

Abstract
ADP-ribosylation refers to the transfer of the ADP-ribose group from NAD+ to target proteins post-translationally, either attached singly as mono(ADP-ribose) (MAR) or in polymeric chains as poly(ADP-ribose) (PAR). Though ADP-ribosylation is therapeutically important, investigation of this protein modification has been limited by a lack of proteomic tools for site identification. Recent work has demonstrated the potential of a tag-based pipeline in which MAR/PAR is hydrolyzed down to phosphoribose, leaving a 212 Dalton tag at the modification site. While the pipeline has been proven effective by multiple groups, a barrier to application has become evident: the enzyme used to transform MAR/PAR into phosphoribose must be purified from the rattlesnake Crotalus adamanteus venom, which is contaminated with proteases detrimental for proteomic applications. Here, we outline the steps necessary to purify snake venom phosphodiesterase I (SVP) and describe two alternatives to SVP—the bacterial Nudix hydrolase EcRppH and human HsNudT16. Importantly, expression and purification schemes for these Nudix enzymes have already been proven, with high-quality yields easily attainable. We demonstrate their utility in identifying ADP-ribosylation sites on Poly(ADP-ribose) Polymerase 1 (PARP1) with mass spectrometry and discuss a structure-based rationale for this Nudix subclass in degrading protein-conjugated ADP-ribose, including both MAR and PAR.
ADP-ribosylation is a post-translational modification (PTM) implicated in a number of disease states, including cancer, diabetes, and a range of neuropathologies1. This protein modification is synthesized by ADP-ribosyl transferases, commonly known as poly(ADP-ribose) polymerases (PARPs), which transfer the ADP-ribose (ADPr) group from NAD+ to protein acceptor amino acids in monomeric (mono(ADPr), MAR) and/or polymeric (poly(ADPr), PAR) form2,3. Identification of specific amino acid acceptors of ADPr group(s), and therefore characterization of the cellular role played by this important protein modification, has been hampered by the lack of a robust, universal method for identifying ADP-ribosylation sites in the proteome. This need has lately been addressed by three different methods4,5,6,7,8,9,10,11,12,13,14, all of which involve the removal of any ADPr subunits beyond the protein-proximal monomer, followed by identification of the ‘tag’ left behind at the ADPr conjugation site (reviewed in ref. 14). One of these methods, hydrolysis of MAR/PAR down to its phosphoribose attachment site, relies upon the pyrophosphatase activity of snake venom phosphodiesterase I (SVP) from Crotalus adamanateus, which can be purchased in a partially purified form that requires further purification for use against ADP-ribosylated proteins3,5,6. Unfortunately, this complicated purification scheme ultimately results in a high level of prep-to-prep variability, likely due to the inherently variable protein source (snake venom) as well as the number of purification steps involved. In an effort to identify a more reliable tool for the degradation of protein-conjugated MAR/PAR to phosphoribose, we describe here the characterization of candidate enzymes from the Nudix hydrolase superfamily.
The Nudix hydrolase superfamily catalyzes hydrolysis of Nucleoside Diphosphates linked to other moieties (“X”). Most Nudix families contribute to cellular ‘housekeeping’ through the breakdown of a wide range of nucleoside diphosphate derivatives15. One of these diphosphate containing compounds is ADPr16,17,18,19, a molecule which is known to accumulate in cells with potentially cytotoxic effects by: (1) altering calcium entry into cells via channel gating, thus affecting membrane depolarization20, (2) serving as a neurotransmitter in primate and murine colons21, and (3) spontaneously modifying proteins22, potentially altering intracellular post-translational signaling. Without Nudix hydrolase activity, free ADPr would amass during the breakdown of PAR23,24, as a side product of tRNA synthesis25, following NAD+ glycohydrolysis26, following deacetylation of O-acetyl-ADPr27, or through the breakdown of the signaling molecule cyclic ADPr28. Accordingly, ADPr degrading Nudix enzymes are broadly conserved, with humans possessing at least six distinct ADPr pyrophosphatases (ADPrases) responsible for hydrolyzing ADPr to AMP and phosphoribose18,29. In light of this known ADPrase activity, we reasoned that Nudix hydrolases could potentially replace SVP for generating phosphoribose tags at ADP-ribosylation sites in the proteomics pipeline described above, and screened bacterial Nudix hydrolases for comparable hydrolysis activity of protein-conjugated ADPr. These efforts lead to the identification of the RNA 5′ pyrophosphohydrolase (RppH) from Escherichia coli (EcRppH) as capable of degrading protein-conjugated ADPr, including both MAR and PAR. From a structural and biological perspective, this finding was unexpected as EcRppH is an RNA decapping enzyme, and not an ADPrase30. While we were characterizing this novel activity of EcRppH, a human Nudix hydrolase NudT16 (HsNudT16) also known to decap RNA31, was shown to degrade protein-conjugated MAR and PAR13. In this study, we provide a structure-based rationale for the inability of Nudix ADPrases to degrade protein-conjugated ADPr, in contrast to Nudix RNA decapping enzymes, and demonstrate the use of both EcRppH and HsNudT16 in the identification of protein ADP-ribosylation sites by mass spectrometry.
Materials and Methods
Mutagenesis of PARP1 to the E988Q catalytically deficient mutant
pET28 6xHis-PARP1 was a gift from Dr. John Pascal and served as the template for mutagenesis into the mono(ADP-ribose) restricted mutant of PARP1, E988Q. Primers used for mutagenesis: Forward: GACACCTCTCTACTATATAACCAGTACATTGTCTATGATATTGC, Reverse: GCAATATCATAGACAATGTACTGGTTATATAGTAGAGAGGTGTC.
Expression and purification of wild-type (WT) and E988Q His-PARP1
WT and E988Q His-PARP1 was expressed and purified as previously described5.
