

Abstract
The human immunodeficiency virus, HIV, is characterized by a tremendously high genetic diversity, leading to the currently known circulating HIV types, groups, subtypes, and recombinant forms. HIV-1 group O is one of the most diverse forms of HIV-1 and has been so far related to Cameroon or individuals originating from Cameroon. In this study, we investigated in Cameroon, the evolution of this viral group from 2006 to 2013, in terms of prevalence, genetic diversity and public health implications. Our results confirmed the predominance of HIV-1 group M (98.5%), a very low prevalence (<0.02%) for HIV-1 group N and P, and HIV-2 in this country. HIV-1 group O was found at around 0.6% (95% confidence interval: 0.4–0.8%), indicating that the frequency of this virus in Cameroon has remained stable over the last decades. However, we found an extensive high genetic diversity within this HIV-1 group, that resulted from previous steady increase on the effective number of HIV-1 group O infections through time, and the current distribution of the circulating viral strains still does not allow classification as subtypes. The frequency of dual infections with HIV-1 group M and group O was 0.8% (95% confidence interval: 0.6–1.0%), but we found no recombinant forms in co-infected patients. Natural resistance to integrase inhibitors was not identified, although we found several mutations considered as natural polymorphisms. Our study shows that infections with HIV-1 group O can be adequately managed in countries where the virus circulates, but this complex virus still represents a challenge for diagnostics and monitoring strategies.
Keywords: HIV-1 group O, Cameroon, Prevalence, Genetic diversity, Dual infections, Resistance
1. Introduction
A recent publication has traced the origin of HIV-1 group O as the result of cross-species transmission of simian lentiviruses (SIVs) infecting lowland gorillas from southern Cameroon – West Central Africa (D’Arc et al., 2015). This finding and data from previous reports clarify the origin of this virus and its epidemic in Cameroon (De Leys et al., 1990; Van Heuverswyn et al., 2006). Thus, contrary to HIV-1 group M epidemic which started in the Democratic Republic of Congo, formerly Zaïre despite the fact that the virus was transmitted from chimpanzee to humans in southern Cameroon (Keele et al., 2006; Van Heuverswyn et al., 2007; Faria et al., 2014), the origin of HIV-1 group O and its epidemic are both located in Cameroon. In addition, group M is known as the leading cause of the AIDS pandemic and represents millions of infections worldwide, while group O has been estimated to have infected a total of around 100,000 individuals. Finally, two other groups of the HIV-1 lineage, N and P, have been reported in less than 20 and 2 individuals respectively, originating from or living in Cameroon, except for one group N infection (Plantier et al., 2009; Vallari et al., 2011; Mourez et al., 2013). The highest HIV-1 group O prevalence is reported from Cameroon, where it represents about 1% of HIV-1 infections (Vessiere et al., 2010b), and the majority of group O infections are traced to Cameroon or to West Central Africa (Peeters et al., 1997). This co-circulation of group M and group O viruses in Cameroon has also resulted in cases of dual M and O infections and emergence of M/O recombinant forms (Peeters et al., 1999; Takehisa et al., 1999; Plantier et al., 2004; Yamaguchi et al., 2004; Vessiere et al., 2010a).
Because of their distinct origins, a significantly high genetic diversity is observed between group O and group M viruses, and DNA sequence identity between the two groups is below 50% in the env region. Moreover, the intra-group diversity between group O viruses is also high, but does not permit classification of group O viruses into subtypes as for the HIV-1 group M (Roques et al., 2002; Mourez et al., 2013). This high genetic variation of HIV-1 group O has significant operational consequences for the diagnosis and monitoring of group O infection, as well as for treatment strategies. Indeed, first cases of group O infection were not detected by routine serological assays (Gurtler et al., 1994; Gould et al., 1996), and despite significant efforts over the last decades to increase the sensitivity of diagnostic assays, we recently showed that many assays used in Cameroon still do not detect group O infection (Aghokeng et al., 2009). Monitoring of this infection has so far been a concern as well, only two commercial viral load assays, respectively from Abbott and Roche, are validated for group O, but they also showed limited performance on this viral group (Etienne et al., 2013). HIV-1 group O is also characterized by natural resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs), because most of the isolates carry the Y181C mutation (Descamps et al., 1997), and the virological response to other drug classes is not well documented.
In this report, we investigated HIV-1 group O infection over the last decade in Cameroon, with the goal to update the prevalence of this highly divergent virus in the country, assess the molecular epidemiology and genetic diversity including the frequency of dual infections and recombinant forms, and characterize natural resistance to new antiretroviral drugs.
2. Material and methods
2.1. Study sites and subjects
For the purpose of this study, we compiled specimens and data collected through our routine and research activities in Cameroon between 2006 and 2013. The study participants included adults aged 18 years and over, who were confirmed HIV positive, and were either ART naïve or ART experienced. The majority of study subjects resided in Yaoundé, the Capital city of Cameroon, or in the neighboring towns and villages. Overall, we collected specimens from 7263 individuals, including 5277 from persons who were receiving ART and 777 from ART naïve subjects. Treatment status was missing for 1209 subjects. The Cameroon National Ethics Committee approved the study.
