

Abstract
Mutations in protein kinase C substrate 80K-H (PRKCSH), which encodes for an 80 KDa protein named hepatocystin (80K-H, PRKCSH), gives rise to polycystic liver disease (PCLD). Hepatocystin functions as the noncatalytic beta subunit of Glucosidase II, an endoplasmic reticulum (ER)-resident enzyme involved in processing and quality control of newly synthesized glycoproteins. Patients harboring heterozygous germline mutations in PRKCSH are thought to develop renal cysts as a result of somatic loss of the second allele, which subsequently interferes with expression of the TRP channel polycystin-2 (PKD2). Deletion of both alleles of PRKCSH in mice results in embryonic lethality before embryonic day E11.5. Here, we investigated the function of hepatocystin during Xenopus laevis embryogenesis and identified hepatocystin as a binding partner of the TRPM7 ion channel, whose function is required for vertebrate gastrulation. We find that TRPM7 functions synergistically with hepatocystin. Although other N-glycosylated proteins are critical to early development, overexpression of TRPM7 in Xenopus laevis embryos was sufficient to fully rescue the gastrulation defect caused by loss of hepatocystin. We observed that depletion of hepatocystin in Xenopus laevis embryos decreased TRPM7 expression, indicating that the early embryonic lethality caused by loss of hepatocystin is mainly due to impairment of TRPM7 protein expression.
In this study we investigated the role of hepatocystin during embryogenesis and its functional relationship to the TRPM7 ion channel, which our studies found to biochemically and functionally interact with hepatocystin. TRPM7 is a divalent-selective channel permeable to Mg2+, Ca2+, as well as trace metal ions such as Zn2+ 1,2. The channel is bifunctional, containing an alpha kinase domain at its COOH-terminus3. Global deletion of TRPM7 from mice results in early embryonic lethality by E7.54. In Xenopus laevis, TRPM7′s channel, but not its kinase, was found to be critical to the regulation of gastrulation and neural fold closure during Xenopus laevis embryogenesis5.
In the ER, hepatocystin functions as the noncatalytic beta subunit of Glucosidase II, which catalyzes glucose trimming of newly synthesized glycoproteins and is involved in ER protein quality control6,7,8. Glucosidase II cleaves the first glucose prior to entry of a glycoprotein into the calnexin/calreticulin cycle9,10,11,12. If a glycoprotein is properly folded, Glucosidase II cleaves off a second glucose residue to allow for transition of the glycoprotein out of the ER to the Golgi apparatus. If however the glycoprotein is improperly folded, it is ultimately targeted for degradation by the ER-associated degradation (ERAD) pathway9,12. Hepatocystin contains an NH2-terminal signal sequence for translocation across the ER membrane, a low-density lipoprotein class A domain, two Ca2+-binding EF hands, a glutamic acid-rich segment, a region with low homology to the mannose 6-phosphate receptor (MRH), and a COOH-terminal His-Asp-Glu-Leu (HDEL) ER retention signal (Fig. 1A)7. Hepatocystin’s ER luminal retention signal is required for the function of the glucosidase II holoenzyme13 Genetic loss of hepatocystin causes autosomal dominant polycystic liver disease (ADPLD), which is characterized by bile duct cyst formation throughout the liver parenchyma14,15. It is hypothesized that liver cyst development is driven by somatic second hit mutations that affect wild type alleles of biliary type cells during early hepatic organogenesis16. Loss of hepatocystin impairs glucosidase II-dependent glucose trimming of critical N-glycoslyated proteins, such as the TRP channel PKD2, which have been shown to function in a genetic interaction network with PRKCSH17,18. Here we report an analogous role for hepatocystin in supporting the expression and function of the TRPM7 ion channel during early embryonic development.
Figure 1. Hepatocystin interacts with TRPM7.

(A) Schematics of the linear structures and domains of TRPM7 and hepatocystin. TRPM7 contains an NH2-terminus melastatin domain, six transmembrane domain, TRP box, as well as coiled-coil (CC), serine/threonine rich domain (ST), and functional alpha kinase (KIN) domains. Hepatocystin contains a leader signal peptide (SP), a low-density lipoprotein class A (LDL) domain, two Ca2+-binding EF hands, a glutamic acid-rich segment (GLUT), a region with low homology to the mannose 6-phosphate receptor (MRH), and a COOH-terminal His-Asp-Glu-Leu (HDEL) ER retention signal. (B) Directed yeast-two-hybrid assay using the indicated fragments of TRPM7 COOH-terminus fused to LexA as bait and Gal4 fused residues 346-528 of hepatocystin as prey. LexA fused to Laminin (Lex-Lam) served as a negative control. (C) Pulldown purification assay using GST fused to residues 346-528 of hepatocystin (GST-HepCT) against HEK-293 cell lysates containing the indicated mfGFP-tagged TRPM7 COOH-terminal fragments. (D) Endogenous hepatocystin co-immunoprecipitates with HA-tagged TRPM7 heterologously expressed in HEK-293T cells. (E) Immunocytochemistry of HEK-293T cells transfected with HA-tagged TRPM7 and FLAG-tagged hepatocystin (Green) show that the two proteins co-localize in the endoplasmic reticulum (ER). A scale bar of 20 microns in length is indicated.