Purification of SVP
SVP was purified as previously described5 from starting material obtained from United States Biological, catalog number P4072, lot number L14030507 C14062702. Briefly, SVP powder from a vial was dissolved into 1 mL of loading buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 10% glycerol) and then loaded onto a pre-equilibrated 1 mL HiTrap blue sepharose column (GE, 17-0412-01), washed with 5 column volumes of loading buffer and then 5 column volumes of elution buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 10% glycerol, 150 mM potassium phosphate). Desired fractions were pooled, dialyzed against loading buffer. Samples were then further dialyzed into size exclusion chromatography buffer (10 mM Tris-HCl pH 7.3, 50 mM NaCl, 15 mM MgCl2, 1% glycerol) and resolved over a SuperDex 200/10/300 GL (GE Healthcare) using an ÄKTA FPLC (GE, 18-1900-26); desired fractions were pooled and stored at −80 °C.
Assessment of contaminating proteolysis activity in SVP prep
For whole cell lysate, 1 mg of proteins from HeLa whole cell lysate was denatured in 8M Urea (Sigma-Aldrich), 50 mM Tris-HCl pH 7.0 for 10 minutes at 37 °C before being reduced in 1 mM Tris(2-carboxyethyl)phosphine (Life Technologies) for 10 minutes and then alkylated in 2 mM 2-chloroacetamide (Sigma-Aldrich) for 10 minutes in the dark. Samples were then diluted to a final concentration of 1M Urea, 50 mM NaCl (Sigma-Aldrich), 15 mM MgCl2 (Quality Biological) and 0.2M Tris-HCl pH 7.3. 5 μg of SVP was added to each sample and incubated for 2 hours at 37 °C. Samples were run on an in-house 6–10% SDS-PAGE gel and transferred to a nitrocellulose membrane. Total protein was visualized by ProAct membrane stain (Amresco) per the manufacturer’s instructions. For purified recombinant His-PARP1, 1 μg of His-PARP1 was automodified as previously published5 and then switched into the same buffer used for the whole cell lysate (described above) with or without 1M urea. 500 ng of SVP was used for each μg of PARP1 and digestion was carried out at 37 °C for 2 hours. Samples were run on in-house 6–10% SDS-PAGE gels and total protein was visualized by SimplyBlue Safe Stain (Life Technologies) per the manufacturer’s instructions. 32P-labeled PAR was visualized on a phosphor-screen (GE, BAS-III 2040) followed by imaging on a Typhoon FLA7000 (GE Healthcare Life Sciences).
Expression and purification of Nudix hydrolases
The expression and purification have been previously published for the following hydrolases: E. coli NudF (EcNudF/ADPRase/UniProtKB Q93K97)18, E. coli NudE (EcNudE/ORF186/UniProtKB J7QI36)32, Bdellovibrio bacteriovorus (Bd3179/BdNDPSase/UniProtKB Q6MIH8), Deinococcus radiodurans Dr1184 (Dr1184/ UniProtKB Q9RV46)33, E.coli RppH (EcRppH/ORF176/UniProtKB P0A77634, Agrobacterium tumefaciens ORF147 (AtORF147/UniProtKB Q7CX66) and Pseudomonas aeruginosa Pa3470 (Pa3470/UniProtKB Q9HYD6)35. Homo sapiens NudT16 (HsNudT16/UniProtKB Q96DE0) was expressed and purified as described in the methods from the Structural Genomics Consortium (http://www.thesgc.org/structures/3cou). For EcRppH, the last step of purification yielded ≥90% homogeneity before being loaded onto a HiLoad 26/60 Superdex 200 prep grade gel filtration column (GE Healthcare), equilibrated in gel filtration buffer (50 mM Tris-HCl pH 8.5, 300 mM NaCl); fractions containing ≥95% homogeneity, as determined by SDS-PAGE, were pooled. EcYfcD was purified using the method described for EcADPRase18.
Structural analysis of Selected Nudix hydrolases
Selected Nudix nucleotide sugar hydrolases were used to test their activity against protein-conjugated ADPr: E. coli NudF, YfcD and NudE, and Bdellovibrio bacteriovorus HD100 NDPSase. The structures were structurally aligned using SSM36 and rendered with PyMOL37. PAR was constructed and minimized in MOE software package (Chemical Computing Group, Montreal, Canada)38. For the sugar hydrolases, PAR was modeled in the active site taking into account the binding preference observed in the structures in complex with sugar nucleoside derivatives18.
Selected known single domain Nudix enzymes were structurally aligned using SSM36 and rendered using PyMOL38. PAR was modeled in the active site using a ‘template guide’, i.e. the mRNA present in the structure of the complex of EcRppH (PDB ID 4S2X)39 and the IMP present in the structure of HsNudT16 (PDB ID 2XSQ)40. Models of the complex structures were built using the selected structures and the PAR. Modeling steps were performed in MOE38. Hydrogen atoms and charges were added automatically to the protein. The binding site was defined as all protein atoms within 4.5 angstrom of any atom of bound ligand. A steepest descent energy minimization was used with a MMFF94x forcefield.
Automodification of WT and E988Q PARP1
Automodification was performed as published previously5 with the following changes: both WT and E988Q PARP1 were incubated with 0.6 μM (1.85 kBq/μL, 37 kBq/sample) 32P-NAD+ for 10 minutes at room temperature, following which WT PARP1 was incubated with 1 mM NAD+ (non-radioactive) for 10 minutes at room temperature to allow for polymer elongation.
Hydrolysis of protein-conjugated ADPr to phosphoribose
For comparative analyses by SDS PAGE: 5 pmoles of 6xHis-PARP1 WT or E988Q mutant were exposed to a hydrolase (various enzymes and amounts) in hydrolysis buffer (50 mM Tris-HCl pH 7.0 (Thermo Scientific), 150 mM NaCl (Sigma-Aldrich), 15 mM MgCl2 (Quality Biological), 1 mM 3-aminobenzamide (Sigma Aldrich)) for two hours at 37 °C.
For comparative analyses by liquid chromatography-tandem mass spectrometry (LC-MS/MS): 60 pmoles of 6xHis-PARP1 was exposed to 120 pmoles of SVP or 3 nmoles of Nudix hydrolase (EcRppH or HsNudT16) in hydrolysis buffer for two hours at 37 °C.