2.2. Sample processing and HIV serotyping
Whole blood specimens were sent to the CREMER virology laboratory, Yaoundé, where they were processed and centrifuged to recover plasma and buffy-coat layer that were aliquoted and stored at −80 °C and −20 °C respectively. We tested all the 7263 plasma specimens with a serotyping assay to discriminate HIV type 1 and HIV type 2, and the four HIV-1 groups (M, N, O, and P). Serotyping is a discriminatory enzyme immunoassay based on antibodies binding to the V3 domain of the gp120 of various HIV types and groups. The method has been previously described and used for the identification and/or discrimination of HIV and simian immunodeficiency virus (SIV) respectively from humans and non-human primates (Simon et al., 2001; Aghokeng et al., 2006). Briefly, wells of microtiter plates were coated with 0.25 μg of each single peptide in 0.05 M bicarbonate buffer, pH 9.6, incubated at 37 °C for 20 h. After washing with phosphate-buffered saline (PBS) containing 0.5% Tween 20, unoccupied sites were blocked with PBS containing 5% fetal calf serum for 2 h at 37 °C. Plasma were diluted 1/100 in a hypertonic PBS solution (0.01 M sodium phosphate buffer, pH 7.4, containing 0.75 M NaCl, 10% fetal calf serum, and 0.5% Tween 20). After incubation for 30 min at room temperature, plates were washed and incubated with peroxidase-conjugated goat anti-human IgG for 30 min at room temperature. After additional washing, the reaction was revealed with hydrogen peroxide–o-phenylendiamine for 15 min at room temperature in the dark. Color development was stopped by adding 2 N H2SO4, and optical densities were read at 492 nm. We included five discriminatory peptides from the V3 domain of the envelope gp120 of various HIV types and groups in this evaluation, most of which were located around the position 7125–7217 of HIV-1 HXB2 reference. These peptides included HIV-2 (GNKTVVPITLMSGLVFHSQPINKRPRQAWC), HIV-1 group M (NNTRK SVRIGPGQAFYATGDIIGDIRQAYC), HIV-1 group N (NNTGGQVQIG PAMTFYNIEKIVGDIRQAYC), HIV-1 group P (NTRRQVQIGPMTWYNMK FYTGDISKAY), and for HIV-1 group O, we used two distinct peptides to cover the high intra-group genetic diversity, V3O-1 (NLTVQEI KIGPMAWYSMGLAAGNGNSRAYC) and V3O-2 (IDIQEMRIGPMAWYS MGIGGTAGNSSRAA).
2.3. Nucleic acid extraction, PCR and sequencing
Viral RNA was extracted from plasma using the QIAamp Viral RNA assay (Qiagen, Courtaboeuf, France), and proviral DNA was extracted from uncultured peripheral blood mononuclear cells present in buffy-coat, with the QIAamp Viral DNA Mini Kit (QIAGEN, Courtaboeuf, France). To confirm HIV-1 group O infection and further characterize both the viral genetic diversity and primary drug resistance to integrase inhibitors (INI), the protease (PR), reverse transcriptase (RT) and integrase (IN) regions were targeted in the pol gene, and the glycoprotein 41 (gp41) sub-unit was targeted in the env gene. Sequences of 1250 base pairs (bp), 1085 bp, and 660 bp were expected respectively from the PR/RT, IN and gp41 regions. HIV-1 group M and O co-infection was investigated by using specific primers that were designed to specifically amplify either group M or group O strains. These specific primers were designed for the pol and env regions respectively. Table 1 describes the new primers that were used to amplify HIV-1 group M and group O strains in different regions. Co-infection was considered if both group M and group O sequences were confirmed in the pol and env regions from the same sample. In case of confirmed dual HIV-1 group M and group O infections, the amplification strategy that was previously reported was used to investigate recombinant forms involving HIV-1 group M and group O viruses (Peeters et al., 1999; Takehisa et al., 1999). This strategy consists of screening PCRs that target the viral region known as spanning the group M/O breakpoint, between vif and vpu accessory genes. The reverse transcription and the nested-PCR were performed using the Qiagen OneStep RT-PCR system and HotStarTaq Master Mix (Qiagen, Courtaboeuf, France), following manufacturer’s recommendations. Amplified products were purified with the QIAquick Gel Extraction kits (Qiagen) and directly sequenced using the BigDye Terminator v3.1 Cycle Sequencing kit (Life Technologies, Saint Aubin, France). Nucleotide sequences were assembled and analyzed using SeqMan II (DNASTAR, Madison, WI).
Table 1.
