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC. Mechanisms of the Hsp70 chaperone system. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2010;88:291–300. doi: 10.1139/o09-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuravleva A, Clerico EM, Gierasch LM. An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell. 2012;151:1296–1307. doi: 10.1016/j.cell.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.