Protein digestions for LC-MS/MS analysis
Proteins were denatured in 8 M Urea, 50 mM Tris-HCl pH 7 for 10 minutes at 37 °C before being reduced in 1 mM Tris-(2-Carboxyethyl)phosphine for 10 minutes and then alkylated in 2 mM 2-chloroacetamide for 10 minutes in the dark. Samples were diluted to: 1M Urea, 50 mM NaCl, 15 mM MgCl2, 0.2 M Tris-HCl pH 7 (7.3 at room temperature), and 1 mM CaCl2. LysC (Wako) and Trypsin (Promega) were added at a 1: 50 enzyme:substrate ratio.
Phosphoenrichment of phosphoribosylated peptides
The ion metal affinity chromatography (IMAC) method was performed as described previously5.
LC-MS/MS analysis of phosphoribosylated peptides
Peptides were separated on a Thermo-Dionex RSLCNano UHPLC instrument with ~10 cm x 75 micron ID fused silica capillary columns with ~10 micron tip opening made in-house with a laser puller (Sutter) and packed with 3 micron reversed phase C18 beads (Reprosil-C18.aq, 120 Angstroms, Dr. Maisch). We applied a 90 min gradient of 10–35% B at 200 nL/min using 0.1% acetic acid as solvent A and solvent B of 0.1% acetic acid, 99.9% acetonitrile. MS data was collected with a Thermo Orbitrap Elite. Data-dependent analysis was applied using Top 5 selection and fragmentation was induced by CID and HCD. Profile mode data was collected in all scans. MS analysis parameters were as previously described5.
Database search of MS/MS spectra for peptide and protein identification
Raw files were analyzed by MaxQuant version 1.5.3.8 using protein, peptide and site FDRs of 0.01 and a score minimum of 40 for modified peptides, 0 for unmodified peptides; delta score minimum of 17 for modified peptides, 0 for unmodified peptides. Sequences were searched against an in-house database containing the proteins of interest as well as UniProt Escherichia coli BL21 DE3 database (definitions updated October 15th, 2014). MaxQuant search parameters: Variable modifications included Oxidation (M), Acetylation (Protein N-term), carbamidomethyl (C), phosphorylation (STY) and phosphoribosylation (DEKRC). Max labeled amino acids was 3, max missed cleavages was 2, enzyme was Trypsin/P, and max charge was 7.
Results
Purification of Snake Venom Phosphodiesterase I from Crotalus adamanteus involves both affinity purification and size exclusion chromatography
Snake venom phosphodiesterase I (SVP) from Crotalus adamanteus was shown to degrade PAR nearly 50 years ago41 and has since proven a valuable tool for the degradation of PAR into its linear, branching and terminal subunits, a technique that yields quantitative information regarding the molecular structure of the intact polymer42,43. The utility of this enzyme, however, is greatly determined by the purification scheme employed to isolate it from the large number of proteases as well as phosphatases and nucleotidases present in the C. adamanteus venom44. Oka et al. successfully isolated the phosphodiesterase activity of commercially available SVP away from the contaminating phosphatase and 5′-nucleotidase activity through affinity purification using blue sepharose, a molecule which mimics NAD+ and therefore interacts with the active domain of SVP45. The results from a simplified version of this method used by our group is shown in Fig. 1a, where 150 mM potassium phosphate pH 7.5 is used as a single step elution off a blue sepharose column. This purification scheme paved the way for development of the quantitative method mentioned above to determine the structure of the intact polymer, but did not address the need to eliminate contaminating protease activity. This protease activity can be problematic when using SVP to hydrolyze protein-conjugated ADPr, either MAR or PAR, for the purpose of creating a phosphoribose ‘tag’ at the otherwise ADP-ribosylated amino acid residue5,6,12. Such protease activity is demonstrated in Fig. 1b wherein a complex mixture of proteins is exposed to blue sepharose purified SVP, resulting in the degradation of the target proteins and the appearance of SVP along with its co-purified proteins. This proteolytic activity is further shown against purified,32P-PARylated PARP1 (both native and denatured) in Fig. 1c. In order to separate the 115 kD SVP from the major contaminating proteins (<30 kD), we subjected the blue sepharose purified product to size exclusion chromatography, yielding a simple mixture of what we hypothesize to be the various glycolytic forms of SVP, based on the knowledge that most secreted proteins are glycosylated46, including those found in snake venom47 (Fig. 1d–f). When tested against 32P-PARylated PARP1 as in Fig. 1c, this highly pure form of SVP displayed phosphodiesterase activity without apparent proteolytic activity (Fig. 1g). Similar results were seen against whole cell lysate, allowing for use of this enzyme for ADP-ribosylation site identification by mass spectrometry5.
Figure 1. Purification of snake venom phosphodiesterase I for the digestion of protein-conjugated PAR.

SVP was affinity purified on a blue sepharose column (boxed fractions in panel (a) producing a protease-contaminated product; (b and c), proteolysis is evident by the loss of protein bands following exposure to SVP). The addition of SVP results in protein degradation within lysates (b) and of purified PARP1 (c). The rightmost section in panel b indicates the size of SVP from the pooled fraction from (a). For (b) 1 mg of whole cell lysate was exposed to 50 pmoles of SVP for 2 hrs at 37 °C, pH 7. For (c) 10 pmoles of PARP1 was exposed to 5 pmoles of SVP for 2 hrs at 37 °C, pH 7. Size exclusion chromatography (d,e) results in a purified SVP fraction (f) that removed the contaminating protease activity (g). Top sections in panels c and g show the autoradiographs of PARylated PARP1 and the bottom sections show the corresponding coomassie staining of the gels. Reaction conditions for (g) were identical to those of (c). ***SDS-PAGE well, **SDS-PAGE interface between stacking and resolving gels, *native size of PARP1; rep = replicate.
While the pipeline presented here is an effective method for isolating SVP from snake venom, we believe the complexity of the purification scheme, along with lot-to-lot variability observed from commercial sources of SVP which serve as the input for this purification (data not shown), stands to be greatly improved by the availability of a recombinant enzyme which could be reliably expressed, purified and scaled to meet the often large material demands of proteomic pipelines. For this reason, we went on to compare the activity of SVP with that of relatively small, stable and well-characterized Nudix hydrolases, which we hypothesized could hydrolyze protein-conjugated ADPr, either MAR or PAR, to phosphoribose with similar specificity.
