

Abstract
Protein phosphatase 2A (PP2A) is a serine/threonine-selective holoenzyme composed of a catalytic, scaffolding, and regulatory subunit. In the heart, PP2A activity is requisite for cardiac excitation-contraction coupling and central in adrenergic signaling. We found that mice deficient in the PP2A regulatory subunit B56α (1 of 13 regulatory subunits) had altered PP2A signaling in the heart that was associated with changes in cardiac physiology, suggesting that the B56α regulatory subunit had an autoinhibitory role that suppressed excess PP2A activity. The increase in PP2A activity in the mice with reduced B56α expression resulted in slower heart rates and increased heart rate variability, conduction defects, and increased sensitivity of heart rate to parasympathetic agonists. Increased PP2A activity in B56α+/− myocytes resulted in reduced Ca2+ waves and sparks, which was associated with decreased phosphorylation (and thus decreased activation) of the ryanodine receptor RyR2, an ion channel on intracellular membranes that is involved in Ca2+ regulation in cardiomyocytes. In line with an autoinhibitory role for B56α, in vivo expression of B56α in the absence of altered abundance of other PP2A subunits decreased basal phosphatase activity. Consequently, in vivo expression of B56α suppressed parasympathetic regulation of heart rate and increased RyR2 phosphorylation in cardiomyocytes. These data show that an integral component of the PP2A holoenzyme has an important inhibitory role in controlling PP2A enzyme activity in the heart.
INTRODUCTION
Protein phosphorylation is tightly regulated through the coordinate activities of kinases and phosphatases. In response to acute stress or chronic disease, increased sympathetic input to the heart tunes cardiac automaticity and contractility through protein phosphorylation. Defects in phosphorylation cascades are directly linked to various cardiac pathologies including sinoatrial node disease, heart failure, and arrhythmia (1–3). In heart failure, increased kinase activity is associated with defects in excitation-contraction coupling, arrhythmias, and metabolic depletion of the heart (2, 4). Clinically, suppression of kinase activity through the use of β-adrenergic receptor blockers in heart failure has remained a mainstay to mitigate morbidity and mortality (3, 5, 6). However, protein kinases represent just one arm of the protein phosphorylation cascade. Kinase activity is countered by the enzymatic action of protein phosphatases that dephosphorylate the target substrates of kinases. The regulatory role of phosphatases in normal cardiac physiology and disease is poorly understood and has emerged as a critical element in regulating cardiac excitability and contractile function.
Protein phosphatase 2A (PP2A) is a serine/threonine phosphatase that is ubiquitously distributed in many tissues, including the heart. Unlike many monomeric enzymes, PP2A is a holoenzyme composed of three subunits: the A structural subunits, the C catalytic subunits, and the B regulatory subunits. In vertebrates, PP2A structural and catalytic subunits are encoded by 2 genes, whereas regulatory subunits are encoded by 13 genes (7). Because of their cell, tissue, and, presumably, target specificity, previous work in myocytes has illustrated that modulation of protein phosphatase subunits may represent a therapeutic avenue to treat aberrant cardiac electrical activity and arrhythmia (8–10). Studies using global phosphatase inhibitors have suggested a role for PP2A and other phosphatases to tune the cardiac inotropic response (11–13). In vitro work in myocytes has led to the proposal that microRNA (miR)–dependent reduction in the PP2A regulatory subunit B56α promotes arrhythmia susceptibility by suppressing dyadic PP2A activity, thus increasing the phosphorylation of the ryanodine receptor (RyR2) and promoting diastolic Ca2+ sparks, waves, and after-depolarizations (14, 15). Because inhibiting PP2A is a potential strategy for the prevention of common forms of arrhythmia associated with increased adrenergic activity, we tested the in vivo role of the B56α regulatory subunit in cardiac signaling and function.
Here, we found that cardiac PP2A-dependent phosphatase activity was directly regulated by the B56α subunit. Specifically, we identified B56α as an autoinhibitor of cardiac PP2A-dependent activity in vivo. B56α+/− and B56α −/− mice displayed an increase in PP2A-dependent phosphatase activity, and consistent with these data, we observe whole-animal, cellular, and molecular phenotypes directly linked with augmented PP2A. At the whole-animal level, mice with decreased B56α expression did not show changes in the abundance of other PP2A subunits and displayed decreased heart rate, heart rate variability, conduction defects, and increased sensitivity to parasympathetic agonists. At the myocyte level, reduced B56α expression was associated with reduced Ca2+ waves and sparks in the presence of sympathetic stimulation. Finally, at the molecular level, B56α deficiency results in reduced phosphorylation of RyR2. In contrast, in vivo B56α overexpression, in the absence of changes in expression of other PP2A subunits, resulted in decreased phosphatase and parasympathetic activity associated with increased phosphorylation of RyR2. These data highlight the role of PP2A regulatory subunits for modulating cardiac signaling and function.
RESULTS
Mice with B56α deficiency do not display compensatory changes in the abundance of other PP2A subunits
To investigate the in vivo mechanisms for PP2A regulation in the heart, we engineered a mouse model with reduced B56α expression through a gene trap strategy, creating an early transcriptional termination sequence after the first exon of Ppp2r5a (fig. S1, A and B). Because our initial goal was to assess the effect of reduced B56α abundance (rather than complete B56α deficiency) on PP2A function and cardiac phenotypes, we first evaluated mice heterozygous for the null Ppp2r5a allele (fig. S1, A to C; B56α+/− mice). B56α+/− mice displayed ~50% reduction of Ppp2r5a in the heart by quantitative real-time fluorescence polymerase chain reaction (qPCR) (fig. S1D). Moreover, B56α+/− mice displayed ~40% reduction of B56α protein by immunoblot (fig. S1, E and F). The abundance of other regulatory subunits was not altered in B56α+/− heart. For example, the abundance of B56γ, the most homologous B56 gene product, and PP2A catalytic and scaffolding subunits was similar in wild-type and B56α+/− hearts (fig. S1, E and G to I). PP2A activity is modified by methylation through leucine carboxymethyltransferase 1 (LCMT-1) and phosphatase methylesterase (PME-1) (7). The abundance of LCMT-1 and PME-1 was similar between genotypes (fig. S1, E, J, and K). Thus, the B56α+/− mouse provides an in vivo model to define the role of reduced B56α deficiency on PP2A regulation and cardiac physiology.
Reduced expression of B56α increases cardiac PP2A activity
The consequence of reduced PP2A regulatory subunit expression on PP2A activity is debated. Some in vitro studies suggest that B56α deficiency may reduce PP2A holoenzyme formation and subsequently decrease cellular PP2A activity (14, 15). However, structural data support a model in which the PP2A scaffolding A and catalytic C subunits form a more active but less regulated enzyme in the absence of regulatory subunit association (16). Further, there is evidence that simply increasing the ratio of catalytic (C) subunit to regulatory (B) subunit may be sufficient to increase PP2A activity (17). To directly address this controversy, we assessed total and PP2A-specific phosphatase activity in B56α+/− mouse hearts. PP2A-specific activity was identified as the fostriecin-sensitive fraction of total phosphatase activity (14). Although global phosphatase abundance did not differ between wild-type and B56α+/− hearts (Fig. 1A), PP2A-dependent phosphatase activity was significantly increased in B56α+/− hearts (Fig. 1B). In B56α−/− hearts, we observed increased PP2A (and total protein phosphatase) activity relative to either wild-type or B56α+/− hearts (Fig. 1, A and B). To test whether these findings were selective to the heart, we analyzed PP2A activity in cerebellar lysates of wild-type and B56α-deficient mice. Similar to the heart, PP2A activity was increased in the cerebellum of B56α-deficient mice compared with wild-type littermates (Fig. 1C). These data identify an inhibitory role of the B56α subunit in phosphatase regulation, such that reduced B56α expression increases PP2A activity.