Results
Hepatocystin is a TRPM7 binding protein
To identify potential regulatory binding partners of TRPM7 we conducted a yeast two hybrid (Y2H) screen of a rat brain library using the COOH-terminus of TRPM7, which contains a coiled-coil domain, serine/threonine rich domain and functional alpha kinase domain (Fig. 1A). The screen identified a number of potential interacting partners (Supplementary Table I), including a COOH-terminal fragment of hepatocystin (residues 346–528) containing the protein’s MRH and HDEL motifs. We then used a directed Y2H assay to confirm the interaction between TRPM7′s COOH-terminus and hepatocystin and showed that TRPM7′s coiled-coil and alpha-kinase domains do not interact with hepatocystin (Fig. 1B). This assay also revealed that a kinase-inactive mutant (K1646R) of TRPM7 is also capable of interacting with hepatocystin, indicating that a functional kinase domain is not required for the protein-protein interaction. We next used a pull-down purification assay using GST fused to residues 346–528 of hepatocystin (GST-hepatocystin) to further delineate the region within TRPM7 COOH-terminus that interacts with hepatocystin. GST-hepatocystin, but not the GST negative control, interacted with the full length TRPM7 COOH-terminus (GFP-CTERM) and the COOH-terminus lacking the coiled-coil (GFP-CTERMΔCC), but interacted more weakly with the isolated coiled-coil domain (GFP-CC) (Fig. 1C) and serine-threonine-rich domains (GFP-ST). Similar to what was observed in the directed Y2H assay, GST-hepatocystin did not interact with the isolated kinase TRPM7 domain (GFP-KIN). These results indicate that hepatocystin interacts with TRPM7′s ST domain. As the ST-domain has been implicated in kinase substrate recognition by TRPM7, we investigated whether TRPM7 phosphorylates hepatocystin19. However, an in vitro kinase assay demonstrated that compared to a commonly used substrate for protein kinases, myelin basic protein (MBP), hepatocystin is not a good substrate for TRPM7′s kinase (Supplementary Fig. 1). We next determined whether the full-length proteins interacted in vivo and found that endogenous hepatocystin co-immunoprecipitated with TRPM7 heterologously expressed in HEK-293 cells (Fig. 1D). Since hepatocystin contains an ER-retention signal and has been shown to be primarily expressed in the ER, we then investigated whether TRPM7 co-localized with hepatocystin to the ER7. Immunocytochemistry analysis of transiently transfected HA-tagged TRPM7 expressed in HEK-293 cells and FLAG-tagged hepatocystin revealed co-localization of both proteins to the ER (Fig. 1E). Collectively, these results indicate that TRPM7 likely encounters and interacts with hepatocystin in the ER. Previous studies have demonstrated that homozygous deletion of either gene from mice is embryonically lethal4,17. As our prior investigation of the role of TRPM7 during early development uncovered a role for TRPM7 channel activity in gastrulation cell movements and neural fold closure, this motivated us to more closely investigate the function of hepatocystin during embryogenesis and to determine whether the function of the two proteins functionally intersect during early development5.
Hepatocystin is required for gastrulation during Xenopus laevis embryogenesis
Deletion of PRKCSH in mice results in embryonic lethality before embryonic day E11.5, however, the reason for this has not been determined17. To investigate the role of hepatocystin during early embryogenesis, we first cloned Xenopus laevis hepatocystin (Xhepatocystin), a protein that shares a sequence identity of 61% with the human protein. Xhepatocystin domain architecture (Fig. 1A) is similar to that of its vertebrate orthologs, exhibiting high sequence identity and similarity within its 6 structural domains or motifs. We next examined the temporal and spatial expression patterns of Xhepatocystin mRNA during Xenopus development. RT-PCR analysis demonstrated ubiquitous expression of Xhepatocystin maternally to the tadpole stage (Fig. 2A). In situ hybridization of gastrula stage embryos (stage 10.5) showed expression of Xhepatocystin in dorsal ectoderm and dorsal mesoderm (Fig. 2B). In neurula stage embryos (stage 18 & 23) Xhepatocystin was highly expressed in the neural plate and anterior neural fold. At the early tadpole stage, Xhepatocystin was expressed in the head, neural tube and notochord. Later in development, strong expression was observed in head, eyes, spinal cord, notochord, liver and kidney. In our previous study, we also observed ubiquitous expression of Xenopus laevis TRPM7 (XTRPM7) maternally to the tadpole stage by RT-PCR and in situ hybridization showed that XTRPM7 is similarly expressed in the neural plate at the neurula stage and in kidney at tadpole stage5. The overlapping expression patterns of Xhepatocystin and XTRPM7 indicated the potential for these two proteins to functionally interact in vivo.
Figure 2. Expression pattern of Xhepatocystin during early development.

(A) Xhepatocystin is expressed throughout embryogenesis as monitored by RT-PCR analysis. Ornithine decarboxylase (ODC) was used as a loading control. (B) Expression pattern of Xhepatocystin at selected developmental stages as analyzed by whole-mount in situ hybridization using a Xhepatocystin anti-sense probe. A sense probe was used as a negative control. Arrows indicate the region where the embryo was vertically sectioned and examined. ST: stage. Xhepatocystin was expressed in dorsal ectoderm and dorsal mesoderm. In neurula stage embryos (stage 18 & 23) Xhepatocystin was highly expressed in the neural plate and anterior neural fold. At the early tadpole stage, Xhepatocystin was found expressed in the head, neural tube and notochord. Later in development, strong expression was observed in head, eyes, spinal cord, notochord, liver and kidney.