New PCR primers designed to amplify HIV-1 group M or group O isolates.
| Primer name | Location on HIV-1 reference strains | Targeted HIV region | Sequence 5′-3′ |
|---|---|---|---|
| Universal primers | HIV-1 B_HXB2 | ||
| PR2 | 1557–1583 | pol (protease-RT) | CCTAGRAAAARGGGCTGTTGGAAATGT |
| PR3 | 1590–1615 | pol (protease-RT) | GARGGACAYCAAATGAAAGAYTGYAC |
| TR2 | 1905–1930 | pol (protease-RT) | TAGAYACAGGAGCAGATGATACAGTA |
| PR2as | 2294–2322 | pol (protease-RT) | TTTATRGCAAAYAYTGGRGTRTTRTATGG |
| TR3as | 2885–2912 | pol (protease-RT) | CTAAYTTYTGTATRTCATTGACAGTCCA |
| TR2as | 2915–2941 | pol (protease-RT) | AATYTGACTTGCCCARTTTARTTTTCC |
| Group M specific primers | |||
| 41UM-1 | 7108–7133 | env (gp41) | GGACAAGCAATGTATGCCCCTCCCAT |
| 41UM-2 | 7231–7256 | env (gp41) | GGAGGARAYATGARGGACAATTGGAG |
| 41UM-1r | 8099–8125 | env (gp41) | GGYGGTAGCTGAAGAGGCACAGGYTCC |
| 41UM-2r | 8016–8044 | env (gp41) | TGYCTTGCTCKCCACCTYCTTCTTCGATT |
| Group O specific primers | HIV-1 O_ANT-70 | ||
| IN-O-1s | 4154–4183 | pol (integrase) | TAGTTCARCAGATAATAGARGAACTRACAA |
| IN-O-2s | 4234–4262 | pol (integrase) | GGAAATGAAAAAATAGATAAATTAGTAAG |
| IN-O-1as | 5380–5408 | pol (integrase) | CTRTCTGCTGTTTCAGGGTCAATCTGTGT |
| IN-O-2as | 5299–5323 | pol (integrase) | TTTCYCCTGGCATYAATCCCCAATA |
| gp41O_1 | 7679–7704 | env (gp41) | GGGGGAGAYATGNRAGATATATGGAG |
| gp41O_2 | 7844–7870 | env (gp41) | CTAAGTGCAGCAGGWAGCACTATGGGC |
| gp41O_1r | 8565–8593 | env (gp41) | TAAGYTGCTCAAGAGGTGGTARNTCCACA |
| gp41O_2r | 8468–8496 | env (gp41) | CTGYCTYCKTCTCCACCTYCTTCTCCTGT |
pol: polymerase; RT: reverse transcriptase; env: envelope; gp41: glycoprotein 41.
2.4. Sequence alignment and phylogenetic analysis
Genomic HIV-1 group O sequences with sampling dates were retrieved from Los Alamos National Laboratory HIV sequence database. We extracted the pol region (PR/RT and IN) and gp41 loci to match newly sequenced data. In addition, we retrieved HIV-1 group O sequences that matched a smaller region within the gp41 locus (HXB2 coordinates 7812–8276). This region was available for several strains and better represents group O diversity. Alignments were performed using Muscle v3.8.31 (Edgar, 2004) and Mafft v7 (Katoh and Standley, 2013) and manually edited in Mesquite v3.02 (http://mesquiteproject.org). We excluded clone sequences and ambiguously aligned regions in the gp41 region datasets to improve the accuracy of phylogenetic inference.
Likelihood mapping analyses for estimation of data quality in each locus were performed using Tree-Puzzle (Quartets ranged between 10,000 and 40,000) (Schmidt et al., 2002). For each alignment we performed recombination screening (RDP, GeneConv, Chimaera, MaxChi, BootScan and SiScan) in RDP4 (Martin and Lemey, 2010).
We estimated the phylogenetic relationships and past population dynamics of HIV-1 group O following partitioned analyses with sequences that belong to the same patient. Each locus was allowed to have its own tree topology, relaxed clock model and nucleotide substitution model values but they shared a Bayesian Skygrid coalescent tree prior, an evolutionary rate prior (gamma prior, shape 0.001 and scale 1000) and a GTR + I + Γ4 nucleotide substitution model that was selected using jModeltest v.2.1.7 (Darriba et al., 2012). To infer the evolutionary parameter values, we employed a Bayesian MCMC approach implemented in BEAST v1.8.1 and performed four MCMC runs with up to 100 million generations to ensure convergence of estimates (Drummond et al., 2012). Tracer was used to check for convergence and mixing (estimated sample size >200) (http://beast.bio.ed.ac.uk/Tracer). The resulting sampling trees were summarized in a maximum clade credibility tree for each locus.
We also performed a similar Bayesian MCMC analysis using the smaller region within gp41. In this case, we encoded an informative gamma prior on the TMRCA (shape 50 and scale 2) to reduce the uncertainty of the estimate.
Sequence accession numbers of new sequences are KT582305–KT582416.
3. Results
3.1. Prevalence of HIV serotypes
Overall, we analyzed 7263 samples using the serotyping method. Two samples out of the 7263, and representing less than 0.03% of the sample size reacted with HIV-2 peptides and where confirmed HIV-2 with a line immunoassay (InnoLIA). 6653 samples, representing 91.6% of the study panel, reacted with HIV-1 peptides. The remaining 608 samples that represented 8.4% of the panel did not react with any peptide and were considered as indeterminate for the serotype status as observed in other reports on serotyping of HIV-1 samples (Vergne et al., 2003). Among the 6653 samples that reacted with HIV-1 peptides, 6556 representing 98.5% (95% confidence interval: 98.2–98.8%) of the HIV-1 panel reacted with the HIV-1 group M peptide, 55 (0.8%; 95% confidence interval: 0.6–1.0%) reacted simultaneously with the HIV-1 group M and group O peptides, 41 (0.6%; 95% confidence interval: 0.4–0.8%) reacted with the HIV-1 group O peptide, one sample reacted with the HIV-1 group N peptide and no reaction was observed with the HIV-1 group P peptide. These results were obtained from prospective analyses from 2006 to 2013 and we did not observe any change of the distribution of these serotypes over time.