Fig. 1. B56α-deficient mice display increased PP2A activity and abnormal heart rate regulation.
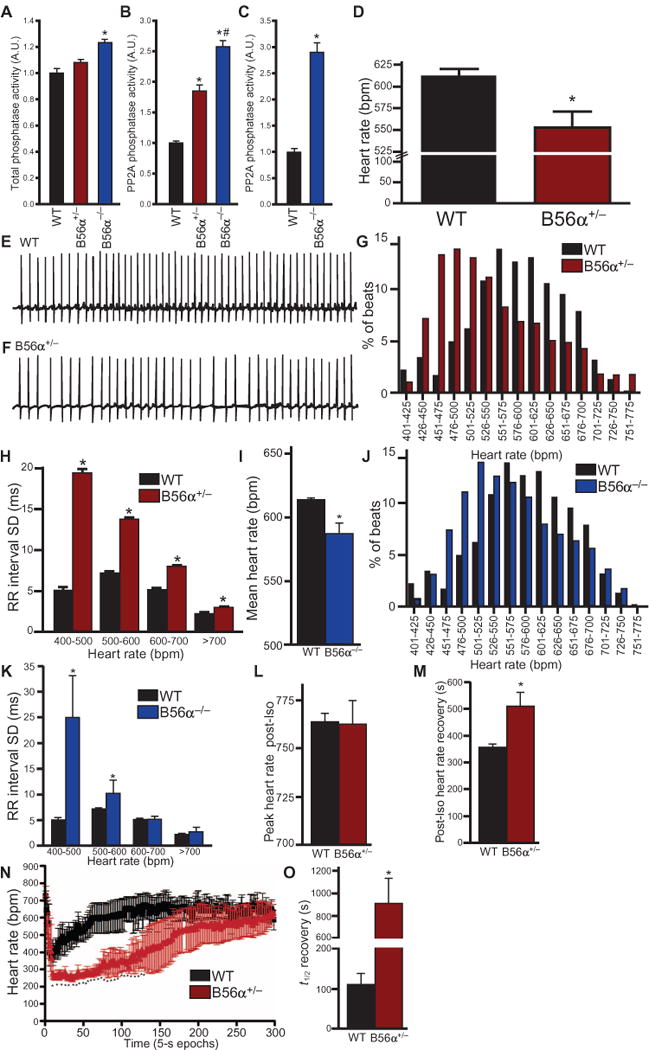
(A) Wild-type (WT) (n = 6) and B56α+/− hearts (n = 6) had no difference in total protein phosphatase activity [P = N.S. (not significant)]. B56α−/− hearts (n = 6) showed increased protein phosphatase abundance compared to WT hearts (*P < 0.05 for B56α−/− compared to WT). A.U., arbitrary unit. (B) PP2A-specific phosphatase activity was significantly increased in B56α+/− and B56α−/− hearts compared to WT hearts (n = 6 hearts per genotype; *P < 0.05 compared to WT; #P < 0.05 of B56α+/− to B56α−/−). (C) PP2A-specific phosphatase activity was significantly higher in B56α−/− than in WT cerebellar lysates (*P < 0.05 compared to WT; n = 4 mice per genotype). (D) At baseline, B56α+/− mice display decreased heart rate compared with WT mice (n = 4 mice per genotype; *P < 0.05). bpm, beats per minute. (E and F) Representative 5-s ECG recordings showing bradycardia and heart rate variability in WT mice (E) compared with B56α+/− (F) littermates. (G) Heart rate distribution of WT and B56α+/− mice demonstrates the leftward shift to lower heart rates in B56α+/− mice when plotted as % of total incidences at defined heart rate intervals (bars represent all data points from n = 4 mice per genotype; *P < 0.05). (H) The average SD from each 5-s RR interval at the indicated heart rates, plotted to demonstrate the increased heart rate variability in B56α+/− compared to WT mice (n = 4 mice per genotype; *P < 0.05). (I) Quantification of mean heart rate for conscious WT and B56α−/− mice (n = 4 mice per genotype; *P < 0.05). (J) Heart rate distribution for WT and B56α−/− mice demonstrates a leftward shift to lower heart rates in B56α−/− mice when plotted as % of total incidences at defined heart rate intervals (bars represent all data points from n = 4 mice per genotype; *P < 0.05). (K) The average SD from each 5-s averaged RR interval at the indicated heart rates, plotted to demonstrate the increased heart rate variability in B56α−/− compared to WT mice (n = 4 mice per genotype; *P < 0.05). (L) WT and B56α+/− mice displayed similar peak heart rates after isoproterenol (Iso) injection (n = 3 mice per genotype; P = N.S.). (M) Heart rate recovery after isoproterenol injection was longer in B56α+/− mice than in WT mice (n = 3 mice per genotype; *P < 0.05). (N and O) After acetylcholine receptor activation with carbachol (CCH), B56α+/− mice had a more pronounced reduction in heart rate and increased period for heart rate recovery compared to WT mice (n = 3 mice per genotype; *P < 0.05 compared to WT).
B56α+/− and B56−/− mice display cardiac phenotypes linked with increased PP2A activity
Heart rate is tightly regulated by sympathetic and parasympathetic tone, and adrenergic imbalance is linked with sinus node disease (18). We found that B56α+/− mice displayed bradycardia at baseline (Fig. 1, D to G), heart rate variability compared with wild-type littermates (Fig. 1H), and no significant difference in QT interval (fig. S2). Similar to B56α+/− mice, B56α−/− mice displayed bradycardia and heart rate variability compared with wild-type mice (Fig. 1, I to K).
Because PP2A modulates cardiac signaling in response to increased sympathetic tone (8), we examined cardiac function in B56α+/− mice after β-adrenergic receptor stimulation. Wild-type and B56α+/− mice showed similar peak heart rates in response to injection of isoproterenol (Fig. 1L) or strenuous exercise (fig. S3). However, in contrast to the rapid recovery of heart rate after isoproterenol treatment in wild-type mice, B56α+/− mice displayed a significant increase in the time for heart rate recovery (Fig. 1M). Further, sympathetic stimulation accentuated heart rate variability and conduction phenotypes in B56α+/− mice. For example, B56α+/− mice regularly had second-degree atrioventricular block, characterized by continual PR interval prolongation followed by a dropped QRS signature (fig. S4, A to D). In addition, we observed sinus node pause or block indicated by the absence of P waves followed by a ventricular escape beat in B56α+/− mice, phenotypes associated with increased parasympathetic stimulation (fig. S4, A to D). B56α+/− or B56α−/− mice did not have ventricular arrhythmias after low- or high-dose isoproterenol treatment or in response to exercise-induced stress. In summary, mice with reduced B56α expression display sinus node and atrioventricular node arrhythmias consistent with increased PP2A activity and lack electrical phenotypes associated with ventricular arrhythmias at rest or after catecholamine stimulation.
