

Abstract
Cortical spreading depression (CSD) has been associated with many pathological entities including migraine, trauma, hemorrhage, and mitochondrial disease. The clinical diagnosis remains challenging without the other concomitant features such as headache because CSD can mimic seizure or acute stroke. Wereport of a 77 year-old right handed man with a left subdural hematoma evacuation that subsequently developed episodic aphasia, slurred speech, right nasolabial fold flattening, and right pronator drift. In this case report, we discuss our multimodal diagnostic approach and treatment in a patient with episodic aphasia and neurological deficits in order to propose the diagnosis of cortical spreading depression. CSD should be considered when focal deficits in brief episodes occur after stroke and seizures have been ruled out. Treatment choices as illustrated by this case report can have an impact on outcome and resolution of episodes.
Keywords: neurohospitalist, clinical specialty, migraine disorders, headache, disorders, imaging, techniques, cerebrovascular trauma, trauma, nervous system, cerebrovascular disorders
Background and Purpose
Cortical spreading depression (CSD) has been associated with many pathological entities including migraine, trauma, hemorrhage, and mitochondrial disease.1,2 The clinical diagnosis remains challenging without the other concomitant features such as headache because CSD can mimic seizure or acute stroke.3,4
In this case report, we discuss our multimodal diagnostic approach and treatment in a patient with episodic aphasia and neurological deficits in order to propose the diagnosis of CSD after subdural hematoma evacuation.
Methods
A 77-year-old right-handed man had a traumatic left subdural hematoma that was managed conservatively until he developed midline shift requiring evacuation. Postoperatively, he had no deficits and was discharged with levetiracetam for seizure prophylaxis. He presented 2 weeks after evacuation with sudden onset of episodic language abnormalities including neologisms and dysarthria without headache. The family reported that he had multiple episodes in succession lasting 20 to 30 minutes with slurred speech. He did not lose consciousness, have urinary or bowel dysfunction, bite his tongue, and had no tonic–clonic movements. He had no memory of the initial events, and during acute evaluation in the emergency department, he was found to be disoriented, aphasic with a receptive greater than expressive component, right nasolabial fold flattening, and a right pronator drift. Administration of intravenous benzodiazepines was not effective. During the first 48 hours of admission, his episodes lasted from 30 minutes to 5 hours with stereotyped deficits but without tonic–clonic movements.
Results
Our workup initially included stroke and seizure evaluation, given that he had no headache. Two initial 30-minute electroencephalograms (EEGs) and a 24-hour EEG captured multiple events with a well-modulated posterior background of 8 to 10 Hz and reactivity to eye opening. During events, EEGs demonstrated no change from his awake baseline of intermittent left greater than right centroparietal region slowing of mixed delta–theta frequency with a superimposed breach pattern secondary to craniotomy. In concurrent EEG recordings, there was no change in the background before, during, or after events. Magnetic resonance imaging (MRI)/magnetic resonance angiography (MRA) of the brain showed no focal lesions and normal vasculature. Diffusion-weighted imaging (DWI) did not demonstrate restriction. Trancranial Doppler ultrasound did not demonstrate any side-side velocity differences, high-intensity transient signals consistent with emboli, or features consistent with vasospasm. Cerebrospinal fluid was unremarkable for signs of infection. Computed tomography perfusion (CTP) during an event demonstrated increased mean transit time (MTT) and decreased cerebral blood flow (CBF) with no focal area of decreased cerebral blood volume (CBV). Subsequent postevent MRI with contrast did not show enhancement or DWI restriction (see Figure 1).
Figure 1.