In our earlier study, we reported a requirement for TRPM7 in controlling gastrulation cell movements and neural fold closure during Xenopus laevis embryogenesis5,20. We found that during gastrulation TRPM7 specifically regulates cell polarity and migration during convergent extension movements by controlling the activation of Rac during non-canonical Wnt signalling. We next sought to determine whether Xhepatocystin possesses a similar regulatory role. To accomplish this we depleted Xhepatocystin protein by injection of anti-sense morpholino oligonucleotide into embryos at the 4-cell stage (Xhepatocystin MO; Fig. 3A) and observed its effect during development. The Xhepatocystin MO effectively inhibited translation of a Myc-tagged Xhepatocystin construct when injected into Xenopus embryos (Fig. 3B). Injection of Xhepatocystin MO into the dorsal marginal zones of the four-cell embryo dose-dependently impaired axis extension and interfered with neural tube closure, while injection of a similar dose of a control MO had no significant effect (Fig. 3C&D). The phenotype produced by dorsal injection of the Xhepatocystin MO was divided into two classes: severe and mild (Fig. 3E). In the severe class, axial extension was impaired, resulting in an extreme dorsal-flexure in the embryos, and the neural tube failed to close. In addition, anterior structures such as the head and eyes were reduced in size. In the mild class, there was delay in neural tube closure and axis extension was mildly impaired, which resulted in short and dorsally curved embryos. The penetrance of the Xhepatostatin MO was relatively mild (approximately 50 percent), which was likely due to the high maternal expression of Xhepatocystin (Fig. 2A). Nevertheless, Xhepatocystin MO induced gastrulation defects that could be rescued by co-injection of mouse hepatocystin RNA, which does not contain the MO binding site (Fig. 3D). Importantly, co-injection of LacZ RNA did not rescue the Xhepatocystin-induced gastrulation defects, indicating the specificity of Xhepatocystin MO. Dosages of the Xhepatocystin MO of 100 ng and above produced non-specific toxic effects, which could not be rescued by the hepatocystin RNA. Collectively, these results demonstrate a critical and specific role for Xhepatocystin for gastrulation and neural fold closure.
Figure 3. Xhepatocystin is required for gastrulation.

(A) Schematic diagram of Xhepatocystin morpholino (MO) binding sites upstream and overlapping the start site of Xhepatocystin. (B) Western blot analysis shows that injection of the Xhepatocystin MO (50 and 75 ng), but not the control MO (75 ng), can effectively inhibit translation of Myc-Xhepatocystin 5′ UTR injected RNA (250 pg). β-tubulin is shown as a loading control. (C) Injection of Xhepatocystin MO into the dorsal blastomeres of the 4-cell stage embryos inhibited gastrulation. The gastrulation defect phenotype was rescued by co-injection of mouse hepatocystin RNA and mouse TRPM7 RNA (125 pg), but not LacZ RNA (125 pg), with the Xhepatocystin MO (75 ng). Injection of the control MO (75 ng) produced no phenotype. (D) Quantification of phenotypic results from (C). Phenotypes were scored according to the severity of the gastrulation defect (GD) at the tadpole stage (E). The injections were repeated at least three times. The ability of co-injection of mouse hepatocystin and mouse TRPM7 RNA with Xhepatocystin MO to rescue the gastrulation phenotype caused by the Xhepatocystin MO was statistically significant (*P<0.05). The collective total number of injected embryos from all experiments is indicated above each bar.
Given the similarity between the spatial and temporal expression pattern of Xhepatocystin and XTRPM7 and the gastrulation defects phenotypes produced by depletion of either protein, we next asked whether two proteins functionally interact. To address this question, embryos were injected with sub-threshold levels of the Xhepatocystin MO or XTRPM7 MOs. Injection of fifteen nanograms of Xhepatocystin MO or of TRPM7 MO with 15 ng Control MO caused mild gastrulation defects in 10–20 percent of the embryos. However, when Xhepatocystin MO and XTRPM7 MOs were co-injected, the number of defected embryos increased to 80 percent – more than the 43 percent that would be expected if the effect were additive (Fig. 4A,B). The synergistic effect produced by co-injection of sub-threshold levels Xhepatocystin and XTRPM7 MOs suggested that Xhepatocystin and TRPM7 function in the same pathway. Thus, we next asked whether expression of TRPM7 could rescue the gastrulation phenotype caused by the Xhepatocystin MO. Surprisingly, given the number of N-glycosylated proteins, such as Frizzled receptors, that function to control gastrulation and neural fold closure, co-injection of mTRPM7 RNA with the Xhepatocystin MO was able to fully rescue the embryonic defect with a similar efficiency as co-injection of murine hepatocystin mRNA with the Xhepatocystin MO (Fig. 3D, lane 3–5)21. These results indicate that the gastrulation and neural fold closure defects caused by depletion of hepatocystin was primarily driven by disruption of TRPM7 function, but the mechanism involved still remained unclear.
Figure 4. TRPM7 and hepatocystin functionally interact.