B56α-deficient mice display aberrant muscarinic regulation
To further evaluate the role of increased PP2A function on cardiac function, we tested B56α+/− mice for increased sensitivity to parasympathetic agonists. The cholinergic agonist carbachol produced a transient decrease in heart rate followed by rapid recovery in wild-type mice. In contrast, B56α+/− mice had an increased parasympathetic heart rate response to carbachol and significantly prolonged recovery period (Fig. 1, N and O). We observed similar findings in B56α−/− mice (fig. S5). Electrocardiograms (ECGs) from wild-type mice showed an initial carbachol-dependent decrease in heart rate that rapidly returned to normal sinus rhythm (Fig. 1, N and O, and fig. S6, A and B). In contrast, after carbachol treatment, B56α+/− mice had sinoatrial node pause and/or atrioventricular block (fig. S6, C and D). During recovery, B56α+/− mice had grouped beating, indicative of sinoatrial node block. We next analyzed heart rate responses after treatment with atropine, a cholinergic receptor antagonist. Wild-type, B56α+/−, and B56α−/− mice showed similar heart rates in response to injected atropine (fig. S7, A and B). Together, these findings support the role of an enhanced parasympathetic response and increased PP2A activity in mice with reduced B56α expression.
B56α+/− myocytes display reduced spark frequency and decreased Ca2+ waves
Cardiac adrenergic regulation is mediated by the integration of cardiac and nervous system signaling. Therefore, we tested the role of B56α deficiency on cardiac-intrinsic pathways using isolated cardiomyocytes. Excitation-contraction coupling is tightly regulated by adrenergic signaling pathways that tune intracellular Ca2+ release and, subsequently, Ca2+ waves. Both cyclic adenosine monophosphate–dependent protein kinase A (PKA) and Ca2+/calmodulin-dependent protein kinase II (CaMKII) pathways augment spontaneous intracellular Ca2+ release through phosphorylation of the cardiac RyR2 at distinct sites (Ser2808 and Ser2814, respectively) (6, 19). Our data demonstrated that B56α reduction resulted in increased PP2A activity that would be expected to decrease spark and Ca2+ wave frequency. Consistent with this hypothesis, spark frequency was reduced in B56α+/− myocytes (Fig. 2, A to E). Moreover, Ca2+ wave frequency after isoproterenol exposure was also significantly reduced in B56α+/− myocytes (Fig. 2F) despite increased sarcoplasmic reticulum (SR) load and increased Ca2+ transient amplitude (Fig. 2, G and H). These data suggest a decrease in Ca2+ sensitivity of the RyR2, typically associated with reduced phosphorylation of RyR2. Thus, B56α is required for normal SR Ca2+ sparks and Ca2+ waves, and reduced abundance of this protein decreases spark frequency and Ca2+ waves.
Fig. 2. Myocytes from B56α-deficient mice display aberrant Ca2+ regulation.
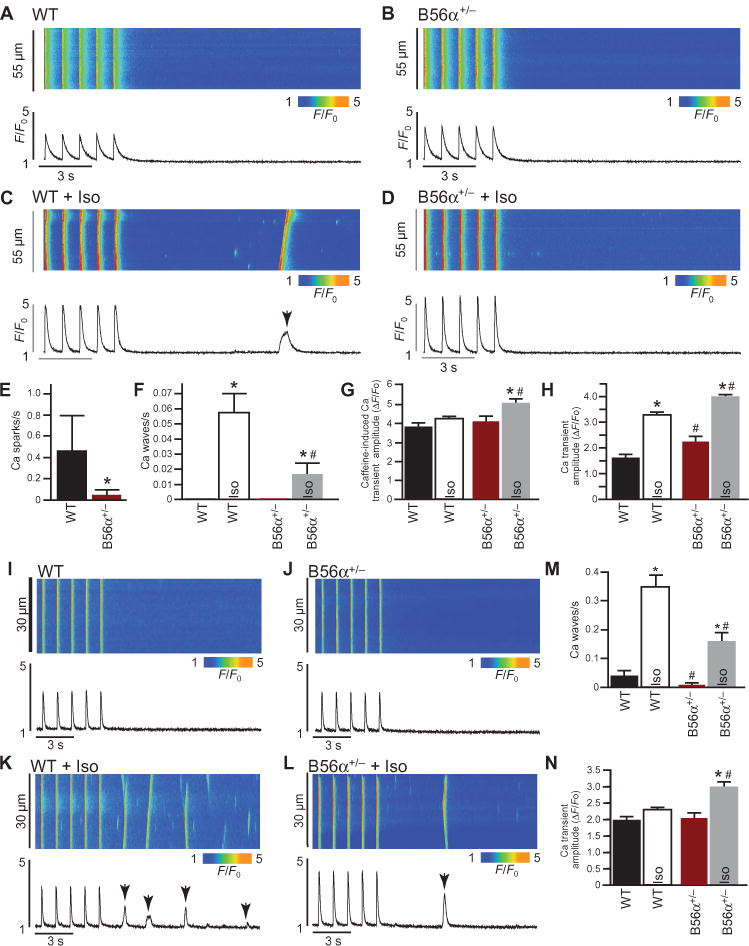
(A to D) Representative line-scan images and corresponding Ca2+ transients normalized to the basal fluorescence (F0) of 1 Hz–stimulated ventricular myocytes, in the absence or presence of Iso. (E) Spark frequency was decreased in B56α+/− ventricular myocytes at baseline compared to WT myocytes (*P < 0.05). (F) Iso increased Ca2+ wave frequency for both WT and B56α+/− ventricular myocytes (*P < 0.05). Ca2+ wave frequency was decreased in B56α+/− + Iso compared to WT + Iso ventricular myocytes (#P < 0.05). (G) Mean SR Ca2+ load in WT and B56α+/− ventricular myocytes at baseline ± Iso (*P < 0.05 B56α+/− compared to B56α+/− + Iso; #P < 0.05 WT + Iso compared with B56α+/− + Iso). (H) Ca2+ transient amplitudes in WT and B56α+/− ventricular myocytes at baseline ± Iso (*P < 0.05 from respective baselines; #P < 0.05 WT + Iso compared to B56α+/− + Iso). (I to L) Representative line-scan images and corresponding Ca2+ transients normalized to F0 of 1 Hz–stimulated atrial myocytes, in the absence or presence of Iso. (M) Iso increased Ca2+ wave frequency for both WT and B56α+/− atrial myocytes (*P < 0.05). Ca2+ wave frequency was decreased in B56α+/− + Iso compared to WT + Iso atrial myocytes (#P < 0.05). (N) Ca2+ transient amplitudes in WT and B56α+/− atrial myocytes at baseline ± Iso (*P < 0.05 from baseline; #P < 0.05 WT + Iso compared to B56α+/− + Iso). For (A) to (M), myocytes were isolated from n = 4 mice per genotype. A minimum of 20 myocytes were analyzed for each independent myocyte preparation.
B56α is critical for the regulation of atrial function and atrial myocyte calcium signaling
PP2A-dependent regulation of RyR2 activity is also critical for atrial function. Because B56α+/− mice showed defects in atrial function (Fig. 1), we tested the effect of reduced B56α expression on atrial function at both the level of the organism as well as the single atrial myocyte. Treatment with atropine plus propranolol [a validated protocol for combined autonomic blockade of sympathetic and parasympathetic signaling (20)] revealed a mild yet significant reduction in the intrinsic heart rate of B56α-deficient mice (fig. S8). Analysis of single isolated atrial myocytes revealed similar PP2A-based defects in Ca2+ regulation as observed in ventricular myocytes, most notably decreased Ca2+ wave phenotypes compared with wild-type atrial myocytes (Fig. 2, I to N). Together, these data demonstrate that altered atrial function in mice with reduced B56α is associated with cardiac-intrinsic effects and altered atrial myocyte Ca2+ regulation.