Nudix ADPrases do not hydrolyze protein-conjugated ADPr
Nudix ADPrases are responsible for the breakdown of free ADPr into its phosphoribose and adenosine monophosphate subunits, thus modulating the levels of free ADPr18,48. This knowledge lead us to test a group of Nudix ADPrases for hydrolase activity against protein-conjugated MAR and PAR: PARP1, an enzyme known to autoPARylate in the presence of NAD+, was exposed to 32P-labeled NAD+ producing either 32P-labeled PARylated (on WT PARP1) or MARylated (on the catalytically deficient PARP1 E988Q mutant) proteins to serve as substrates for hydrolysis by candidate Nudix enzymes or the positive control, SVP (Fig. 2a–c). As Fig. 2b shows, the various Nudix enzymes tested did not significantly hydrolyze ADPr from its conjugated proteins when compared to the positive control SVP. From a structural perspective, the lack of activity towards ADPr could be explained by the dimeric structure of ADPrases, where each dimer is formed by monomers of an N-terminal β-sheet domain and a C-terminal Nudix domain (Fig. 2d–g). The N-terminal domains are swapped, creating two active sites where both monomers contribute to substrate recognition (each active site is composed of the N-terminal β-sheet of one monomer and the C-terminal Nudix domain of the other monomer). As shown in Fig. 2h,i, ADPr is nested in the active site of the ADPRase EcNudF so that the 1′-hydroxyl of the terminal ribose group is completely buried by the protein dimer (white arrowhead in Fig. 2i), preventing conjugation to another ADPr group (or a protein). This explanation could likely be extended to the other three nucleoside sugar hydrolases tested in this study as they display the same quaternary arrangement and have a high structural homology with a pairwise root mean square deviation ranging from 0.9 to 2.0 Å (Fig. 2d–g; rmsd calculated with SSM36).
Figure 2. Nudix ADPrases are ineffective against protein-conjugated ADPr.

(a–c) Autoradiograph showing 32P-labeled mono- or poly(ADP-ribose) conjugated to PARP1 following exposure to canonical nucleotide sugar hydrolases: EcNudF, EcYfcD and EcNudE and Bd3179. For (a), 5 pmoles of PARylated PARP1 was exposed to 10 pmoles of hydrolase for 2 hrs at 37 °C; for (b), 100 pmoles of hydrolases were utilized, while in (c) 5 pmoles of PARP1 E988Q, a mutant only capable of synthesizing mono(ADP-ribose), was exposed to 100 pmoles of hydrolases for 2 hrs at 37 °C. Ribbon diagrams show the structure of each of the enzymes used: (d) EcNudF/ADPrase (PDB ID 1KHZ)18, (e) EcYfcD (PDB ID 2FKB), (f) EcNudE (PDB ID 1VHG) and (g) Bd3179 (PDB ID 5C7Q)52. Panel (h) shows a surface representation of EcNudF with modeled PAR polymer based on the complex with a nonhydrolyzable ADPr48. Panel (i) shows a surface representation of EcNudF with the modeled PAR polymer, the inset shows how the terminal ribose is protected and buried within the enzyme. The white arrowhead indicates where protein conjugation would occur on ADPr. ***SDS-PAGE well, **SDS-PAGE interface between stacking and resolving gels, *native size of PARP1, ^co-purified PARP1 protein fragments.
Single domain Nudix hydrolases are capable of hydrolyzing protein-conjugated ADPr
In order to consider Nudix enzymes with active sites more open to fit the target ADPr group bound to either a PAR polymer or protein, we turned to Nudix enzymes representative of families which are not swapped dimers and lack N- or C-terminal domain insertions, hypothesizing that enzymes with just the Nudix fold would have a more open active site. We chose four Nudix enzymes known to be monomeric by gel filtration35 (e.g. RppH as shown in Fig. 3). Two of them (Dr1184/CoAse from Deinococcus radiodurans and EcRppH from Escherichia coli) degraded the 32P-labeled PAR on the model protein PARP1 (Fig. 4a), while only EcRppH showed slight activity against 32P-labeled MAR at the tested concentration (Fig. 4b). Structural analysis and modeling revealed that the active site within these enzymes could accommodate protein-conjugated ADPr (Figs. 4c,d), as opposed to the dimeric ADPrases (c.f. Fig. 2d–g).
Figure 3. Sizing chromatography of the Nudix enzyme EcRppH.
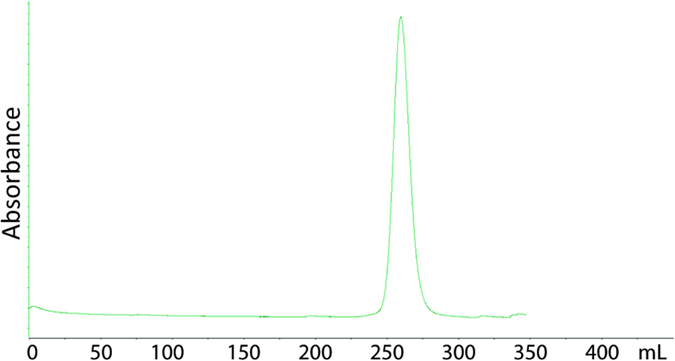
EcRppH size exclusion chromatography uses 50 mM Tris-HCl pH 7.5, 1 mM EDTA, 0.30 M NaCl at a flow rate of 2.0 mL/min; peak at 259.4 mL on a HiPrep 26/60 Superdex 200 prep grade. Molecular weight standards thyroglobulin Mr 670,000, peak 140.6 mL; γ-globulin Mr 158,000, peak 184.5 mL; ovalbumin Mr 44, 000, peak 226.2 mL; myoglobin Mr 17,000, peak 253.4 mL; vitamin B12 Mr 1350, peak 297.3.
Figure 4. Single domain Nudix hydrolases EcRppH and Dr1184 show activity against protein-conjugated ADPr.