3.2. Confirmation of HIV-1 group O and HIV-1M/O co-infections
Results of molecular analyses confirmed HIV-1 group O infection for all the 41 isolates that reacted only with group O peptides. Indeed, a group O sequence was generated for these samples in at least one of the three genomic regions that were targeted and which included pol (PR and partial RT), pol (IN) and env (gp41). Nucleotide sequences were generated in the three regions for 32 isolates. Three samples were confirmed only in PR/RT and gp41, two only in PR/RT and IN, two only in IN and gp41, one only in PR/RT and one was confirmed only in the gp41 region.
Fifty-five samples from our panel reacted simultaneously with HIV-1 group M and group O peptides and we carried out investigations for 52 of them to confirm possible M/O recombinant forms. Enough material for PCR analyses was not available for the three remaining samples. We confirmed only HIV-1 group M infection for the majority of these samples, 38 out of 52 (73.1%), and only group O virus for 5/52 (9.6%). For 6/52 (11.5%), we confirmed group M and group O co-infection, and all attempts to identify M/O recombinant strains failed. Results obtained for the three remaining samples did not confirm M/O co-infection or recombination, but did not exclude these scenarios. For two of them we confirmed group M infection in the env region, but all PCRs failed in pol; and for one sample, a group O sequence was confirmed in pol, but all PCRs failed in the env region.
3.3. HIV-1 group O genetic diversity
We conducted phylogenetic analyses to update the genetic diversity of HIV-1 group O strains that circulate in Cameroon. Overall, analyses included 38 new sequences for PR/RT (967 pb) and gp41 (595 pb), and 36 new sequences for the integrase (864 pb). The likelihood mapping showed that all datasets contained phylogenetic signal since none of them had more than 25% of unresolved quartets. Conclusive evidence of recombination within regions of newly identified group O was not detected.
For the inferred phylogenetic trees for each region, we confirmed the high genetic diversity within HIV-1 group O (Fig. 1). We observed nearly the same clustering of strains in each tree but the branching pattern, and sometimes the associated posterior probability value, varied widely. Such degree of diversity does not allow the distinction in subtypes like those seen in HIV-1 group M and to a certain extent, to assess recombination. For example, further recombination tests done with strains showing recognizable tree discordance (e.g. ZAXA), showed that there was no preferable topology as we moved along the full sequence alignment. Given so, these results may point out that some individual sequences may represent distinct lineages with substantial divergence but we should not exclude the possibility that the recombination could have been very high and deep-rooted for them.
Fig. 1.
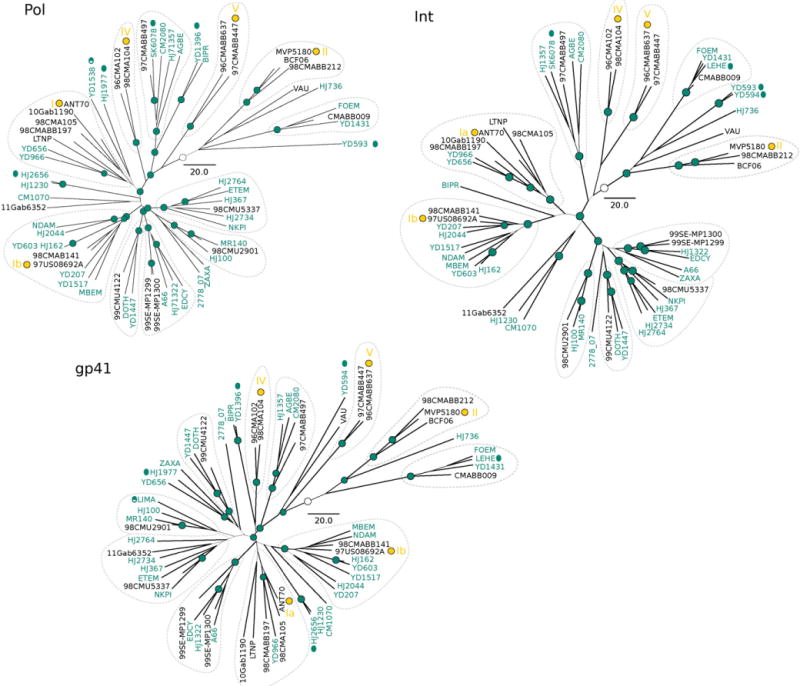
Maximum clade credibility trees for the partitioned analysis. New sequences are colored in dark green. The width of the branches represents Bayesian posterior probabilities (values above 0.9 are symbolized with dark green nodes circles). Clustering of sequences are indicated with dotted lines for the sake of comparisons. Yamaguchi et al. (2002) clusters were indicated with yellow roman numerals. The white circles represent the inferred root. Scale bar for years.