B56α+/− and B56α−/− hearts display reduced phosphorylation of RyR2
As a first step to establish the cellular and molecular mechanisms for the B56α-dependent phenotypes, we examined the phosphorylation status of PP2A targets associated with excitation-contraction coupling. Consistent with increased PP2A activity and decreased Ca2+ spark and wave frequency in B56α+/− myocytes, B56α+/− and B56α−/− ventricles showed decreased phosphorylation of RyR2 of Ser2808 and Ser2814 (Fig. 3, A and B, and fig. S9) without changes in the abundance of the PKA catalytic subunit, CaMKII, PP1, SERCA2, or PLB, or the phosphorylation of PLB at Ser16 or Thr17 (Fig. 3, A and B). Coimmunoprecipitation experiments demonstrated that B56α was associated with the PP2A/C and RyR2 complex (fig. S10), but B56α was not required for the association between PP2A/C and RyR2 (fig. S10). Finally, consistent with findings in ventricle, and with atrial phenotypes at the level of the animal and single atrial myocyte, phosphorylation of RyR2 at Ser2808 and Ser2814 was decreased in B56α+/− and B56α−/− atria (fig. S11).
Fig. 3. B56α+/− hearts display reduced RyR2 phosphorylation.
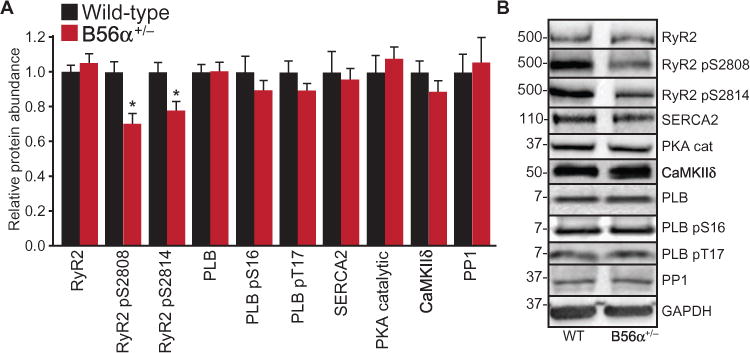
(A) Quantification of myocyte proteins from WT and B56α+/− heart lysates. Total RyR2 abundance was unchanged, and B56α+/− mice showed reduced phosphorylation (p) of RyR2 at Ser2808 and Ser2814 (*P < 0.05; abundance was normalized to total RyR2). The total abundance of PLB, SERCA2, PKA catalytic (cat) subunit, CaMKIId, or protein phosphatase 1 (PP1), and the phosphorylation of PLB at Ser16 or Thr17 (normalized to total PLB) did not differ. n = 4 mouse hearts per genotype; *P < 0.05. (B) Representative immunoblots of WT and B56α+/− lysates.
B56α+/− hearts do not show changes in the phosphorylation status of select myofilament proteins
PP2A has other targets in addition to the excitation-contraction coupling machinery in the heart, and its activity is tied to cardiac contractile function, metabolism, and with cardiac stress pathways likely unrelated to B56α (21, 22). We therefore tested the impact of B56α reduction on the phosphorylation status of key myofilament proteins. The phosphorylation of myofilament PP2A target proteins including myosin, myosin-binding protein C, troponin T, tropomyosin, troponin I, and myosin light chain were not significantly different between wild-type and B56α+/− hearts (fig. S12).
B56α+/− and B56α−/− myocytes display increased perinuclear and nuclear PP2A localization
B56α regulates the targeting of PP2A in myocytes (23). We therefore examined the localization of PP2A core complex in B56α+/− and B56α−/− myocytes. As shown by our group and others, PP2A core enzyme subunits are distributed throughout the myocyte, in the cytosol, myofilaments, and nuclear envelope (fig. S13A) (7, 24). However, in contrast to wild-type myocytes, localization of PP2A/A to the nuclear envelope and nucleus was increased in B56α+/− and B56α−/− myocytes (fig. S13, B and C). All PP2A/A localization was not dependent on B56α because we observed secondary cytosolic populations of PP2A/A in B56α−/− myocytes (fig. S13C). Defining additional pathways for PP2A core subunit targeting will be an important future area of research. These pathways may include other PP2A regulatory subunits as proposed by Rogers and colleagues (24) or by the direct association of PP2A core enzyme subunits with PP2A targets (5, 25).
B56α abundance is increased in mouse model of cardiac ankyrin-B deficiency
B56α is targeted to the cardiac dyad by association with the membrane adapter ankyrin-B (23). Loss of ankyrin-B in myocytes results in aberrant B56α targeting in myocytes, CaMKII-dependent hyperphosphorylation of RyR2, altered RyR2 open probability, afterdepolarizations, and arrhythmia (23, 26, 27). In patients harboring ANK2 loss-of-function variants, this mechanism underlies a form of potentially fatal catecholaminergic polymorphic ventricular arrhythmia (26, 27). On the basis of our data, a major question was the basis for differential phenotypes observed between ankyrin-B+/− and B56α+/− mice. To address this question, we used an animal model of cardiac-selective ankyrin-B deficiency (aMHC-Cre; Ank2f/f; “ankyrin-B−/− mice”). These mice showed increased B56α abundance (fig. S14). Thus, mice with ankyrin-B and B56α deficiencies display distinct cellular phenotypes resulting in differential cardiac pathologies.
In vivo B56α expression augments cardiac sympathetic signaling
Reduced B56α expression results in increased cardiac PP2A activity and augmented parasympathetic function, supporting the role of B56α as an inhibitor of PP2A signaling. To explore this mechanism, we examined the in vivo sympathetic and parasympathetic balance of mice with increased B56α expression (B56α overexpression). B56α expression was targeted to the myocardium of wild-type mice by AAV9 [with internal ribosomal entry site (IRES) mCherry reporter]. The identical construct lacking the B56α complementary DNA (cDNA) (AAV9 mCherry alone) was used to control for the effect of viral transduction on cardiac function. Transgene expression was confirmed in the heart by mCherry expression in atrial and ventricular myocytes (Fig. 4, A and B), and immunoblot of whole-heart lysates (Fig. 4C) 4 weeks after expression. Transgene expression was equivalent between atria and ventricle (Fig. 4, B and D). Consistent with the proposed role of B56α as an inhibitor of PP2A activity, PP2A-dependent phosphatase activity was decreased in B56α-transduced hearts compared with wild-type hearts (Fig. 4E). Moreover, these mice showed altered cardiac physiology, including a higher resting heart rate (Fig. 4F), increased peak heart rate in response to either exercise or isoproterenol stimulation (Fig. 4G), and altered responses to carbachol (Fig. 4, G to I). Specifically, we observed a significantly blunted heart rate response to carbachol treatment (Fig. 4, G and H). Further, expression of AAV9 B56α was sufficient to restore a normal heart rate response to carbachol in B56α+/− mice (Fig. 4, H and I). Finally, consistent with reduced cardiac parasympathetic signaling, phosphorylation of RyR2 at Ser2808 and Ser2814 was increased in wild-type mice transduced with AAV9 B56α compared with wild-type hearts (Fig. 4, J and K) without changes in PP2A core subunit abundance. Together, our findings illustrated that expression of B56α (in the absence of altered abundance of PP2A catalytic and scaffolding subunits) was sufficient to alter cardiac parasympathetic signaling.