(a,b) Autoradiograph showing the 32P-labeled PAR (a) or MAR (b) conjugated to PARP1 following exposure to Nudix hydrolases: AtORF147, Pa3470, Dr1184, and EcRppH. For (a), 5 pmoles of PARylated PARP1 was exposed to either 20 or 100 pmoles of hydrolase for 2 hrs at 37 °C. For (b), 5 pmoles of MARylated PARP1 E988Q mutant was exposed to 100 pmoles of hydrolase for 2 hrs at 37 °C. (c,d) show surface representations of Dr1184/CoAse (teal, PDB ID 1NQY) (c) and EcRppH (green, PDB ID 4S2Y) (d) with PAR modeled into their active sites based on the orientation of capped mRNA bound to EcRppH in the reported structure39. A dark shadow shows the possible orientation of the attached PARylated protein. ***SDS-PAGE well, **SDS-PAGE interface between stacking and resolving gels, *native size of PARP1, ^co-purified PARP1 protein fragments.
Both EcRppH and HsNudT16 can degrade protein-conjugated PAR and MAR to a phosphoribose tag for mass spectrometry
A recent study by Palazzo et al.13 has revealed that HsNudT16, a human Nudix (deoxy)inosine diphosphatase49 which is also known to decap small nucleolar RNAs50 as well as cytoplasmic mRNAs51, has the ability to degrade protein-conjugated ADPr. As shown in Fig. 5a–f HsNudT16 has a high structural similarity to both EcRppH and Dr1184 (which showed activity against protein-conjugated ADPr, see Fig. 4a,b) and also possesses an open active site which would allow for the target ADPr to be conjugated to a protein or additional ADPr unit(s). Based on these similarities, we postulated that the activity against protein-conjugated ADPr would be comparable for all three Nudix enzymes. To test this,32P-PARylated or MARylated PARP1 was exposed to increasing amounts of SVP, EcRppH, Dr1184 or HsNudT16. As shown via autoradiographs in Fig. 5g,h, both HsNudT16 and EcRppH are able to hydrolyze both protein-conjugated PAR and MAR.
Figure 5. HsNudT16 degrades protein-conjugated ADPr.

(a) A ribbon model of the structure of Dr1184/CoAse (teal, PDB ID 1NQY). (b) A ribbon model of the structure of EcRppH (green, PDB ID 4S2Y). (c) A ribbon model of HsNudT16 (PDB ID 2XSQ), (d) Surface representation of HsNudT16 with PAR modeled (sticks) and a protein depicted at the conjugation site of ADPr. Panel (e) shows a structural alignment between HsNudT16 and EcRppH, while (f) shows a structural alignment of HsNudT16, EcRppH and Dr1184. The grey hexagons in panels (a), (b), (c) and (e) show the helix that moves upon substrate binding. Panels (g) and (h) show the removal of 32P-labeled ADPr from 5 pmoles of PARylated (g) or MARylated (h) PARP1 by increasing amounts of SVP (0.1, 1 and 10 pmoles) or Dr1184, EcRppH or HsNudT16 (50, 250 and 750 pmoles). ***SDS-PAGE well, **SDS-PAGE interface between stacking and resolving gels, *native size of PARP1.
To validate that EcRppH and HsNudT16 are degrading protein-conjugated ADPr down to its phosphoribose attachment site, we treated 60 pmoles of automodified WT PARP1 with 120 pmoles of SVP, 3 nmoles of EcRppH or 3 nmoles of HsNudT16 (either 2x or 50x molar excess over PARP1, as per the activity assays shown in Fig. 5) before digesting the proteins to peptides and subjecting them to phosphoenrichment on an IMAC matrix5. As shown in Table 1, phosphoribosylation sites were confidently identified by mass spectrometry on PARP1 following treatment by each enzyme, demonstrating that all three enzymes are hydrolyzing the ADPr modification down to its phosphoribose attachment site. Notable differences were observed between the site identifications that were achieved following the use of these three enzymes — only two of the 14 phosphoribosylated peptide species were found in all samples, and all enzymes produced at least one unique peptide species. Taken together, these data suggest that these enzymes have an inherent bias for PTM structure, location, or length and therefore may be used in parallel to achieve greater coverage of the ADP-ribosylated proteome. Representative spectra from these runs have been annotated in Fig. 6, further confirming the evidence of phosphoribose modifications on these peptides.
Table 1. Phosphoribosylated PARP1 peptides identified by LC-MS/MS following hydrolysis of PARylated PARP1 by SVP, EcRppH and HsNudT16.
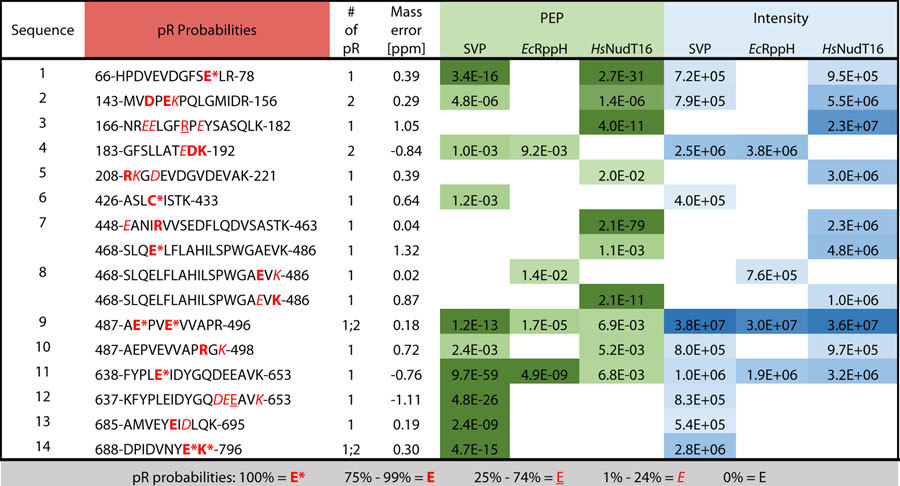
Phosphoribosylated peptides reported with their corresponding posterior error probabilities (PEP) and summed intensities from 60 pmoles of the model protein PARylated PARP1 after the conversion of ADPr to phosphoribose by 120 pmoles of SVP, 3 nmoles of EcRppH or 3 nmoles of HsNudT16. All elements of the table are found in the pR (DEKRC)Sites.txt output table from MaxQuant. Summed intensities include all forms of the peptide (for example, both the singly and doubly modified forms of peptides 9 and 14), PEPs are the most confident reported for each peptide, and mass errors [ppm] are the largest mass errors reported for each peptide identification included in this table.