We performed additional phylogenetic analysis in a single region, the partial gp41, but with substantially larger sample size to assess in more detail the genetic diversity of newly identified group O isolates and currently published sequences and to minimize issues related with incomplete taxon sampling (Zwickl and Hillis, 2002) (Fig. 2). The general tree configuration closely matches that of the partitioned analysis (Fig. 2A). We notice that even with more sequences, the uncertainty of the phylogenetic association among several clusters remains high (Fig. 2B). On the other hand, most non-Cameroonian sequences are annotated as France and scattered all over the tree.
Fig. 2.
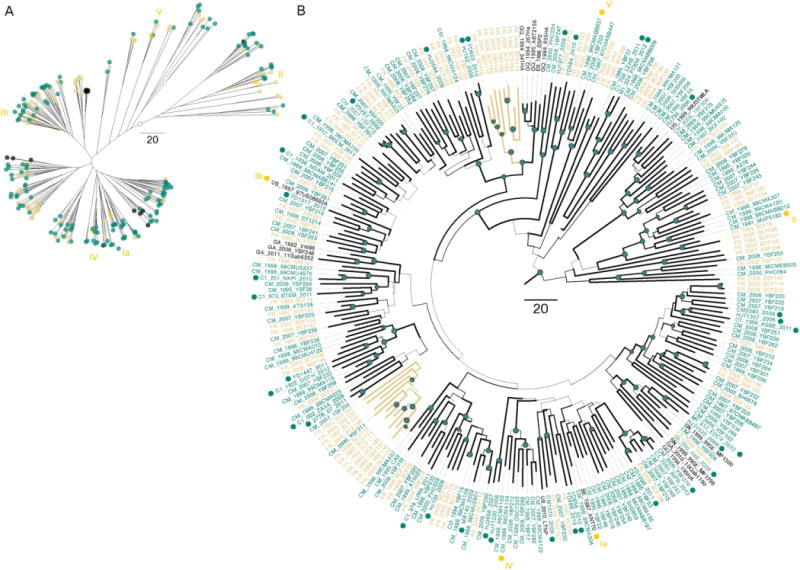
Maximum clade credibility tree for the gp41 locus. (A) Radial tree layout with tips names removed for clarity. (B) Polar tree layout with the width of the branches representing Bayesian posterior probabilities (values above 0.9 are symbolized with dark green nodes circles). In both layouts the accessions annotated as Cameroon are colored in dark green and sequences annotated as France are colored in orange. Dark green tip circles denote new sequences. Yamaguchi et al. (2002) clusters were indicated with yellow roman numerals. Scale bar for years.
3.4. HIV-1 group O demographic history
We performed the demographic analyses to provide an estimate of the effective number of HIV-1 group O infections over time. The partitioned analysis exhibits a steady growth rate with only limited fluctuations, slight increase between 1985 and 1995 (Fig. 3). The demographic history was also inferred using only gp41 sequences to include a sufficient number of individuals so that the diversity of the population can be captured. Overall, the result agrees with that of the partitioned analysis but exhibits a slightly better delineated dynamics and a reduced epidemic growth in the 2000s.
Fig. 3.
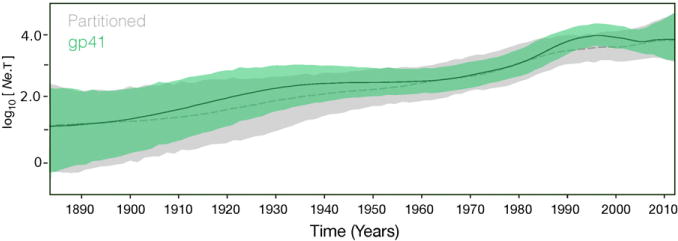
Bayesian skygrid estimates of past population dynamics. The left y-axis represents the effective number of infections (Ne) multiplied by the mean viral generation time (t). Dashed and solid lines correspond respectively to the median values for the partitioned and gp41 estimates. The 95% HPD estimates are colored in gray and green respectively for the partitioned and gp41 analyses.
3.5. HIV-1 group O natural resistance to integrase inhibitors
Some of the group O sequences (15/41) were derived from patients on first-line ART regimen that includes NNRTIs, and therefore we studied only natural resistance to integrase inbibitors which are not used yet in Cameroon. Full integrase sequences were successfully generated for 36 group O isolates and analyzed for the presence of INI resistance mutations known for raltegravir (RAL), elvitegravir (EVG) and dolutegravir (DTG). None of the currently reported major INI resistance mutations was identified, but we found 3 mutations that occurred at significantly high frequency, and included: L74I for 36/36 (100%); T124A for 34/36 (94.4%) and T206S for 33/36 (91.7%). These mutations are all considered as natural polymorphisms for HIV-1 group M with no impact on currently approved INIs. One mutation T97A, however, considered as inducing a reduced susceptibility to RAL according to the Stanford HIVDB interpretation rule, was identified in one isolate. The overall drug resistance interpretation varied very slightly according to the algorithm considered. The ANRS algorithm did not select any mutation associated with minor or major resistance to INIs; both HIVDB and Rega selected T97A, while L74I was only selected by HIVDB and both T124A and T206S were selected only by Rega.