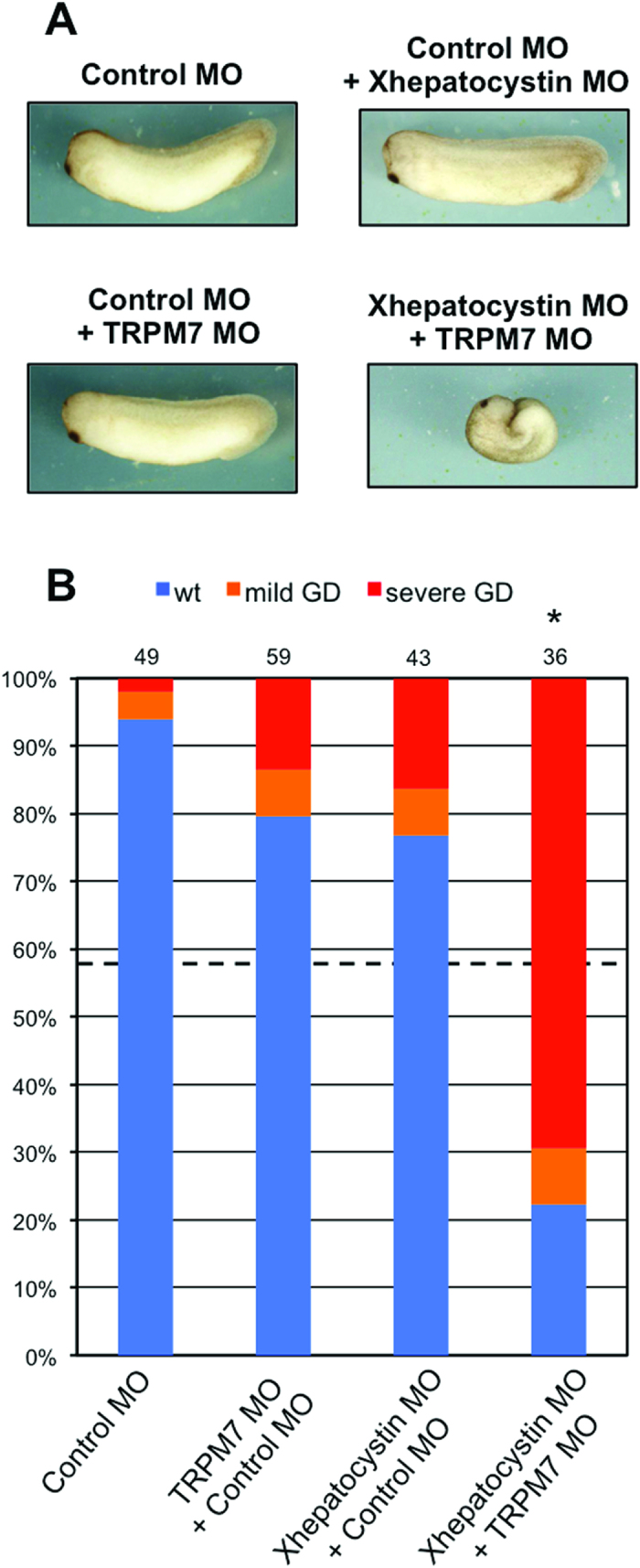
(A,B) Co-injection of Xhepatocystin MO (15 ng) and XTRPM7 MO (15 ng) synergistically inhibit gastrulation, but have little or no effect when injected separately. (B) Phenotypes were scored according to the severity of the gastrulation defect (GD) at the tadpole stage. The dotted line in (B) indicates the amount of gastrulation defects that would be expected if the effects were additive (43%). The injections were repeated three times. Statistic analysis indicated that the number of embryos exhibiting gastrulation defects (77%) by co-injection of Xhepatocystin and XTRPM7 MOs was significantly different (P=0.05) from what would be expected if the effects were additive (43%). The collective total number of injected embryos from all three experiments is indicated above each bar.
Hepatocystin augments TRPM7 protein expression
Glucosidase II is a soluble ER-resident heterodimer composed of two subunits6,8. Glucosidase II’s α subunit (GIIα) contains the glycosyl hydrolase active site, whereas Glucosidase II’s β subunit (hepatocystin) is required to sustain efficient N-glycan trimming by the interaction of its MRH domain and mannoses in the glycans13. The glycan structure in glycoproteins determines whether a glycoprotein is retained in the ER, sorted to its final normal destination or retrotranslocated to the cytosol to be degraded by proteasomes10,11,12,22,23. Thus, hepatocystin plays a determinative role in the quality control of glycoprotein folding in the ER. For example, loss of hepatocystin function impaired glucose trimming of PKD2, which likely accounts for its decreased expression in Prkcsh−/− mice and the genetic interaction observed between PKD2 and PRKCSH in ADPLD17,18.
TRPM7 is reported to be N-glycosylated24. Given hepatocystin role in biogenesis of N-glycosylated proteins, we hypothesized that cellular depletion of hepatocystin could negatively impact TRPM7 protein expression. To examine the role of N-glycosylation in controlling TRPM7 expression and activity, we applied 5 μg/ml of tunicamycin to HEK-293 cells expressing TRPM7. The antibiotic tunicamycin selectively inhibits N-glycoslyation of membrane proteins and has previously been reported to interfere with TRPM7 N-glycosylation24,25. Tunicamycin treatment of 293-TRPM7 cells, transiently expressing TRPM7 in response to tetracycline, reduced channel expression (Fig. 5A,B), indicating that TRPM7 N-glycoslyation is important for the channel’s stability. We next investigated whether depletion of Xhepatocystin affects TRPM7 expression in Xenopus laevis embryos. Co-injection of Xhepatocystin MO with TRPM7 mRNA reduced TRPM7 protein expression in a dose dependent manner (Fig. 5C,D). Collectively, these results indicate a supportive role for Xhepatocystin in promoting TRPM7 protein expression and function during Xenopus laevis embryogenesis.
Figure 5. Hepatocystin and N-glycosylation affect the abundance of TRPM7.