Fig. 4. Ectopic B56α expression decreases PP2A and parasympathetic activity.
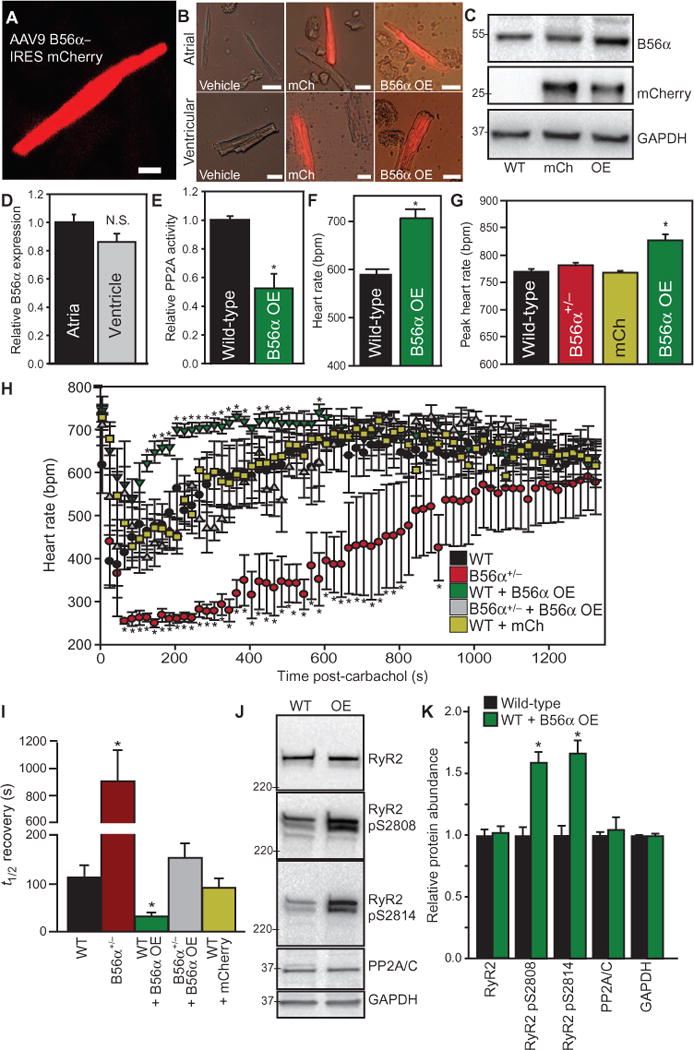
(A) Four weeks after injection of AAV9 B56α–IRES mCherry into mice, ventricular myocytes were isolated to confirm mCherry expression. (B) Example images of control WT myocytes, as well as AAV9 mCherry (mCh) and AAV9 mCherry B56α-transduced atrial and ventricular cardiomyocytes. Data are representative of three independent experiments per genotype. Scale bars, 20 μm. (C) Representative immunoblot of B56α, mCherry, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in three independent experiments of WT and B56α–overexpressing (OE) hearts. (D) Similar abundance of B56α in atria and ventricle of mouse hearts transduced with AAV9 mCherry B56α (n = 3 hearts per genotype; P = N.S.). (E) Relative PP2A activity in WT and B56α OE hearts (n = 4 hearts per genotype; *P < 0.05 compared to WT). (F) Conscious B56α-OE mice had higher resting heart rates than did WT mice (n = 3 mice per experimental group; *P < 0.05). (G) Conscious B56α OE mice had higher peak heart rates after isoproterenol injection than WT, B56α+/−, or control mCherry-transduced mice (n = 3 mice per experimental group; *P < 0.05). (H and I) Heart rates and heart rate recovery [t1/2 (half-time)] of WT mice transduced with mCherry or mCherry B56α were compared to WT and B56α+/− mice (reported in Fig. 1) after injection of carbachol to activate acetylcholine receptors. B56α OE mice displayed a blunted heart rate response compared with WT, B56α+/−, and mCherry-transduced mice (n = 3 mice per experimental group; *P < 0.05). Heart rate recovery was more rapid in B56α OE mice than in WT mice, B56α+/− mice, or control mice transduced with AAV9 mCherry (n = 3 mice per experimental group; *P < 0.05). B56α expression in B56α+/− mice (gray symbols) restored carbachol heart rate response to levels similar to WT mice (n = 3 mice per experimental group; P = N.S. between WT and B56α+/− + B56α OE). (J and K) Representative immunoblots and quantification of WT and B56α OE mouse cardiac lysates illustrating increased phosphorylation of RyR2 at Ser2808 and Ser2814 (n = 3 hearts per experimental group; *P < 0.05; phosphorylation was normalized to total RyR2). The total abundance of RyR2, GAPDH, or PP2A/C did not differ between experimental groups (n = 3 hearts per experimental group; P = N.S.).
DISCUSSION
Attempts to modulate PP2A function in vivo through altering either scaffolding or catalytic subunits results in mice with substantial phenotypes. Overexpression of PP2A/C in mice results in cardiac dilation and hypertrophy, reduced L-type Ca2+ current, and impaired contractility (17). Similarly, overexpression of a dominant-negative PP2A/A subunit in mice that is unable to bind PP2A regulatory subunits causes dilated cardiomyopathy and decreased fractional shortening (28). As an alternative approach, as well as to investigate the mechanisms underlying phosphatase regulation, we focused on the PP2A regulatory subunit family because of select subunit distribution patterns within cardiac chambers, cell types, and subcellular domains (7). For example, the regulatory subunit B56α is localized to transverse tubules by association with the adapter molecule ankyrin-B (23). Modulation of B56α at these sites alters excitability in cultured cardiomyocytes (14) and has been linked with human arrhythmias associated with ankyrin-B dysfunction (29). However, despite its potentially important role for modulation of electrical function, the in vivo role of B56α is unknown.
Contrary to previous in vitro reports (14, 15), in vivo B56α reduction did not promote phenotypes associated with ventricular arrhythmia. Our in vivo and myocyte findings demonstrate that chronic B56α reduction, in the absence of alterations in the abundance of PP2A catalytic and scaffolding subunits, increases PP2A-dependent phosphatase activity in the heart, thus reducing phenotypes linked with ventricular arrhythmia. In line with these findings, B56α+/− mice displayed augmented parasympathetic regulation of heart rate and conduction defects. In contrast to previous publications manipulating B56α abundance in acutely isolated cardiomyocytes, in vivo cardiac B56α deficiency does not promote RyR2 hyperphosphorylation, increased Ca2+ spark and wave frequency, afterdepolarizations, and/or arrhythmia. Rather, we found that B56α+/− and B56α−/− atrial and ventricular myocytes showed reduced phosphorylation of RyR2 at Ser2808 and Ser2814, reduced Ca2+ waves and sparks, and resistance to catecholamine and/or exercise stress–induced arrhythmias. We hypothesize that differences between this study and past work are likely rooted in the experimental model (in vivo animal model compared with acutely transfected or transduced myocytes) and approach (direct B56α regulation compared to miR-dependent regulation with potentially multiple downstream targets).