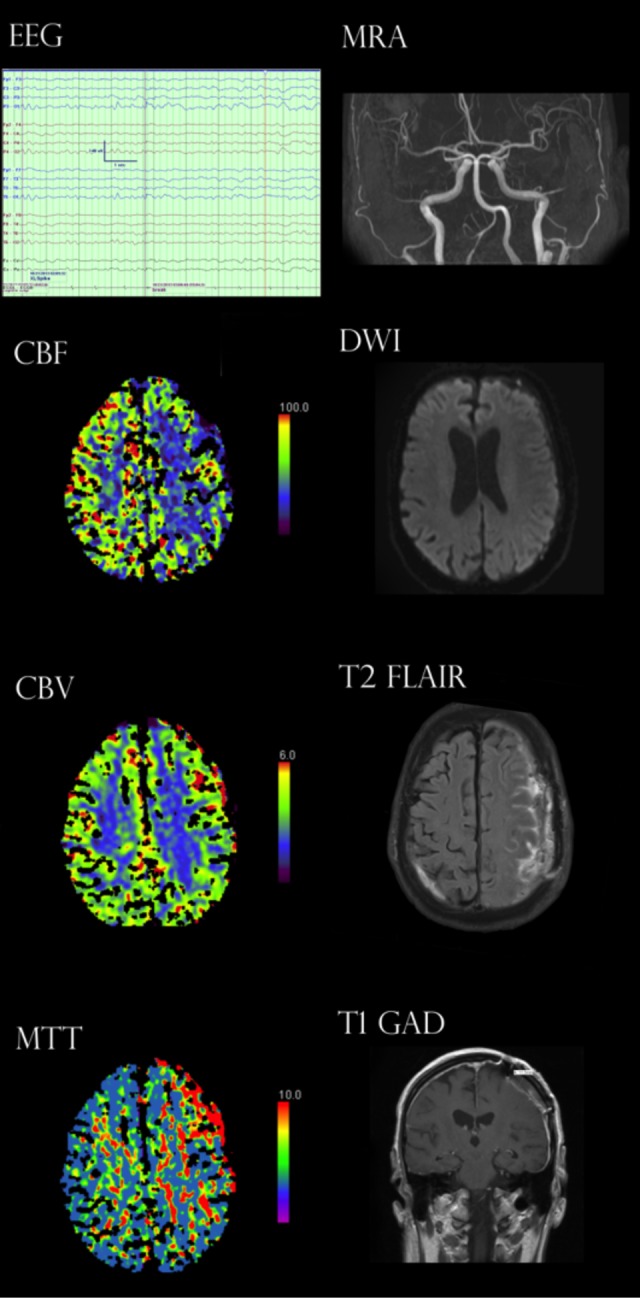
Electroencephalogram (EEG) demonstrated left predominant centroparietal region slowing. Intracranial vasculature patent bilaterally on magnetic resonance angiography. Computed tomography perfusion (CTP) demonstrated decreased cerebral blood flow (CBF) with increased mean transit time (MTT) but no decrease in cerebral blood volume (CBV) indicating no infarct. Diffusion-weighted imaging (DWI) was negative for restriction. T2-Flair only demonstrated hyperintensity consistent with subdural evacuation postsurgical changes. T1 with gadolinium contrast did not demonstrate intra-axial enhancement.
Initial management included empiric seizure treatment with intravenous benzodiazepines. However, the events increased in frequency to multiple times a day in the face of 2 antiepileptics, carbamazepine and levetiracetam, with escalating doses over 3 days. After completion of the diagnostic assessment, we discontinued previous antiepileptics and initiated oral magnesium and topiramate, which resulted in resolution of episodes within 24 hours.
At 6-month follow-up, the patient had had no more events and had resumed daily activities.
Discussion
The distinction between seizure and stroke was challenging initially, given the patient’s history of subdural hematoma evacuation and fluctuating event periods. After the administration of antiepileptics, titrating their maintenance doses, evaluating EEGs, and eliminating acute stroke diagnosis, we considered other entities such as vasospasm and infection. During an episode, the patient was taken to CTP which demonstrated a decrease in CBF and an increase in MTT in the left frontal lobe, normal CBV, and therefore without evidence of infarct. Repeat MRI demonstrated no infarct or gadolinium enhancement. These changes are consistent with CSD and may explain this clinical presentation.