Figure 6. Treatment of PARylated PARP1 protein with SVP, EcRppH and HsNudT16 results in the identification of phosphoribosylation sites by mass spectrometry.

(a) E491 (red, identified as 212 Dalton mass shift) on PARP1 was identified as a PARP1 automodification site following SVP, EcRppH and HsNudT16 treatment. (b) E190 and K192 were identified as PARP1 automodification sites on PARP1 following EcRppH digestion. (c) Identification of R452 as a PARP1 automodification site following HsNudT16 treatment.
Discussion
This work, along with that of Palazzo et al.13, has revealed new substrates for the RNA decapping Nudix hydrolases EcRppH and HsNudT16, as well as the CoAse Dr1184, though it must be noted that the existence of this activity against protein-conjugated ADPr in vivo has yet to be explored. In an attempt to determine the structural basis for this unique protein-conjugated ADPr hydrolase activity, we aligned these three proteins with respect to their known structures and identified a short, novel 310 helix downsteam from the helix of the Nudix signature sequence (Fig. 7a, red box), suggesting a possible significance to these structures for the degradation of protein-conjugated ADPr. We caution, however, that the structure is not known of the other two single domain Nudix hydrolases tested which lack the activity of interest: AtORF147 and Pa3470 (c.f. Fig. 4a,b). Moreover, the sequence homology between EcRppH/HsNudT16/Dr1184 and AtORF147/Pa3470 is not high beyond the Nudix signature sequence, so it is difficult to predict if the presence or absence of such short helices are the determining structural factor to facilitate the hydrolysis of protein-conjugated ADPr. Modeling of a trimeric PAR in the binding site of EcRppH positions the ribose towards the front of the enzyme allowing ample space for a protein to bind (Fig. 7b). Notably, the bond to be cleaved to leave a phosphoribose attached to a protein is in an ideal position to be hydrolyzed by the residues of the catalytic site of the Nudix enzyme (Fig. 7b). This model gives compelling structural arguments to the biochemistry data collected.
Figure 7. Alignment of Nudix enzymes with activity against protein-conjugated PAR reveals a novel 310 helix.

(a) Alignment of the sequences of EcRppH, HsNudT16 and Dr1184. The red background indicates sequence identity, red letters sequence indicates conservation of charge, and white background indicates non-homologous residues. The secondary structure of each enzyme is shown in blue. The box indicates a short 310 helix not observed in other Nudix enzymes such as the ADPRases. The secondary structure elements are more conserved between the two enzymes with activity towards PAR and MAR (EcRppH and HsNudT16) compared to Dr1184 that can only hydrolyze PAR. (b) Surface representation of the model of EcRppH with PAR modeled and minimized using the software package MOE. The diphosphate bond of the terminal ADP-ribose attached to a protein is in the active site, well positioned to be hydrolyzed and released as a phosphoribosylated protein product, where the scissile bond is marked.
Mass spectrometry-based proteomics represents the gold standard for the study of post-translational modifications, and the field of ADP-ribosylation will surely benefit from increased access to the suite of proteomic tools that have been developed for other PTMs, such as phosphorylation, acetylation and ubiquitylation. The advent of tag-based approaches for identifying ADP-ribosylation sites has begun to provide access to these tools, but adoption has been relatively low due to technical difficulties which accompany the current methods. The work presented here promises to streamline one of the major methods for ADPr site identification: the simplification of PAR or MAR to its phosphoribose attachment. Recombinant HsNudT16 and EcRppH can be synthesized and purified from E. coli, allowing for low-cost, high yield production which can be performed in most proteomic laboratories. Furthermore, since the structure is known for both of these enzymes, it is possible to predict mutations and truncations which could potentially increase the enzyme’s activity towards protein-conjugated ADPr; for example, introducing mutations which will further open up the active site to allow larger ADP-ribosylated substrates access.
The work presented here has the potential to greatly aid in the elucidation of the ADP-ribosylated proteome by providing researchers the necessary tools for generating phosphoribose as a mass spectrometry friendly site localization tag of protein ADP-ribosylation sites.
Additional Information
How to cite this article: Daniels, C. M. et al. Nudix hydrolases degrade protein-conjugated ADP-ribose. Sci. Rep. 5, 18271; doi: 10.1038/srep18271 (2015).
Acknowledgments
The authors would like to thank Dr. John Pascal for kindly sharing the human PARP-1 expression plasmid used to generate the WT and E988Q mutant proteins used in this study. We thanked Dr. Cynthia Wolberger for scientific discussions on this project. Work was funded by an NIH grant R01-GM104135 (A.K.L.L), the Safeway Research Foundation (A.K.L.L), the Patrick C. Walsh Prostate Cancer Research Fund (A.K.L.L.), the Allegheny Health Network–Johns Hopkins Cancer Research Fund (A.K.L.L and S.B.G), the Johns Hopkins Catalyst Award (A.K.L.L.), a Joy Cappel Young Investigator Award (C.M.D), an NCI training grant 5T32CA009110 (C.M.D), an NIAMS grant R01AR065459 (S-E.O), and an NCI grant P50 CA062924-SPORE in Gastrointestinal Cancer (S.B.G).
Footnotes
A pending US patent application for the utilization of recombinant EcRppH and its homologs in the identification of ADP-ribosylation sites has been filed (no. US 62/173,554 to C.M.D., A.K.L.L. and S.B.G.).
Author Contributions C.M.D., A.K.L.L. and S.B.G. conceived the project, designed the experiments and wrote the manuscript. C.M.D. and S.B.G. prepared the figures. S.E.O. and C.M.D. performed mass spectrometry analyses. S.B.G. performed structural analyses. S.B.G. and P.T. prepared all Nudix proteins. C.M.D. prepared PARP1 and SVP proteins. C.M.D. designed and performed biochemical screens. All authors reviewed the manuscript.