Our data provide evidence that an integral component of the PP2A holoenzyme serves an autoinhibitory role to tune and/or temper increased PP2A enzyme activity in vivo. Although unexpected, our data illustrate the complexity of phosphatase signaling pathways in the heart. The PP2A B56α complex is tightly regulated by multiple pathways both basally and in disease states. On the basis of our current and past data (7, 26), and the in vivo work of others using PP2A subunit overexpression models (17, 28), we hypothesize that the PP2A B56α molecule serves multiple roles in regulating local PP2A function. First, on the basis of findings in humans and mice, B56α facilitates the targeting of the PP2A holoenzyme complex to the cardiac dyad to modulate RyR2 phosphorylation (5). Second, because PP2A catalytic and scaffolding subunits may associate and display PP2A activity independent of regulatory subunit integration (28), we hypothesize that B56α may sequester and suppress free PP2A core enzyme that has higher activity than the holoenzyme (28). Thus, in the B56α-deficient models, reduction of B56α regulatory subunit results in active, unregulated PP2A core enzyme activity, resulting in increased PP2A-dependent phosphatase activity, reduced RyR2 phosphorylation, and reduced Ca2+ wave and spark frequency. In contrast, in the ankyrin-B model (fig. S14), we surmise that although B56α abundance was increased, global PP2A abundance was normal due to formation of the mature PP2A B56α holoenzyme. However, the holoenzyme might have been less efficiently localized to the cardiac dyad, resulting in increased phosphorylation of RyR2 at Ser2814 and increased arrhythmia burden. It is unexpected that 50% loss of a single regulatory subunit out of 13 could have such a substantial effect on cardiac function. However, these data demonstrate the key role of the B56α subunit as a nodal point for cardiac phosphatase activity in the regulation of cardiac signaling. Given the complexity of PP2A signaling in the heart, it will be interesting to compare potential differences in B56α regulation across cardiac pathologies, to define additional cardiac PP2A targets, and to test for susceptibility of heart failure and arrhythmia phenotypes in B56α-deficient and overexpression models in response to aging and/or stress (such as transaortic banding).
MATERIALS AND METHODS
Animal studies
All animal studies were performed in accordance with the American Physiological Society Guiding Principles for Research Involving Animals and Human Beings, and approved by Ohio State University Institutional Animal Care and Use Committee. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
B56α+/− and B56α−/− mouse models
B56α+/− and B56α−/− mice were created by inserting a gene trap vector, containing a premature stop codon, into the first intron of Ppp2r5a (mouse accession NM_144880) and backcrossed onto a pure C57BL6 background before the study. For verification of genotype, DNA was isolated and analyzed by PCR. B56α deficiency was further confirmed by reverse transcription PCR (RT-PCR) and immunoblot using B56α-specific primers and antibodies. B56α+/− and B56α−/− mice were born healthy in expected ratios. For all experiments in the manuscript, 8- to 10-week-old male and female mice were used for experiments. No gender-based differences in phenotypes were observed. Control experiments were performed using wild-type littermates from B56α+/− × B56α−/− crosses. For AAV9 transduction experiments, 4-week-old animals were injected with AAV9 and analyzed for phenotypes for 4 weeks.
ECG experiments
ECG recordings of ambulatory mice were collected by subcutaneously implanted radiotelemeters (Data Sciences International) as previously described (30). Briefly, recordings were collected in conscious mice at baseline, post-isoproterenol (0.4 or 0.05 mg/kg), post-carbachol (0.1 mg/kg), and post-atropine (1.0 mg/kg). Data points shown consist of data from 5-s epochs (n = 3 to 5 mice per experimental condition).
Immunoblots and antibodies
Immunoblots of whole-ventricle tissue lysates were analyzed on Mini-PROTEAN Tetra Cell (Bio-Rad) on a 4 to 15% TGX precast gel (Bio-Rad) in tris/glycine/SDS buffer (Bio-Rad) as described (30). Antibodies included the following: PP2A/A (Calbiochem 539509), PP2A–C subunit (1:500, Millipore 05-421), PPP2R5A (1:500, Abcam ab72028), PPP2R5C (1:500, Abcam ab94633), PME-1 (1–500, Abcam ab154569), LCMT-1 (1:500, OriGene clone 209), CaMKIId (1:1000, Santa Cruz Biotechnology SC-13082), PKA catalytic (1:2500, R&D Systems MAB5908), SERCA2 (1:2000, Pierce Antibodies IID8), PLB (1:10,000, Abcam ab2865), PLB Ser16 (1:20,000, Badrilla A010-12), PLB Ser17 (1:20,000, Badrilla A010-13) (31), RyR2 (1:1000, Millipore), and GAPDH (1:5000, Fitzgerald). The polyclonal anti-RyR2 immunoglobulins directed against phosphorylated Ser2808 (1:1000) or Ser2814 (1:1000) have been previously described (32). Secondary antibodies included donkey anti-mouse horseradish peroxidase (HRP) and donkey anti-rabbit HRP (Jackson Laboratories). Densitometry was performed using Adobe Photoshop software, and data were normalized to GAPDH or total RyR2 or PLB present in each sample.
Confocal microscopy
Intracellular Ca2+ handling, SR load, Ca2+ sparks, and waves were assessed from isolated myocytes using the Ca2+-sensitive dye Fluo-4. Myocytes were paced at 1 and 4 Hz using field stimulation, and when indicated, cells were treated with 1 μM isoproterenol (33). Immunostaining was performed as previously described (34).
Statistics
Data are presented as means ± SEM. SigmaPlot 12.0 was used for statistical analysis. The Wilcoxon–Mann-Whitney U test was used to determine P values for single comparisons. One-way analysis of variance (ANOVA) was used for multiple comparisons with the Bonferroni test for post hoc testing. If the data distribution failed normality tests with the Shapiro-Wilk test, a Kruskal-Wallis one-way ANOVA on ranks was applied with a Dunn’s multiple comparison test for significant P values. Categorical data were compared using χ2 test.
Cardiomyocyte preparations
Adult cardiomyocytes were prepared as previously described (35, 36).
Real-time RT-PCR
Total RNAwas isolated from the hearts of 8-week-old male mice using the Qiagen RNeasy Mini Kit following the manufacturer’s instructions (Qiagen). RNA was treated with TURBO DNase (Invitrogen). First-strand cDNA was synthesized from 200 ng of RNA as a template using the SuperScript III First-Strand Synthesis System (Invitrogen). Real-time PCR was conducted on fast optical 96-well reaction plates using the StepOne Plus Real-Time PCR System (Invitrogen) in a 10-μl final volume, using 0.5 μl of TaqMan primer (FAM-labeled), 2 μl of PCR-grade H2O, and 5.0 μl of TaqMan Master Mix per well. A 1:5 cDNA (2.5 μl) dilution was added to each well. The oligonucleotide primer for Ppp2r5a (Mm00523125_m1) was from Applied Biosystems. Gene expression was calculated relative to both Rn18S (Mm03928990_g1) and β-actin (Mm00607939_s1) and are reported as fold change . Ct values were normalized to wild-type samples to simplify data presentation.
Pro-Q Diamond staining
Tissue lysatewas loaded on a 13% SDS–polyacrylamide gel electrophoresis gel and run for 45 min at 175 V, fixed overnight, and stained using the Pro-Q Diamond phosphoprotein basic staining protocol (Invitrogen). The gel was imagedin a Typhoon variable mode scanner (GEHealthcare) using an excitation wavelength of 532 and 610 nm 30 bandpass emission filter at a photomultiplier tube setting of 500. Gel was stained with SYPRO Ruby and imaged to determine total protein. Densitometric analysis was performed on each band, and the ratio of Pro-Q stain intensity to total protein intensity was calculated.