Cortical spreading depression is a depolarization wave in cerebral gray matter that propagates across the brain at a velocity of 2 to 5 mm/min.5 For many years, CSD was believed to be an artifact produced in animal experiments and without significance for human neurological conditions, but now it is implicated in migraine, stroke, subarachnoid hemorrhage, and traumatic brain injury. Cortical spreading depression and depolarization waves have been associated with loss of brain ion homeostasis, efflux of excitatory amino acids from nerve cells, changes in energy metabolism, and changes in cerebral blood flow.6 In injured brains, vasoconstriction followed by hypoperfusion are seen which is suspected to contribute to permanent damage.7 Cerebral blood flow of patients during migraine attacks has shown a wave of reduced blood flow that moves across the brain at the same rate as CSD and with the same signs of vascular impairment.8
Our choice in medical management was influenced by animal studies that have demonstrated magnesium and topiramate decrease CSDs as there have been no clinical studies of nonanalgesics to date.9-12 Levetiracetam has not been reported to suppress CSD, and carbamazepine has been found not to be effective at CSD suppression.13 Observational data of acute brain injured patients have shown that ketamine was associated with less CSDs, however, ketamine was not given as an intervention for CSD and higher doses were correlated with worse outcomes at follow-up.14 Subsequent animal studies have demonstrated ketamine is indeed effective at reducing CSDs.15 Additional analgesics such as midazolam, isoflurane, and propofol may be promising for treating CSD in the neurocritical care setting.14,16,17
Cortical spreading depression can be difficult to detect due to its brief and focal pathology. Electroencephalogram should be done to exclude seizure during the event. Intracranial probe electrode studies have demonstrated correlation with scalp EEG demonstration of ultraslow potentials.18 However, this analysis requires the processing of the power spectra and while promising, the correlates of gold standard subdural electrocorticography for detection of CSD and scalp CSD were arbitrarily defined, requiring further clinical study.18,19 We were unable to identify those patterns in this patient’s recordings due to technical limitations of data postprocessing, however, in the future, the use of real-time power spectra EEG could be considered. Magnetic resonance imaging/MRA should be done to exclude stroke. Perfusion-based imaging may be done as an ancillary test.
Our patient’s remarkable response to medication changes is noteworthy for clinical consideration. In the correct clinical context, diagnosis and aggressive management may prevent infarct, as CSD’s vascular hypoperfusion is postulated to be an etiology of stroke in migraineurs with aura due to CSD.20
Conclusion
Cortical spreading depression should be considered when focal deficits in brief episodes occur after stroke and seizures have been ruled out. Treatment choices illustrated by this case report can have an impact on outcome and resolution of episodes.
Footnotes
Authors’ Note: This work has not been previously published, but has been presented in abstract form at the American Academy of Neurology Annual Meeting in 2014. Dr David Adams and I have been involved in all parts of the work presented herein. We both contributed substantively to the conception, design, or analysis and interpretation of the data. We both contributed substantively to the drafting of the manuscript or critical revision for important intellectual content and gave final approval of the version to be published. We both agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Informed consent was obtained per institutional criteria and family has been informed that the case will be presented for publication, which they acknowledged. Dr Shah drafted/revised the manuscript for content, analysis, and interpretation of data and contributed to concept and design, clinical evaluation, and management. Dr Adams drafted/revised the manuscript for content, study concept, and design and contributed to analysis or interpretation of data and study supervision.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Hartings JA, Gugliotta M, Gilman C, Strong AJ, Tortella FC, Bullock MR. Repetitive cortical spreading depolarizations in a case of severe brain trauma. Neurol Res. 2008;30(8):876–882. [DOI] [PubMed] [Google Scholar]