References
- Curtin N. J. & Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Molecular aspects of medicine 34, 1217–1256, 10.1016/j.mam.2013.01.006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger M. O., Hassa P. O., Luscher B., Schuler H. & Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends in biochemical sciences 35, 208–219, 10.1016/j.tibs.2009.12.003 (2010). [DOI] [PubMed] [Google Scholar]
- Vyas S. et al. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nature communications 5, 4426, 10.1038/ncomms5426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wang J., Ding M. & Yu Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nature methods 10, 981–984, 10.1038/nmeth.2603 (2013). [DOI] [PubMed] [Google Scholar]
- Daniels C. M., Ong S. E. & Leung A. K. A phosphoproteomic approach to characterize protein mono and poly(ADP-ribosyl)ation sites from whole cell lysate. Journal of proteome research, 10.1021/pr401032q (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman J. D., Gagne J. P., Poirier G. G. & Goodlett D. R. Mapping PARP-1 auto-ADP-ribosylation sites by liquid chromatography-tandem mass spectrometry. Journal of proteome research, 10.1021/pr301219h (2013). [DOI] [PubMed] [Google Scholar]
- Hengel S. M., Shaffer S. A., Nunn B. L. & Goodlett D. R. Tandem mass spectrometry investigation of ADP-ribosylated kemptide. Journal of the American Society for Mass Spectrometry 20, 477–483, 10.1016/j.jasms.2008.10.025 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oetjen J., Rexroth S. & Reinhold-Hurek B. Mass spectrometric characterization of the covalent modification of the nitrogenase Fe-protein in Azoarcus sp. BH72. The FEBS journal 276, 3618–3627, 10.1111/j.1742-4658.2009.07081.x (2009). [DOI] [PubMed] [Google Scholar]
- Tao Z., Gao P. & Liu H. W. Studies of the expression of human poly(ADP-ribose) polymerase-1 in Saccharomyces cerevisiae and identification of PARP-1 substrates by yeast proteome microarray screening. Biochemistry 48, 11745–11754, 10.1021/bi901387k (2009). [DOI] [PubMed] [Google Scholar]
- Messner S. et al. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic acids research 38, 6350–6362, 10.1093/nar/gkq463 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal F. et al. Identification of distinct amino acids as ADP-ribose acceptor sites by mass spectrometry. Methods in molecular biology 780, 57–66, doi: 10.1007/978-1-61779-270-0_4 (2011). [DOI] [PubMed] [Google Scholar]
- Hengel S. M. & Goodlett D. R. A Review of Tandem Mass Spectrometry Characterization of Adenosine Diphosphate-Ribosylated Peptides. International journal of mass spectrometry 312, 114–121, 10.1016/j.ijms.2011.06.003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo L. et al. Processing of Protein ADP-ribosylation by Nudix Hydrolases. The Biochemical journal, 10.1042/BJ20141554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels C. M., Ong S. E. & Leung A. K. The Promise of Proteomics for the Study of ADP-Ribosylation. Molecular cell 58, 911–924, 10.1016/j.molcel.2015.06.012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessman M. J., Frick D. N. & O’Handley S. F. The MutT proteins or “Nudix” hydrolases, a family of versatile, widely distributed, “housecleaning” enzymes. The Journal of biological chemistry 271, 25059–25062 (1996). [DOI] [PubMed] [Google Scholar]
- Mildvan A. S. et al. Structures and mechanisms of Nudix hydrolases. Archives of biochemistry and biophysics 433, 129–143, 10.1016/j.abb.2004.08.017 (2005). [DOI] [PubMed] [Google Scholar]
- Tong L., Lee S. & Denu J. M. Hydrolase regulates NAD + metabolites and modulates cellular redox. The Journal of biological chemistry 284, 11256–11266, 10.1074/jbc.M809790200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelli S. B., Bianchet M. A., Bessman M. J. & Amzel L. M. The structure of ADP-ribose pyrophosphatase reveals the structural basis for the versatility of the Nudix family. Nature structural biology 8, 467–472, 10.1038/87647 (2001). [DOI] [PubMed] [Google Scholar]
- Dunn C. A., O’Handley S. F., Frick D. N. & Bessman M. J. Studies on the ADP-ribose pyrophosphatase subfamily of the nudix hydrolases and tentative identification of trgB, a gene associated with tellurite resistance. The Journal of biological chemistry 274, 32318–32324 (1999). [DOI] [PubMed] [Google Scholar]
- Perraud A. L. et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411, 595–599, 10.1038/35079100 (2001). [DOI] [PubMed] [Google Scholar]
- Durnin L., Hwang S. J., Ward S. M., Sanders K. M. & Mutafova-Yambolieva V. N. Adenosine 5-diphosphate-ribose is a neural regulator in primate and murine large intestine along with beta-NAD(+). The Journal of physiology 590, 1921–1941, 10.1113/jphysiol.2011.222414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson E. L., Cervantes-Laurean D. & Jacobson M. K. Glycation of proteins by ADP-ribose. Molecular and cellular biochemistry 138, 207–212 (1994). [DOI] [PubMed] [Google Scholar]
- Miwa M. & Sugimura T. Splitting of the ribose-ribose linkage of poly(adenosine diphosphate-robose) by a calf thymus extract. The Journal of biological chemistry 246, 6362–6364 (1971). [PubMed] [Google Scholar]
- Steffen J. D. & Pascal J. M. New players to the field of ADP-ribosylation make the final cut. The EMBO journal 32, 1205–1207, 10.1038/emboj.2013.83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull N. P., Spinelli S. L. & Phizicky E. M. A highly specific phosphatase that acts on ADP-ribose 1"-phosphate, a metabolite of tRNA splicing in Saccharomyces cerevisiae. Nucleic acids research 33, 650–660, 10.1093/nar/gki211 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolle C., Rack J. G. & Ziegler M. NAD and ADP-ribose metabolism in mitochondria. The FEBS journal 280, 3530–3541, 10.1111/febs.12304 (2013). [DOI] [PubMed] [Google Scholar]