Protein phosphatase activity
Protein phosphatase activity was examined using the SensoLyte Protein Phosphatase Assay Kit (AnaSpec). Enzyme activity (nanomole per minute) was calculated using the following formula: (V × vol)/(ɛ × l), where V is the reaction velocity [OD405 (optical density at 405 nm) per minute], vol is the reaction volume in liters, ɛ is the extinction coefficient of pNPP (1.78 × 104 M−1 cm−1), and l is the path length of light through the sample in centimeters (for the 100 ml sample, l = 0.5 cm). Enzyme activity was normalized to protein concentration.
Assessment of recovery of heart rate after carbachol injection
The time to recovery to baseline heart after stimulation with carbachol was determined by fitting raw heart rate data to the best-fit curve using the modified Hill equation:
where HRmax is heart rate at maximum recovery, t is time in seconds, t1/2 is time to recovery of half HRmax, and k is a constant that is equivalent to minimum heart rate in response to carbachol. Best-fit curves were assessed in GraphPad with no constraints applied to any variable.
Supplementary Material
Fig. S1. B56α+/− mice display reduced B56α abundance without changes in the abundance of PP2A core subunits.
Fig. S2. Wild-type and B56α+/− mice have similar QT intervals.
Fig. S3. Wild-type and B56α+/− mice have similar peak heart rates after exercise.
Fig. S4. B56α+/− mice display an aberrant response to adrenergic stimulation.
Fig. S5. B56α−/− mice display phenotypes associated with increased parasympathetic activity.
Fig. S6. B56α+/− mice display exaggerated cholinergic response to carbachol administration.
Fig. S7. B56α+/− and B56α−/− mice display normal ECG responses after inhibition of muscarinic acetylcholine receptors.
Fig. S8. B56α-deficient mice display reduced heart rate after inhibition of sympathetic and parasympathetic signaling.
Fig. S9. B56α−/− hearts display reduced phosphorylation of RyR2.
Fig. S10. B56α associates with PP2A/C and RyR2 but is not required for the interaction between RyR2 and PP2A/C.
Fig. S11. B56α+/− and B56α−/− atria display reduced phosphorylation of RyR2.
Fig. S12. Myofilament proteins are phosphorylated to a similar extent in wild-type and B56α+/− mice.
Fig. S13. The PP2A core enzyme is differentially localized in B56α+/− and B56α−/− myocytes.
Fig. S14. B56α abundance is increased in ankyrin-B–deficient hearts.
Acknowledgments
We thank E. M. Hade and J. Peng in The Ohio State University Center for Biostatistics.
Funding: This work was supported by the NIH (HL084583, HL083422, and HL114383 to P.J.M.; HL114893 to T.J.H.; HL098039 to S.C.L.; HL089598, HL091947, and HL117641 to X.H.W.; HL114940 to B.J.B.), the American Heart Association (P.J.M. and X.H.W.), and the Fondation Leducq (Alliance for CaMKII Signaling in Heart; P.J.M., T.J.H., and X.H.W.).
Footnotes
Author contributions: S.C.L., J.C., C.F.K., M.A.M., H.-T.H., Z.X., X.W., I.P., H.M., J.P.D., and A.M.M. performed the experiments and analyzed the data; S.C.L., C.A.C., B.J.B., N.W., S.G., X.H.W., T.J.H., and P.J.M. designed the study, wrote the manuscript, and directed the project.
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Müller FU, Schmitz W, Schotten U, Anderson ME, Valderrábano M, Dobrev D, Wehrens XH. Calmodulin kinase II–mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adán V, Crown LA. Diagnosis and treatment of sick sinus syndrome. Am Fam Physician. 2003;67:1725–1732. [PubMed] [Google Scholar]
- 3.Reiken S, Gaburjakova M, Gaburjakova J, He Kl KL, Prieto A, Becker E, Yi Gh GH, Wang J, Burkhoff D, Marks AR. β-Adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation. 2001;104:2843–2848. doi: 10.1161/hc4701.099578. [DOI] [PubMed] [Google Scholar]
- 4.Mishra S, Gupta RC, Tiwari N, Sharov VG, Sabbah HN. Molecular mechanisms of reduced sarcoplasmic reticulum Ca2+ uptake in human failing left ventricular myocardium. J Heart Lung Transplant. 2002;21:366–373. doi: 10.1016/s1053-2498(01)00390-4. [DOI] [PubMed] [Google Scholar]
- 5.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 6.Reiken S, Wehrens XH, Vest JA, Barbone A, Klotz S, Mancini D, Burkhoff D, Marks AR. β-Blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation. 2003;107:2459–2466. doi: 10.1161/01.CIR.0000068316.53218.49. [DOI] [PubMed] [Google Scholar]
- 7.DeGrande ST, Little SC, Nixon DJ, Wright P, Snyder J, Dun W, Murphy N, Kilic A, Higgins R, Binkley PF, Boyden PA, Carnes CA, Anderson ME, Hund TJ, Mohler PJ. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J Biol Chem. 2013;288:1032–1046. doi: 10.1074/jbc.M112.426957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heijman J, Dewenter M, El-Armouche A, Dobrev D. Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J Mol Cell Cardiol. 2013;64:90–98. doi: 10.1016/j.yjmcc.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Chiang DY, Li N, Wang Q, Alsina K, Quick A, Reynolds JO, Wang G, Skapura D, Voigt N, Dobrev D, Wehrens XH. Impaired local regulation of ryanodine receptor type 2 by protein phosphatase 1 promotes atrial fibrillation. Cardiovasc Res. 2014;103:178–187. doi: 10.1093/cvr/cvu123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reiken S, Gaburjakova M, Guatimosim S, Gomez AM, D’Armiento J, Burkhoff D, Wang J, Vassort G, Lederer WJ, Marks AR. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts: Role of phosphatases and response to isoproterenol. J Biol Chem. 2003;278:444–453. doi: 10.1074/jbc.M207028200. [DOI] [PubMed] [Google Scholar]
- 11.Hescheler J, Mieskes G, Rüegg JC, Takai A, Trautwein W. Effects of a protein phosphatase inhibitor, okadaic acid, on membrane currents of isolated guinea-pig cardiac myocytes. Pflugers Arch. 1988;412:248–252. doi: 10.1007/BF00582504. [DOI] [PubMed] [Google Scholar]
- 12.Neumann J, Boknik P, Herzig S, Schmitz W, Scholz H, Gupta RC, Watanabe AM. Evidence for physiological functions of protein phosphatases in the heart: Evaluation with okadaic acid. Am J Physiol. 1993;265:H257–H266. doi: 10.1152/ajpheart.1993.265.1.H257. [DOI] [PubMed] [Google Scholar]
- 13.Neumann J, Bokník P, Herzig S, Schmitz W, Scholz H, Wiechen K, Zimmermann N. Biochemical and electrophysiological mechanisms of the positive inotropic effect of calyculin A, a protein phosphatase inhibitor. J Pharmacol Exp Ther. 1994;271:535–541. [PubMed] [Google Scholar]