4. Discussion
Since the first cases were reported in early 1990s, HIV-1 group O infection is restricted to Cameroon and neighboring countries, and the majority of infections identified outside this area are linked to West Central Africa. Group O infections have been very rarely detected outside this region, but a few have been detected and it is possible that there is a slightly higher spread of this virus that has been missed due to false negative results. In Cameroon, group O is the most frequent HIV-1 strain after group M, is estimated at around 1% of all HIV-1 infections (Vessiere et al., 2010b), and thus representing 5000 to 10,000 individuals in the actual epidemic. Understanding the epidemiology, diversity and evolution of this virus is essential to improve the diagnosis, monitoring and treatment of this infection not only in Cameroon, but also in other regions of the world where this virus circulate.
In this report, we analyzed up to 7263 HIV samples collected in Cameroon from 2006 to 2013, and confirmed the predominance of HIV-1, and an extremely low prevalence of HIV-2. Within the HIV-1 lineage, we confirmed the high predominance of group M viruses and the low or very low prevalence of the other groups. In fact, we found a very low frequency group N strain, confirming its very low prevalence. Group P was not identified in our panel, despite the use of specific peptides and our findings correlate with current knowledge, since only two isolates of this group have been identified to date (Plantier et al., 2009; Vallari et al., 2011). Recent studies have confirmed the origin of group P in Cameroon through cross-species transmissions of SIVs infecting gorillas to humans (D’Arc et al., 2015), but the reasons of its extremely low prevalence are still to be identified. HIV-1 group P could be a more recent cross-transmission and the virus did not have the opportunity yet to spread efficiently in the human population. We cannot also exclude the possibility that the current diagnostic and/or discriminatory tools are missing this infection, and identification of additional isolates will be essential to better understand the epidemiology, the genetic diversity of this group and dating of the zoonotic transmission event. HIV-1 group O represented the second group identified in our panel. However, compared to the 1% prevalence recently reported (Vessiere et al., 2010a), we found a prevalence of 0.6%, which is relatively lower. Nonetheless, our data together with previous reports indicated a sustained low prevalence of HIV-1 group O and we confirmed that aspect of this infection with the reconstructed demographic history which showed a steady effective number of HIV-1 group O infections during recent years. A study conducted between 1986 and 1998 also indicated a decreasing prevalence of group O in Cameroon, and reported prevalence between 0.4% and 0.8% (Ayouba et al., 2001). Our results showed similar prevalence and indicated that the frequency of group O has remained stable over the last two decades in Cameroon, fluctuating between 0.5 and 1.0%. Nonetheless, despite the low prevalence and limited geographical range, our findings support the fact that group O exhibits high diversity and a median substitution rate comparable to that of HIV-1 group M (Worobey et al., 2008). The fact that we cannot clearly identify subtypes or subgroups as previously suggested by Roques and colleagues (Roques et al., 2002) likely reflects the absence of founder effects of particular strains in locations within and outside Cameroon. Indeed, since the publication of these possible phylogenetic clades of HIV-1 group O, clade O:A, O:B and O:C, no other studies have clearly confirmed such distribution and we did not find any similar diversity pattern in our study.
We noted that the sequences reported from France (FR) intermix with sequences annotated as Cameroon (CM) in Fig. 2, but the former strains coded as BCF originated by birth in Cameroon (Roques et al., 2002), and there is limited information about the origin of group O infections identified by the French RES-O network that were denoted as RBF (Depatureaux et al., 2010). Consequently, more studies are required to fully address the HIV-O transmission in France, even with a well-supported monophyletic clade in terms of country (e.g. upper colored clade in Fig. 2).
The co-circulation of group M and group O viruses in Cameroon has led to co-infection with both viruses and emergence of recombinant M/O strains (Peeters et al., 1999; Takehisa et al., 1999; Vergne et al., 2003). Although a few reports confirmed the circulation of these M/O recombinant forms in Cameroon, and recently outside of Cameroon (Vessiere et al., 2010a), little is known about the epidemiology of M/O infections. In this study, we found a relatively high prevalence (0.8%) of M/O dual reactivity based on serology. However, molecular assessment confirmed only dual M/O infections for a few cases and we did not identify any M/O recombinant. This may indicate that dual M/O infections could be common in Cameroon, and that group M becomes the predominant strain in the patient. This is in agreement with previous studies showing that group O strains have a lower fitness (Arien et al., 2005). We also show that the occurrence of recombinant forms is less frequent than believed.
Despite the low prevalence we here reported, group O remains a major public health issue, especially in Cameroon where the majority of infected patients lives. Major challenges include diagnosis and monitoring as already showed, but also treatment efficacy. Group O natural resistance to NNRTIs is a major concern because of the extensive use of this drug class in Cameroon as recommended by WHO for developing countries. The Y181C mutation is known as being responsible for the natural resistance of group O to NNRTIs, and we confirmed its high frequency in this viral group, since 79% (30/38) the sequences analyzed carried the mutation. We cannot confirm all represented natural resistance since we had limited data on treatment status, but other studies reported similar prevalence of this mutation among ART naïve group O patients (Depatureaux et al., 2010). Therefore, assessing the efficacy of other drug classes is essential to provide more treatment options. Integrase inhibitors have been shown as efficient potential options for group O treatment, but with limited clinical data (Aghokeng et al., 2013). Our results indicated the absence of natural resistance of HIV-1 group O to the currently FDA approved INIs, including RAL, EVG and DTG. We identified few mutations considered as natural polymorphisms by most algorithms. These are L74I, T97A, T124A and T206S. Only T97A, identified in one isolate, was considered as inducing a low level resistance to RAL. Our results confirm previous findings (Depatureaux et al., 2010; Depatureaux et al., 2011), and indicate that INIs can represent effective treatment option for group O infected patients.