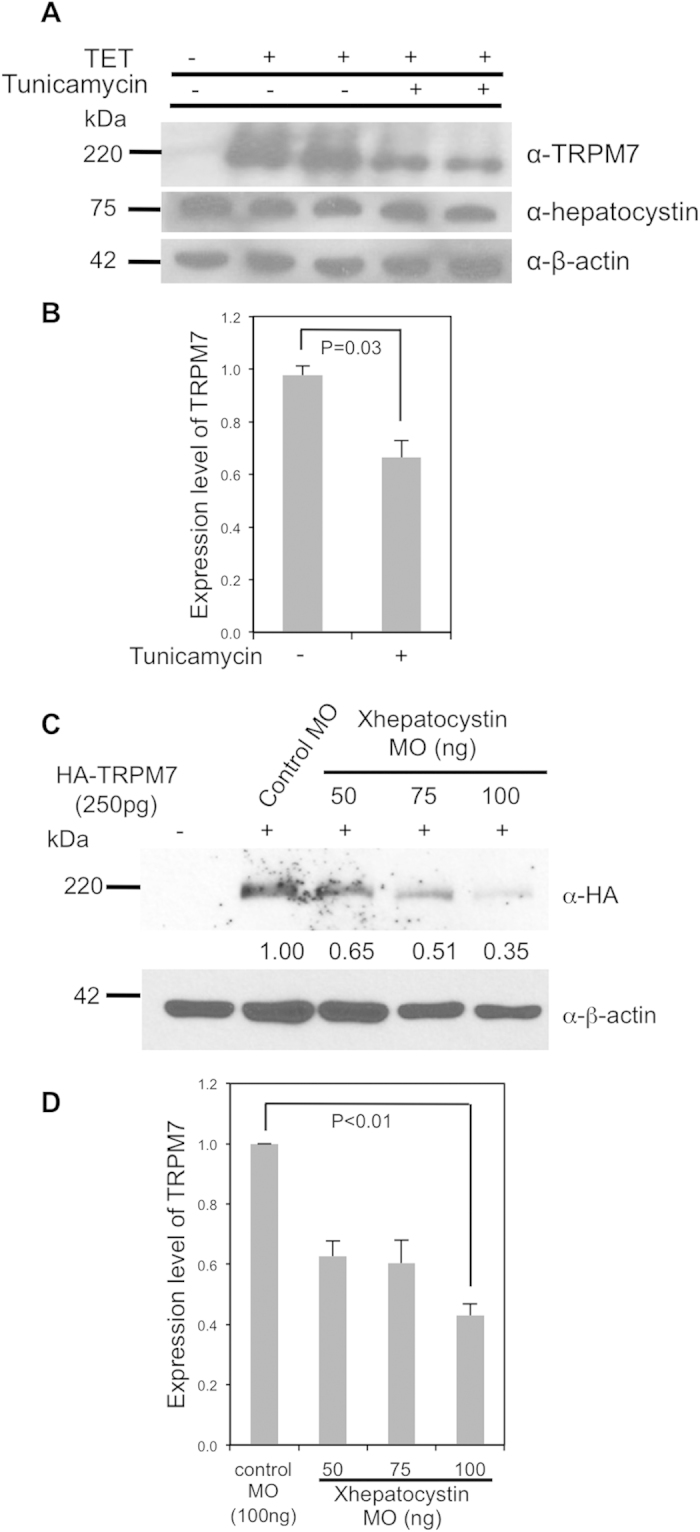
(A) Treatment of cells with tunicamycin (5 μg/ml) for 24 hours decreased tetracycline-induced TRPM7 protein expression in 293-TRPM7 cells. (B) Quantification of data from (A). (C) Co-injection of increasing dosages of the Xhepatocystin MO with 250 pg mRNA of HA-tagged mTRPM7 (HA-TRPM7) decreases TRPM7 expression in Xenopus embryos at stage 17. β-actin is shown as a loading control. (D) Quantification of data from (C). The experiments were repeated at least three times.
Discussion
Our studies have uncovered a critical role for hepatocystin in supporting the function of the TRPM7 ion channel during embryogenesis. Like TRPM7, Xhepatocystin is ubiquitously expressed maternally to the tadpole stage and displayed a similar expression pattern as TRPM7 throughout development. In addition, depletion of Xhepatostatin in Xenopus laevis also produced gastrulation and neural fold closure defects, which offers explanation for the embryonic lethality of homozygous deletion of PRKCSH from mice. These results are also consistent with studies of PRKCSH in Zebrafish, where depletion of PRKCSH causes dorsal-flexure of the embryo, indicative of convergent extension defects, and situs inversus and pronephric cysts later in development26. The overlapping expression patterns of Xhepatocystin and XTRPM7 indicated the potential for these two proteins to functionally interact. Indeed, depletion of either Xhepatocystin or XTRPM7 interfered with gastrulation and neural fold closure and gastrulation phenotype produced by depletion of Xhepatocystin could be rescued by co-injection of mTRPM7 RNA. In addition, simultaneous depletion of XTRPM7 and Xhepatocystin by co-injection of subthreshold levels of their respective morpholinos produced a synergistic effect on the gastrulation phenotype, indicating that PRKCSH and TRPM7 are epistatic and that the function of TRPM7 is dependent upon that of hepatocystin.
Depletion of Xhepatocystin in Xenopus embryos interfered with TRPM7 protein expression. Based on what is known regarding hepatocystin’s established function as the β-subunit of Glucosidase II, N-glycosylation and glycan trimming through Glucosidase II likely regulates the abundance of TRPM7 by limiting the channel’s turnover. Indeed, treatment of HEK-293 cells with tunicamycin decreased TRPM7 expression. Together these results provide a basis for further studies addressing the role of glycosylation in TRPM7 function.
Mutations in PRKCSH lead to Autosomal Dominant Polycystic Liver Disease (ADPLD), a rare hereditary disorder characterized by slowly progressive cyst formations. In addition to defects in PRKCSH, mutations in SEC63 can also produce ADPLD27,28. SEC63 is an ER protein involved in mediating the translocation of nascent or newly synthesized polypetides across the ER membrane of cells. Thus both proteins implicated in ADPLD are involved in translocation through the ER membrane and in the oligosaccharide processing of newly synthesized glycoproteins16. Interestingly, only a few hepatic cysts are detectable in affected individuals in the first four decades of life, but hundreds of individual cysts can be found at a later stage16. It has been hypothesized that ADPLD cysts arise as a result of a cellular recessive two-hit mechanisms16. In the case of a germline PRKCSH mutation in ADPLD, the second hit could involve a somatic PRKCSH or SEC63 mutation on the other allele, or somatic hits on genes implicated in Autosomal Dominant Polycystic Kidney Disease (ADPKD), such as PKD1 and PKD216. Mutations in PRKCSH or SEC63 only account for 16–22% of cases of ADPLD, indicating that other genes contribute to this disease29.