- 14.Terentyev D, Belevych AE, Terentyeva R, Martin MM, Malana GE, Kuhn DE, Abdellatif M, Feldman DS, Elton TS, Györke S. miR-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56α and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res. 2009;104:514–521. doi: 10.1161/CIRCRESAHA.108.181651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belevych AE, Sansom SE, Terentyeva R, Ho HT, Nishijima Y, Martin MM, Jindal HK, Rochira JA, Kunitomo Y, Abdellatif M, Carnes CA, Elton TS, Györke S, Terentyev D. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLOS One. 2011;6:e28324. doi: 10.1371/journal.pone.0028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature. 2007;445:53–57. doi: 10.1038/nature05351. [DOI] [PubMed] [Google Scholar]
- 17.Gergs U, Boknik P, Buchwalow I, Fabritz L, Matus M, Justus I, Hanske G, Schmitz W, Neumann J. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J Biol Chem. 2004;279:40827–40834. doi: 10.1074/jbc.M405770200. [DOI] [PubMed] [Google Scholar]
- 18.Dobrzynski H, Boyett MR, Anderson RH. New insights into pacemaker activity: Promoting understanding of sick sinus syndrome. Circulation. 2007;115:1921–1932. doi: 10.1161/CIRCULATIONAHA.106.616011. [DOI] [PubMed] [Google Scholar]
- 19.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 20.Gehrmann J, Hammer PE, Maguire CT, Wakimoto H, Triedman JK, Berul CI. Phenotypic screening for heart rate variability in the mouse. Am J Physiol Heart Circ Physiol. 2000;279:H733–H740. doi: 10.1152/ajpheart.2000.279.2.H733. [DOI] [PubMed] [Google Scholar]
- 21.Deshmukh PA, Blunt BC, Hofmann PA. Acute modulation of PP2a and troponin I phosphorylation in ventricular myocytes: Studies with a novel PP2a peptide inhibitor. Am J Physiol Heart Circ Physiol. 2007;292:H792–H799. doi: 10.1152/ajpheart.00225.2006. [DOI] [PubMed] [Google Scholar]
- 22.Wijnker PJ, Boknik P, Gergs U, Müller FU, Neumann J, dos Remedios C, Schmitz W, Sindermann JR, Stienen GJ, van der Velden J, Kirchhefer U. Protein phosphatase 2A affects myofilament contractility in non-failing but not in failing human myocardium. J Muscle Res Cell Motil. 2011;32:221–233. doi: 10.1007/s10974-011-9261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhasin N, Cunha SR, Mudannayake M, Gigena MS, Rogers TB, Mohler PJ. Molecular basis for PP2A regulatory subunit B56α targeting in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;293:H109–H119. doi: 10.1152/ajpheart.00059.2007. [DOI] [PubMed] [Google Scholar]
- 24.Gigena MS, Ito A, Nojima H, Rogers TB. A B56 regulatory subunit of protein phosphatase 2A localizes to nuclear speckles in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2005;289:H285–H294. doi: 10.1152/ajpheart.01291.2004. [DOI] [PubMed] [Google Scholar]
- 25.Davare MA, Horne MC, Hell JW. Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2000;275:39710–39717. doi: 10.1074/jbc.M005462200. [DOI] [PubMed] [Google Scholar]
- 26.DeGrande S, Nixon D, Koval O, Curran JW, Wright P, Wang Q, Kashef F, Chiang D, Li N, Wehrens XH, Anderson ME, Hund TJ, Mohler PJ. CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome. Heart Rhythm. 2012;9:2034–2041. doi: 10.1016/j.hrthm.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogné K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 28.Brewis N, Ohst K, Fields K, Rapacciuolo A, Chou D, Bloor C, Dillmann W, Rockman H, Walter G. Dilated cardiomyopathy in transgenic mice expressing a mutant A subunit of protein phosphatase 2A. Am J Physiol Heart Circ Physiol. 2000;279:H1307–H1318. doi: 10.1152/ajpheart.2000.279.3.H1307. [DOI] [PubMed] [Google Scholar]
- 29.Mohler PJ, Bennett V. Ankyrin-based cardiac arrhythmias: A new class of channelopathies due to loss of cellular targeting. Curr Opin Cardiol. 2005;20:189–193. doi: 10.1097/01.hco.0000160372.95116.3e. [DOI] [PubMed] [Google Scholar]
- 30.Curran J, Makara MA, Little S, Musa H, Liu B, Wu X, Polina I, Alecusan J, Wright PJ, Li J, Billman G, Boyden PA, Gyorke S, Band H, Hund T, Mohler PJ. EHD3-dependent endosome pathway regulates cardiac membrane excitability and physiology. Circ Res. 2014;115:68–78. doi: 10.1161/CIRCRESAHA.115.304149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for b2-adrenergic regulation. Proc Natl Acad Sci USA. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho HT, Liu B, Snyder JS, Lou Q, Brundage EA, Velez-Cortes F, Wang H, Ziolo MT, Anderson ME, Sen CK, Wehrens XH, Fedorov VV, Biesiadecki BJ, Hund TJ, Györke S. Ryanodine receptor phosphorylation by oxidized CaMKII contributes to the cardiotoxic effects of cardiac glycosides. Cardiovasc Res. 2014;101:165–174. doi: 10.1093/cvr/cvt233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith SA, Sturm AC, Curran J, Kline CF, Little SC, Bonilla IM, Long VP, Makara M, Polina I, Hughes LD, Webb TR, Wei Z, Wright P, Voigt N, Bhakta D, Spoonamore KG, Zhang C, Weiss R, Binkley PF, Janssen PM, Kilic A, Higgins RS, Sun M, Ma J, Dobrev D, Zhang M, Carnes CA, Vatta M, Rasband MN, Hund TJ, Mohler PJ. Dysfunction in the bII spectrin–dependent cytoskeleton underlies human arrhythmia. Circulation. 2015;131:695–708. doi: 10.1161/CIRCULATIONAHA.114.013708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A βIV-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120:3508–3519. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Kline CF, Hund TJ, Anderson ME, Mohler PJ. Ankyrin-B regulates Kir6.2 membrane expression and function in heart. J Biol Chem. 2010;285:28723–28730. doi: 10.1074/jbc.M110.147868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. B56α+/− mice display reduced B56α abundance without changes in the abundance of PP2A core subunits.
Fig. S2. Wild-type and B56α+/− mice have similar QT intervals.
Fig. S3. Wild-type and B56α+/− mice have similar peak heart rates after exercise.
Fig. S4. B56α+/− mice display an aberrant response to adrenergic stimulation.
Fig. S5. B56α−/− mice display phenotypes associated with increased parasympathetic activity.
Fig. S6. B56α+/− mice display exaggerated cholinergic response to carbachol administration.
Fig. S7. B56α+/− and B56α−/− mice display normal ECG responses after inhibition of muscarinic acetylcholine receptors.
Fig. S8. B56α-deficient mice display reduced heart rate after inhibition of sympathetic and parasympathetic signaling.
Fig. S9. B56α−/− hearts display reduced phosphorylation of RyR2.
Fig. S10. B56α associates with PP2A/C and RyR2 but is not required for the interaction between RyR2 and PP2A/C.
Fig. S11. B56α+/− and B56α−/− atria display reduced phosphorylation of RyR2.
Fig. S12. Myofilament proteins are phosphorylated to a similar extent in wild-type and B56α+/− mice.
Fig. S13. The PP2A core enzyme is differentially localized in B56α+/− and B56α−/− myocytes.
Fig. S14. B56α abundance is increased in ankyrin-B–deficient hearts.