In conclusion, HIV-1 group O still represents the second highly prevalent strain of HIV-1 in Cameroon after group M infection and we confirmed the relatively low prevalence of this group in the country, around 0.6%. This low prevalence is however associated with an extensive high intra HIV-1 group O genetic diversity of circulating strains, with no evidence of existing sub-groups or subtypes as defined for HIV-1 group M. Public health challenges associated with HIV-1 group O still include diagnosis, monitoring and treatment, but the increased availability of NNRTIs free regimens such as PI- and INI-based regimens, in countries where group O circulates may minimize treatment challenges.
Acknowledgments
We thank all the patients who kindly accepted to participate in this study, the medical staffs and Cameroon public health authorities.
Funding
This work was supported by IRD and grants from the National Institutes of Health R37 AI50529 and the Agence Nationale de Recherches sur le SIDA, France (ANRS 12125, ANRS 12182, ANRS 12255 and ANRS 12325). CJVA was supported by a fellowship from the Labex EpiGenMed, via the National Research Agency, Programme for Future Investment “ANR-10-LABX-12-01 and University of Montpellier.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Contributor Information
Christian Julian Villabona-Arenas, Email: christian-julian.villabona-arenas@univ-montp1.fr.
Jenny Domyeum, Email: jdomyem@yahoo.fr.
Fatima Mouacha, Email: fatima.mouacha@ird.fr.
Christelle Butel, Email: christelle.butel@ird.fr.
Eric Delaporte, Email: Eric.Delaporte@ird.fr.
Martine Peeters, Email: martine.peeters@ird.fr.
Eitel Mpoudi-Ngole, Email: empoudi2001@yahoo.co.uk.
Avelin Fobang Aghokeng, Email: avelin.aghokeng@ird.fr.
References
- Aghokeng AF, Liu W, et al. Widely varying SIV prevalence rates in naturally infected primate species from Cameroon. Virology. 2006;345(1):174–189. doi: 10.1016/j.virol.2005.09.046. [DOI] [PubMed] [Google Scholar]
- Aghokeng AF, Mpoudi-Ngole E, et al. Inaccurate diagnosis of HIV-1 group M and O is a key challenge for ongoing universal access to antiretroviral treatment and HIV prevention in Cameroon. PLoS One. 2009;4(11):e7702. doi: 10.1371/journal.pone.0007702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghokeng AF, Kouanfack C, et al. Successful integrase inhibitor-based highly active antiretroviral therapy for a multidrug-class-resistant HIV type 1 group O-infected patient in Cameroon. AIDS Res Hum Retrovir. 2013;29(1):1–3. doi: 10.1089/aid.2012.0196. [DOI] [PubMed] [Google Scholar]
- Arien KK, Abraha A, et al. The replicative fitness of primary human immunodeficiency virus type 1 (HIV-1) group M, HIV-1 group O, and HIV-2 isolates. J Virol. 2005;79(14):8979–8990. doi: 10.1128/JVI.79.14.8979-8990.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayouba A, Mauclere P, et al. HIV-1 group O infection in Cameroon, 1986 to 1998. Emerg Infect Dis. 2001;7(3):466–467. doi: 10.3201/eid0703.010321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arc M, Ayouba A, et al. Origin of the HIV-1 group O epidemic in western lowland gorillas. Proc Natl Acad Sci U S A. 2015;112(11):E1343–E1352. doi: 10.1073/pnas.1502022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, et al. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leys R, Vanderborght B, et al. Isolation and partial characterization of an unusual human immunodeficiency retrovirus from two persons of west-central African origin. J Virol. 1990;64(3):1207–1216. doi: 10.1128/jvi.64.3.1207-1216.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depatureaux A, Leoz M, et al. Specific diagnosis and follow-up of HIV-1 group O infection: RES-O data. Med Mal Infect. 2010;40(12):669–676. doi: 10.1016/j.medmal.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Depatureaux A, Charpentier C, et al. Impact of HIV-1 group O genetic diversity on genotypic resistance interpretation by algorithms designed for HIV-1 group M. J. Acquir Immune Defic Syndr. 2011;56(2):139–145. doi: 10.1097/QAI.0b013e318201a904. [DOI] [PubMed] [Google Scholar]
- Descamps D, Collin G, et al. Susceptibility of human immunodeficiency virus type 1 group O isolates to antiretroviral agents: in vitro phenotypic and genotypic analyses. J Virol. 1997;71(11):8893–8898. doi: 10.1128/jvi.71.11.8893-8898.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, et al. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne L, Eymard-Duvernay S, et al. Single real-time reverse transcription-PCR assay for detection and quantification of genetically diverse HIV-1, SIVcpz, and SIVgor strains. J Clin Microbiol. 2013;51(3):787–798. doi: 10.1128/JCM.02792-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria NR, Rambaut A, et al. HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science. 2014;346(6205):56–61. doi: 10.1126/science.1256739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould K, Britvan L, et al. HIV-1 group O infection in the USA. Lancet. 1996;348(9028):680–681. doi: 10.1016/S0140-6736(05)65100-8. [DOI] [PubMed] [Google Scholar]