Deletion of TRPM7 from mouse kidney leads to kidney cysts and a defect in nephrogenesis30. Whether, loss of TRPM7 function in liver contributes to the pathogenesis of ADPLD in response to PRKCSH deletion remains unknown. Recently, disruption of the Wnt pathway via mutations in LRP5 was shown to be associated with hepatic cystogenesis31. In our previous work we demonstrated that TRPM7 functions within the non-canonical Wnt pathway during gastrulation and neural fold closure to regulate convergent extension cell movements5. This cellular process has also been shown to be required for tubulogenesis32. It has been speculated that malfunction of the non-canonical Wnt pathway could potentially result in tubule enlargement rather than tubule elongation33. Therefore, more studies are needed to assess the role of both the non-canonical Wnt pathway and TRPM7 in cystogenesis.
Around 30% of proteins are reported to undergo ER translocation and are subject to folding and quality control by hepatocystin and Sec63p34. This raises the question of the extent of the fidelity of hepatocystin’s interaction with TRPM7. TRPM7 is among only a select number of proteins, including TRPV4 and the inositol 1,4,5-trisphosphate receptor, that have been identified as hepatocystin binding partners via a yeast two-hybrid screen23,35,36. One possibility is that proteins that more specifically interact with hepatocystin may be subject to an additional layer of signalling control. Alternatively, binding of hepatocystin to TRPM7 may help target Glucosidase II activity towards the channel. Future studies will focus on how glycosylation affects TRPM7 function and how Glucosidase II is specifically impacting the channel.
Methods
Reagents
All cell culture reagents, unless otherwise stated, were from Life Technologies (Carlsbad, CA). All chemicals, unless otherwise stated were from Sigma Aldrich (St. Louis, MO). Carbenicillin, kanamycin, 5-Bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-gal), isopropyl-beta-D-thiogalactopyranoside (IPTG) were purchased from Gold Biotechnology (St. Louis, MO).
Plasmids and oligonucleotides
The pOG1 vector encoding human TRPM7 was previously described37. Murine hepatocystin in the pCMV-SPORT6 vector (Accession #: BC009816) was from GE Life Sciences (Piscataway, NJ). COOH-terminus FLAG-tagged hepatocystin was cloned into pcDNA5/FRT/TO (Life Technologies, Inc; Grand Island, NY) from the pCMV-SPORT6 vector using primers described in Supplementary Methods file.) To conduct pulldown purification assays we fused fragments of TRPM7′s COOH-terminus to a multifunctional Green Fluorescent Protein (mfGFP) in which GFP was engineered to contain a streptavidin binding peptide (SBP) tag, octa-histidine-tag, and c-Myc-tag in tandem into a loop of GFP38. mfGFP was subcloned into the NheI and HindIII sites of pcDNA6-V5-HisB to make pcDNA6-mfGFP. mfGFP-fusion proteins of TRPM7-COOH terminal fragments were subcloned by PCR into pcDNA6-mfGFP using pcDNA5/FRT/TO-HA-TRPM7 as a template39. The list of primers employed to make the mfGFP constructs are described in the Supplementary Methods file.
Yeast two-hybrid (Y2H) screen and directed Y2H assay
The COOH-terminus of TRPM7 (residues 1120–1862) was subcloned into pBMT116 LexA vector to make the “bait” vector LexA-CTERM. A rat brain prey library in the pACT2 AD vector was purchased from Clontech Laboratories, Inc (Mountainview, CA). A yeast-two-hybrid screen was conducted following the manufacturer’s protocol for large-scale transformation. Positive interactions were identified by growth on selective media and a beta galactosidase assay before sequencing of purified plasmids. To conduct the directed Y2H beta galactosidase assay, the coiled-coil (CC) domain (residues 1120-1288) and kinase (KIN) domain (residues 1597–1862) of TRPM7 were subcloned into pBMT116 to make LexA-CC and LexA-KIN. The kinase-inactive LexA-CTERM (K1646R) mutant was made using QuikChange (Agilent Technologies, Santa Clara, CA) following the manufacturer’s protocol. The primers used to make the Y2H “bait” vectors are described in Supplementary Methods.
Immunofluorescence and Confocal Microscopy
For immunocytochemical analysis of TRPM7 and hepatocystin, HEK-293T cells were plated onto coverslips and transfected with the pcDNA5/FRT/TO-HA-TRPM7 and pcDNA6-FLAG-hepatocystin plasmids. Cells were fixed at room temperature for 10 minutes in phosphate buffered saline (PBS) at pH 7.4 with 4% paraformaldehyde (Electron Microscopy Sciences; Hatfield, PA), and permeabilized with PBS containing 10% fetal bovine serum and 0.1% saponin. The rat monoclonal antibody Anti-HA antibody (clone 3F10, Roche Life Sciences; Indianapolis, IN) was used to visualize HA-tagged TRPM7 and mouse monoclonal Anti-FLAG M2 antibody (Sigma Aldrich; St. Louis, MO) was used to detect FLAG-tagged hepatocystin. Alexa Fluor 488 and Alexa Fluor 568 goat antibodies to rat and mouse (Life Technologies, Inc; Grand Island, NY) were used as secondary antibodies. Images were obtained at the Rutgers RWJMS CORE Confocal facility using a Yokogawa CSUX1-5000 microscope using 488-nm and 561-nm excitation wavelengths.