- 2. Betts J, Jaros E, Perry RH, et al. Molecular neuropathology of MELAS: level of heteroplasmy in individual neurones and evidence of extensive vascular involvement. Neuropathol Appl Neurobiol. 2006;32(4):359–373. [DOI] [PubMed] [Google Scholar]
- 3. Mishra NK, Rossetti AO, Ménétrey A, Carota A. Recurrent Wernicke’s aphasia: migraine and not stroke! Headache. 2009;49(5):765–768. [DOI] [PubMed] [Google Scholar]
- 4. Brunot S, Osseby GV, Rouaud O, et al. Transient ischaemic attack mimics revealing focal subarachnoid haemorrhage. Cerebrovasc Dis Basel Switz. 2010;30(6):597–601. [DOI] [PubMed] [Google Scholar]
- 5. Leao AA. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol. 1947;10(6):409–414. [DOI] [PubMed] [Google Scholar]
- 6. Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31(1):17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ayata C. Spreading depression and neurovascular coupling. Stroke. 2013;44(6 suppl 1):S87–S89. [DOI] [PubMed] [Google Scholar]
- 8. Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol. 1981;9(4):344–352. [DOI] [PubMed] [Google Scholar]
- 9. Akerman S, Goadsby PJ. Topiramate inhibits cortical spreading depression in rat and cat: impact in migraine aura. Neuroreport. 2005;16(12):1383–1387. [DOI] [PubMed] [Google Scholar]
- 10. Unekawa M, Tomita Y, Toriumi H, Suzuki N. Suppressive effect of chronic peroral topiramate on potassium-induced cortical spreading depression in rats. Cephalalgia. 2012;32(7):518–527. [DOI] [PubMed] [Google Scholar]
- 11. Van der Hel WS, van den Bergh WM, Nicolay K, Tulleken KA, Dijkhuizen RM. Suppression of cortical spreading depressions after magnesium treatment in the rat. Neuroreport. 1998;9(10):2179–2182. [DOI] [PubMed] [Google Scholar]
- 12. Tepe N, Filiz A, Dilekoz E, et al. The thalamic reticular nucleus is activated by cortical spreading depression in freely moving rats: prevention by acute valproate administration. Eur J Neurosci. 2015;41(1):120–128. [DOI] [PubMed] [Google Scholar]
- 13. Costa C, Tozzi A, Rainero I, et al. Cortical spreading depression as a target for anti-migraine agents. J Headache Pain. 2013;14:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hertle DN, Dreier JP, Woitzik J, et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain J Neurol. 2012;135(pt 8):2390–2398. [DOI] [PubMed] [Google Scholar]
- 15. Sánchez-Porras R, Santos E, Schöll M, et al. The effect of ketamine on optical and electrical characteristics of spreading depolarizations in gyrencephalic swine cortex. Neuropharmacology. 2014;84:52–61. [DOI] [PubMed] [Google Scholar]
- 16. Takagaki M, Feuerstein D, Kumagai T, Gramer M, Yoshimine T, Graf R. Isoflurane suppresses cortical spreading depolarizations compared to propofol--implications for sedation of neurocritical care patients. Exp Neurol. 2014;252:12–17. [DOI] [PubMed] [Google Scholar]
- 17. Kudo C, Toyama M, Boku A, et al. Anesthetic effects on susceptibility to cortical spreading depression. Neuropharmacology. 2013;67:32–36. [DOI] [PubMed] [Google Scholar]
- 18. Drenckhahn C, Winkler MK, Major S, et al. Correlates of spreading depolarization in human scalp electroencephalography. Brain J Neurol. 2012;135(pt 3):853–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dreier JP, Woitzik J, Fabricius M, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain J Neurol. 2006;129(pt 12):3224–3237. [DOI] [PubMed] [Google Scholar]
- 20. Øygarden H, Kvistad CE, Bjørk M, Thomassen L, Waje-Andreassen U, Naess H. Diffusion-weighted lesions in acute ischaemic stroke patients with migraine. Acta Neurol Scand Suppl. 2014;(198):41–46. doi:10.1111/ane.12236. [DOI] [PubMed] [Google Scholar]