- Peterson F. C. et al. Orphan macrodomain protein (human C6orf130) is an O-acyl-ADP-ribose deacylase: solution structure and catalytic properties. The Journal of biological chemistry 286, 35955–35965, 10.1074/jbc.M111.276238 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Jacobson E. L. & Jacobson M. K. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science 261, 1330–1333 (1993). [DOI] [PubMed] [Google Scholar]
- McLennan A. G. The Nudix hydrolase superfamily. Cellular and molecular life sciences : CMLS 63, 123–143, 10.1007/s00018-005-5386-7 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deana A., Celesnik H. & Belasco J. G. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451, 355–358, 10.1038/nature06475 (2008). [DOI] [PubMed] [Google Scholar]
- Li Y., Song M. & Kiledjian M. Differential utilization of decapping enzymes in mammalian mRNA decay pathways. Rna 17, 419–428, 10.1261/rna.2439811 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Handley S. F., Frick D. N., Dunn C. A. & Bessman M. J. Orf186 represents a new member of the Nudix hydrolases, active on adenosine(5′)triphospho(5′)adenosine, ADP-ribose, and NADH. The Journal of biological chemistry 273, 3192–3197 (1998). [DOI] [PubMed] [Google Scholar]
- Kang L. W. et al. Structure of a coenzyme A pyrophosphatase from Deinococcus radiodurans: a member of the Nudix family. Journal of bacteriology 185, 4110–4118 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessman M. J. et al. The gene ygdP, associated with the invasiveness of Escherichia coli K1, designates a Nudix hydrolase, Orf176, active on adenosine (5′)-pentaphospho-(5′)-adenosine (Ap5A). The Journal of biological chemistry 276, 37834–37838, 10.1074/jbc.M107032200 (2001). [DOI] [PubMed] [Google Scholar]
- Xu W., Shen J., Dunn C. A. & Bessman M. J. A new subfamily of the Nudix hydrolase superfamily active on 5-methyl-UTP (ribo-TTP) and UTP. The Journal of biological chemistry 278, 37492–37496, 10.1074/jbc.M307639200 (2003). [DOI] [PubMed] [Google Scholar]
- Krissinel E. & Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta crystallographica. Section D, Biological crystallography 60, 2256–2268, 10.1107/S0907444904026460 (2004). [DOI] [PubMed] [Google Scholar]
- Schrodinger L. L. C. The PyMOL Molecular Graphics System, Version 1.3r1 (2010).
- Molecular Operating Environment (MOE) 2013.08. Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7 (2015).
- Vasilyev N. & Serganov A. Structures of RNA Complexes with the Escherichia coli RNA Pyrophosphohydrolase RppH Unveil the Basis for Specific 5′-End-dependent mRNA Decay. The Journal of biological chemistry 290, 9487–9499, 10.1074/jbc.M114.634824 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresaugues L. et al. Structural Basis for the Specificity of Human NUDT16 and Its Regulation by Inosine Monophosphate. PloS one 10, e0131507, 10.1371/journal.pone.0131507 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y., Ueda K., Nakazawa K. & Hayaishi O. Studies on the polymer of adenosine diphosphate ribose. I. Enzymic formation from nicotinamide adenine dinuclotide in mammalian nuclei. The Journal of biological chemistry 242, 3164–3171 (1967). [PubMed] [Google Scholar]
- Kawaichi M., Ueda K. & Hayaishi O. Multiple autopoly(ADP-ribosyl)ation of rat liver poly(ADP-ribose) synthetase. Mode of modification and properties of automodified synthetase. The Journal of biological chemistry 256, 9483–9489 (1981). [PubMed] [Google Scholar]
- Desmarais Y., Menard L., Lagueux J. & Poirier G. G. Enzymological properties of poly(ADP-ribose)polymerase: characterization of automodification sites and NADase activity. Biochimica et biophysica acta 1078, 179–186 (1991). [DOI] [PubMed] [Google Scholar]
- Rokyta D. R., Lemmon A. R., Margres M. J. & Aronow K. The venom-gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC genomics 13, 312, 10.1186/1471-2164-13-312 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka J., Ueda K. & Hayaishi O. Snake venom phosphodiesterase: simple purification with Blue Sepharose and its application to poly(ADP-ribose) study. Biochemical and biophysical research communications 80, 841–848 (1978). [DOI] [PubMed] [Google Scholar]
- Cao J. et al. Identification of N-glycosylation sites on secreted proteins of human hepatocellular carcinoma cells with a complementary proteomics approach. Journal of proteome research 8, 662–672, 10.1021/pr800826u (2009). [DOI] [PubMed] [Google Scholar]
- Soares S. G. & Oliveira L. L. Venom-sweet-venom: N-linked glycosylation in snake venom toxins. Protein and peptide letters 16, 913–919 (2009). [DOI] [PubMed] [Google Scholar]
- Gabelli S. B. et al. Mechanism of the Escherichia coli ADP-ribose pyrophosphatase, a Nudix hydrolase. Biochemistry 41, 9279–9285 (2002). [DOI] [PubMed] [Google Scholar]
- Abolhassani N. et al. NUDT16 and ITPA play a dual protective role in maintaining chromosome stability and cell growth by eliminating dIDP/IDP and dITP/ITP from nucleotide pools in mammals. Nucleic acids research 38, 2891–2903, 10.1093/nar/gkp1250 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh T., Peterson B., Tomasevic N. & Peculis B. A. Xenopus U8 snoRNA binding protein is a conserved nuclear decapping enzyme. Molecular cell 13, 817–828 (2004). [DOI] [PubMed] [Google Scholar]
- Song M. G., Li Y. & Kiledjian M. Multiple mRNA decapping enzymes in mammalian cells. Molecular cell 40, 423–432, 10.1016/j.molcel.2010.10.010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff K. A. et al. A redox regulatory system critical for mycobacterial survival in macrophages and biofilm development. PLoS pathogens 11, e1004839, 10.1371/journal.ppat.1004839 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]