Cell lines
The 293T cell line (CRL-3216) was purchased from American Type Culture Collection (Manassas, VA). The HEK-293 cell line expressing recombinant Hemaglutinin (HA)-tagged TRPM7 in tetracycline-inducible manner (293-TRPM7 cells) was previously described21. 293T and 293-M7 cells were maintained in a Dulbecco’s Modified Eagle Medium (DMEM), high glucose media with 10% FBS in a humidified 37 °C, 5% CO2 incubator
Kinase Assay
To conduct the in vitro kinase assay, a GST-fusion protein containing TRPM7′s kinase domain (GST-KIN) was purified on a glutathione-agarose (Sigma Aldrich, St. Louis, MO) and dialyzed into 1X kinase buffer without ATP (20 mM Mops (pH 7.2), 100 mM NaCl, 20 mM MgCl2, 0.5 mM ATP, and 2 μCi of [γ-32P]ATP). A His-patch (HP) thioredoxin-fusion protein (Thio-hepatocystin) of hepatocystin (residues 346–528) was expressed in the pET102/D-TOPO vector (Life Technologies, Inc) and purified using HA agarose (Qiagen; Valencia, CA). HP thioredoxin-hepatocystin was dialyzed into 1X KIN Buffer and then subjected to an in vitro kinase assay using myelin basic protein (MBP) as previously described3.
Embryo manipulations
Embryo manipulations were performed as previously described40,41. Embryo injections were performed using in vitro transcribed RNAs or Morpholino oligonucleotides (MO). The Xenopus laevis hepatocystin morpholino (Xhepatocystin MO) was 5- GCAGCAGCGCAAGCAAAGCCTTCAT-3 and was synthesized by Gene-Tools (Philomath, OR). The XTRPM7 MO2 was previously described5.
Detection of protein expression
To detect HA-TRPM7 and actin expression in Xenopus laevis, embryos were lysed with lysis buffer (50 mM TRIS (pH 7.4), 150 mM NaCl, and 1% Igepal 630) and the proteins resolved by SDS-PAGE and western blotting using standard protocols. For detection of proteins in cultured cells lines, cells were lysed in the lysis buffer described above. The rat monoclonal antibody Anti-HA antibody (clone 3F10, Roche Life Sciences; Indianapolis, IN) was used to detect HA-tagged TRPM7. Anti-β-actin (sc-47778; Santa Cruz Biotechnology, Inc, Dallas, TX) was used to detect β-actin as loading control. Anti-GFP (sc-9996; Santa Cruz Biotechnology, Inc, Dallas, TX) was used to detect mf-GFP in pulldown purification assays. The anti-hepatocystin antibodies (catalogue # sc-374453, sc-10774; Santa Cruz Biotechnology, Inc. Dallas, TX) were used to detect endogenous hepatocystin in HEK-293 cells. HRP-linked secondary antibodies were goat anti-rat (sc-2006) and goat-anti rabbit (sc-2004). The sheep anti-mouse secondary antibody was purchased from GE Life Sciences (Piscataway, NJ). Transient transfection of cDNAs was conducted using TurboFect Transfection Reagent (Life Technologies, Inc; Grand Island, NY). For the co-immunoprecipitation experiment, 293-TRPM7 cells were treated with tetracycline (TET) at 1 μg/ml for 24 hrs to induce expression of HA-TRPM7. Following cell lysis, HA-TRPM7 was immunoprecipitated with monoclonal anti-HA-agarose (Sigma Aldrich, St. Louis, MO) overnight and the proteins resolved by SDS-PAGE and western blotting. For the pulldown purification assays using GST-hepatocystin, cDNAs for mfGFP-tagged TRPM7 fragments were transiently transfected into 293T cells and the proteins expressed for 24 hrs. Cells were lysed with lysis buffer and subjected to a GST-pulldown purification assay and the proteins resolved by SDS page and western blotting. For the tunicamycin experiment, 2 × 106 TRPM7 cells were plated onto polylysine coated 60 mm dishes. The following day, tunicamycin (5 μg/ml) and tetracycline (5 μg/ml) were added. The cells were harvested 24 hours later and the cell lysate was subjected to SDS-PAGE and western blotting.
Additional Information
How to cite this article: Overton, J. D. et al. Hepatocystin is Essential for TRPM7 Function During Early Embryogenesis. Sci. Rep. 5, 18395; doi: 10.1038/srep18395 (2015).
Supplementary Material
Acknowledgments
This work was supported by the generous support of the National Institutes of Health, NIGMS (GM080753) to LWR and (GM078172) to RH. We are also grateful to Dr. David Clapham (Harvard Medical School) for his initial support of the project and to Dr. Stefan Somlo (Yale University School of Medicine) for reagents and helpful comments about the manuscript.
Footnotes
Author Contributions J.O. and Y.K. designed and carried out most of the experiments, analyzed data, co-wrote the paper, and contributed equally to the work. C.M., K.N., N.C. and L.L. performed experiments. S.F. created reagents and advised on some of the experiments. L.W.R. and R.H. conceived and supervised the work and co-wrote the paper.