- Gurtler LG, Hauser PH, et al. A new subtype of human immunodeficiency virus type 1 (MVP-5180) from Cameroon. J Virol. 1994;68(3):1581–1585. doi: 10.1128/jvi.68.3.1581-1585.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele BF, Van Heuverswyn F, et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science. 2006;313(5786):523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Lemey P, et al. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26(19):2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourez T, Simon F, et al. Non-M variants of human immunodeficiency virus type 1. Clin Microbiol Rev. 2013;26(3):448–461. doi: 10.1128/CMR.00012-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters M, Gueye A, et al. Geographical distribution of HIV-1 group O viruses in Africa. AIDS. 1997;11(4):493–498. doi: 10.1097/00002030-199704000-00013. [DOI] [PubMed] [Google Scholar]
- Peeters M, Liegeois F, et al. Characterization of a highly replicative intergroup M/O human immunodeficiency virus type 1 recombinant isolated from a Cameroonian patient. J Virol. 1999;73(9):7368–7375. doi: 10.1128/jvi.73.9.7368-7375.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantier JC, Lemee V, et al. HIV-1 group M superinfection in an HIV-1 group O-infected patient. AIDS. 2004;18(18):2444–2446. [PubMed] [Google Scholar]
- Plantier JC, Leoz M, et al. A new human immunodeficiency virus derived from gorillas. Nat Med. 2009;15(8):871–872. doi: 10.1038/nm.2016. [DOI] [PubMed] [Google Scholar]
- Roques P, Robertson DL, et al. Phylogenetic analysis of 49 newly derived HIV-1 group O strains: high viral diversity but no group M-like subtype structure. Virology. 2002;302(2):259–273. doi: 10.1006/viro.2002.1430. [DOI] [PubMed] [Google Scholar]
- Schmidt HA, Strimmer K, et al. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18(3):502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Simon F, Souquiere S, et al. Synthetic peptide strategy for the detection of and discrimination among highly divergent primate lentiviruses. AIDS Res Hum Retrovir. 2001;17(10):937–952. doi: 10.1089/088922201750290050. [DOI] [PubMed] [Google Scholar]
- Takehisa J, Zekeng L, et al. Human immunodeficiency virus type 1 intergroup (M/O) recombination in Cameroon. J Virol. 1999;73(8):6810–6820. doi: 10.1128/jvi.73.8.6810-6820.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallari A, Holzmayer V, et al. Confirmation of putative HIV-1 group P in Cameroon. J Virol. 2011;85(3):1403–1407. doi: 10.1128/JVI.02005-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Heuverswyn F, Li Y, et al. Human immunodeficiency viruses: SIV infection in wild gorillas. Nature. 2006;444(7116):164. doi: 10.1038/444164a. [DOI] [PubMed] [Google Scholar]
- Van Heuverswyn F, Li Y, et al. Genetic diversity and phylogeographic clustering of SIVcpzPtt in wild chimpanzees in Cameroon. Virology. 2007;368(1):155–171. doi: 10.1016/j.virol.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Vergne L, Bourgeois A, et al. Biological and genetic characteristics of HIV infections in Cameroon reveals dual group M and O infections and a correlation between SI-inducing phenotype of the predominant CRF02_AG variant and disease stage. Virology. 2003;310(2):254–266. doi: 10.1016/s0042-6822(03)00167-3. [DOI] [PubMed] [Google Scholar]
- Vessiere A, Leoz M, et al. First evidence of a HIV-1 M/O recombinant form circulating outside Cameroon. AIDS. 2010a;24(7):1079–1082. doi: 10.1097/QAD.0b013e3283355659. [DOI] [PubMed] [Google Scholar]
- Vessiere A, Rousset D, et al. Diagnosis and monitoring of HIV-1 group O-infected patients in Cameroun. J Acquir Immune Defic Syndr. 2010b;53(1):107–110. doi: 10.1097/QAI.0b013e3181b97ec1. [DOI] [PubMed] [Google Scholar]
- Worobey M, Gemmel M, et al. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature. 2008;455(7213):661–664. doi: 10.1038/nature07390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi J, Vallari AS, et al. Evaluation of HIV type 1 group O isolates: identification of five phylogenetic clusters. AIDS Res Hum Retroviruses. 2002;18(4):269–282. doi: 10.1089/088922202753472847. [DOI] [PubMed] [Google Scholar]
- Yamaguchi J, Bodelle P, et al. HIV infections in northwestern Cameroon: identification of HIV type 1 group O and dual HIV type 1 group M and group O infections. AIDS Res Hum Retrovir. 2004;20(9):944–957. doi: 10.1089/aid.2004.20.944. [DOI] [PubMed] [Google Scholar]
- Zwickl DJ, Hillis DM. Increased taxon sampling greatly reduces phylogenetic error. Syst Biol. 2002;51(4):588–598. doi: 10.1080/10635150290102339. [DOI] [PubMed] [Google Scholar]