References
- Monteilh-Zoller M. K. et al. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol 121, 49–60 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler M. J. et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 411, 590–595 (2001). [DOI] [PubMed] [Google Scholar]
- Runnels L. W., Yue L. & Clapham D. E. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 291, 1043–1047 (2001). [DOI] [PubMed] [Google Scholar]
- Jin J. et al. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 322, 756–760 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. et al. TRPM7 regulates gastrulation during vertebrate embryogenesis. Developmental biology 350, 348–357 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns D. M. & Touster O. Purification and characterization of glucosidase II, an endoplasmic reticulum hydrolase involved in glycoprotein biosynthesis. The Journal of biological chemistry 257, 9990–10000 (1982). [PubMed] [Google Scholar]
- Drenth J. P., Martina J. A., Te Morsche R. H., Jansen J. B. & Bonifacino J. S. Molecular characterization of hepatocystin, the protein that is defective in autosomal dominant polycystic liver disease. Gastroenterology 126, 1819–1827 (2004). [DOI] [PubMed] [Google Scholar]
- Trombetta E. S., Simons J. F. & Helenius A. Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound noncatalytic HDEL-containing subunit. The Journal of biological chemistry 271, 27509–27516 (1996). [DOI] [PubMed] [Google Scholar]
- Hammond C., Braakman I. & Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proceedings of the National Academy of Sciences of the United States of America 91, 913–917 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A. & Aebi M. Intracellular functions of N-linked glycans. Science 291, 2364–2369 (2001). [DOI] [PubMed] [Google Scholar]
- Helenius A. & Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73, 1019–1049 (2004). [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Bonifacino J. S., Yuan L. C. & Klausner R. D. Degradation from the endoplasmic reticulum: disposing of newly synthesized proteins. Cell 54, 209–220 (1988). [DOI] [PubMed] [Google Scholar]
- Stigliano I. D., Caramelo J. J., Labriola C. A., Parodi A. J. & D’Alessio C. Glucosidase II beta subunit modulates N-glycan trimming in fission yeasts and mammals. Mol Biol Cell 20, 3974–3984 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenth J. P., te Morsche R. H., Smink R., Bonifacino J. S. & Jansen J. B. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet 33, 345–347 (2003). [DOI] [PubMed] [Google Scholar]
- Li A. et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet 72, 691–703 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenth J. P., Martina J. A., van de Kerkhof R., Bonifacino J. S. & Jansen J. B. Polycystic liver disease is a disorder of cotranslational protein processing. Trends Mol Med 11, 37–42 (2005). [DOI] [PubMed] [Google Scholar]
- Fedeles S. V. et al. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet 43, 639–647 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofherr A., Wagner C., Fedeles S., Somlo S. & Kottgen M. N-glycosylation determines the abundance of the transient receptor potential channel TRPP2. The Journal of biological chemistry 289, 14854–14867 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K. et al. Massive autophosphorylation of the Ser/Thr-rich domain controls protein kinase activity of TRPM6 and TRPM7. PLoS One 3, e1876; 10.1371/journal.pone.0001876 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L. T. et al. TRPM7 regulates polarized cell movements. Biochem J 434, 513–521 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald B. T. & He X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harbor perspectives in biology 4, 10.1101/cshperspect.a007880 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L., Molinari M. & Helenius A. Setting the standards: quality control in the secretory pathway. Science 286, 1882–1888 (1999). [DOI] [PubMed] [Google Scholar]
- Hodgkinson C. P., Mander A. & Sale G. J. Identification of 80K-H as a protein involved in GLUT4 vesicle trafficking. Biochem J 388, 785–793 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K. et al. TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J 25, 290–301 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuki A. & Tamura G. Effect of tunicamycin on the synthesis of macromolecules in cultures of chick embryo fibroblasts infected with Newcastle disease virus. J Antibiot (Tokyo) 24, 785–794 (1971). [DOI] [PubMed] [Google Scholar]
- Gao H. et al. PRKCSH/80K-H, the protein mutated in polycystic liver disease, protects polycystin-2/TRPP2 against HERP-mediated degradation. Hum Mol Genet 19, 16–24 (2010). [DOI] [PubMed] [Google Scholar]
- Davila S. et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet 36, 575–577 (2004). [DOI] [PubMed] [Google Scholar]
- Waanders E., te Morsche R. H., de Man R. A., Jansen J. B. & Drenth J. P. Extensive mutational analysis of PRKCSH and SEC63 broadens the spectrum of polycystic liver disease. Hum Mutat 27, 830, 10.1002/humu.9441 (2006). [DOI] [PubMed] [Google Scholar]
- Cnossen W. R. & Drenth J. P. Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J Rare Dis 9, 69; 10.1186/1750-1172-9-69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J. et al. The channel kinase, TRPM7, is required for early embryonic development. Proceedings of the National Academy of Sciences of the United States of America 109, E225–233; 10.1073/pnas.1120033109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnossen W. R. et al. Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. Proceedings of the National Academy of Sciences of the United States of America 111, 5343–5348; 10.1073/pnas.1309438111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienkamp S. S. et al. Vertebrate kidney tubules elongate using a planar cell polarity-dependent, rosette-based mechanism of convergent extension. Nat Genet 44, 1382–1387; 10.1038/ng.2452 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strazzabosco M. & Somlo S. Polycystic liver diseases: congenital disorders of cholangiocyte signaling. Gastroenterology 140, 1855–1859 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills E. S., Roepman R. & Drenth J. P. Polycystic liver disease: ductal plate malformation and the primary cilium. Trends Mol Med 20, 261–270 (2014). [DOI] [PubMed] [Google Scholar]
- Gkika D., Mahieu F., Nilius B., Hoenderop J. G. & Bindels R. J. 80K-H as a new Ca2+ sensor regulating the activity of the epithelial Ca2+ channel transient receptor potential cation channel V5 (TRPV5). The Journal of biological chemistry 279, 26351–26357 (2004). [DOI] [PubMed] [Google Scholar]
- Kawaai K. et al. 80K-H interacts with inositol 1,4,5-trisphosphate (IP3) receptors and regulates IP3-induced calcium release activity. The Journal of biological chemistry 284, 372–380 (2009). [DOI] [PubMed] [Google Scholar]
- Chubanov V. et al. Hypomagnesemia with secondary hypocalcemia due to a missense mutation in the putative pore-forming region of TRPM6. J Biol Chem 282, 7656–7667 (2007). [DOI] [PubMed] [Google Scholar]
- Kobayashi T. et al. Engineering a novel multifunctional green fluorescent protein tag for a wide variety of protein research. PLoS One 3, e3822; 10.1371/journal.pone.0003822 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L. T. et al. TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain. The Journal of biological chemistry 281, 11260–11270 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khadka D. K., Liu W. & Habas R. Non-redundant roles for Profilin2 and Profilin1 during vertebrate gastrulation. Developmental biology 332, 396–406 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A. et al. Profilin is an effector for Daam1 in non-canonical Wnt signaling and is required for vertebrate gastrulation. Development 133, 4219–4231 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.