

Abstract
A series of four BODIPYs containing one or two ortho- or para-carborane clusters were synthesized using palladium(0)-catalyzed Suzuki cross-coupling or nucleophilic substitution reactions, at the 2,6- or the 8-positions of halogenated boron dipyrromethenes (BODIPYs). The spectroscopic, structural (including one X-ray) and in vitro BBB permeability of the BODIPYs using hCMEC/D3 brain endothelial cells were investigated.
Keywords: BODIPY, carborane, BNCT, fluorescence, BBB model
Graphical Abstract
Two synthetic routes to four BODIPYs containing one or two ortho- or para-carborane clusters, are described. The most promising compound for application as boron delivery agent for BNCT is 8-(ortho-carboranyl-1-thio)-1,3,5,7-tetramethyl-BODIPY, due to its higher permeability across hCMEC/D3 cell monolayers.
1. Introduction
BNCT is a binary treatment that operates through the reaction of low energy thermal or epithermal neutrons with 10B-containing drug molecules localized within tumor cells. Upon capture of a neutron, the stable isotope of boron-10 is converted into the highly reactive boron-11 isotope which readily undergoes fission, releasing high linear energy transfer (high-LET) alpha and lithium-7 particles, γ radiation and approximately 2.4 MeV of kinetic energy [1,2]. The high-LET particles have limited path lengths in tissue (less than 10 μm), therefore limiting the devastation effects to cells containing boron-10. As a result, BNCT potentially functions as a very selective and localized therapy, able to destroy boron-containing tumors without damaging the remaining healthy tissues. Consequently, research over the last few decades has focused on the development of tumor-selective boronated molecules capable of delivering the necessary therapeutic dose of 15–30 μg boron-10/g tumor to infected cells with minimal damage to normal tissues [3,4]. Although the natural abundance of boron-10 is 20%, this isotope can be incorporated into BNCT agents at the 95% level from the corresponding boron-10 enriched starting material.
BNCT is particularly promising for the treatment of malignant brain tumors due to the deep penetration of epithermal neutron beams (up to 10 cm). However, treating diseases of the brain imposes its own challenges, predominantly as a result of the selective permeability of the blood brain barrier (BBB). The BBB is a highly selective physical and metabolic protective barrier surrounding the brain, which serves to protect the homeostatic environment of the brain. Necessary nutrients for proper brain function bypass the BBB via several transport mechanisms, including paracellular diffusion, ion symports and antiports, transcellular diffusion, cation channels, active transport, and receptor-adsorptive mediated endocytosis [5,6]. Tight junctions limit paracellular diffusion of molecules and fluid into and out of the brain, preventing diseases and viruses from attacking the sensitive brain tissue. The investigation of several types of molecules, including peptides, sugars, liposomes and porphyrins, using hCMEC/D3 cell monolayers as BBB model [7–9], has shown that several structural and physical factors play a role in their ability to permeate the BBB, including size, conformation, lipophilicity, and molecular weight [10–12].
Two boronated compounds are approved for clinical use as BNCT agents, the sodium salt of the sulfhydryl boron hydride Na2B12H11SH known as BSH, and L-4-dihydroxy-borylphenylalanine known as BPA [4]. Although BSH and BPA have demonstrated low toxicity and efficacy in BNCT clinical trials [13,14], improved BNCT agents with higher tumor selectivity, including boronated amino acids, antibodies, nucleosides, sugars, lipids, liposomes, and porphyrin derivatives, have been the focus of research. Herein we report the synthesis, spectroscopic properties, and BBB permeability of a new type of low molecular weight BNCT agent, known as BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) [15,16], containing one or two ortho- or para-carborane clusters. The neutral isomeric carboranes, ortho-, meta-, and para-C2B10H12, are generally the clusters of choice for attachment to boron delivery agents, because of their high boron content, lipophilic properties, and their high kinetic and hydrolytic stabilities [17]. Two synthetic routes were explored for the synthesis of carborane-substituted BODIPYs: a Suzuki cross-coupling reaction between a carborane-containing boronic acid and a 2,6-diiodo- or a 8-chloro-BODIPY, and a nucleophilic substitution reaction between a thiol-containing carborane and a 8-chloro-BODIPY.
2. Materials and Methods
2.1 Synthesis and characterization
2.1.1 General
Reagents and solvents were acquired from Sigma-Aldrich, Fisher Scientific, or TCI and used without further purification. Reactions were monitored by TLC using 0.2 mm silica plates with UV indicator (UV254). Column chromatography was performed using Sorbent Technologies 60Å silica gel (230 – 400 mesh). Merck TLC silica gel 60 glass plates were used for preparative thin layer chromatography. The 1H NMR and 13C NMR spectra were obtained using a Bruker DPX-400 spectrometer (400 MHz for 1H, 100 MHz for 13C) with samples dissolved in deuterated chloroform using tetramethylsilane as an internal indicator. Chemical shifts (δ) are given in ppm and coupling constants (J) in Hertz (Hz). High resolution ESI mass spectra were obtained using an Agilent Technologies 6210 ESI-TOF mass spectrometer at the Louisiana State University Mass Spectrometry Facility.
BODIPYs 1 [18], 4 [19], 7 [19] and methyl-o-carborane phenyl boronic acid 2 [20] were prepared as previously reported.
2.1.2. BODIPY 3
BODIPY 1 (0.02 g, 0.03 mmol), methyl-o-carborane phenyl boronic acid 2 (0.04 g, 0.13 mmol), and Pd(PPh3)4 (0.004 g, 0.003 mmol) were dissolved in dry toluene (2 mL) over 3Å molecular sieves. A solution of 1M Na2CO3 (0.5 mL) was added and the mixture was stirred at reflux for 15 hours. The reaction was quenched with water and extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and the solvent was evaporated. The resulting residue was purified by silica gel column chromatography using a gradient from hexanes to 50:50 hexanes/CH2Cl2 as eluent to obtain a red crystalline solid (0.025 g) in 89% yield. 1H-NMR (400 MHz, CDCl3) δ 8.21-8.20 (d, J = 8, 2H), 7.50-7.48 (d, J = 8, 2H), 7.23-7.21 (d, J = 7.8, 4H), 7.15-7.13 (d, J = 7.8, 4H) 3.96 (s, 3H), 3.48 (s, 4H), 2.8-1.5 (br m, 22H), 2.56 (s, 6H), 2.18 (s, 6H), 1.30 (s, 6H); 13C-NMR (100 MHz, CDCl3) δ 166.40, 157.74, 140.79, 140.09, 139.11, 133.99, 133.35, 133.27, 130.95, 130.89, 130.53, 130.34, 130.31, 128.42, 77.44, 74.86, 52.45, 40.94, 23.73, 13.50, 13.03; MS (ESI-TOF) m/z 874.6523 [M+H]+, calcd for C41H57B21F2N2O2: 874.6488.
2.1.3. BODIPY 5
BODIPY 4 (0.01 g, 0.035 mmol), methyl-o-carborane phenyl boronic acid 2 (0.022 g, 0.074 mmol), and Pd(PPh3)4 (0.004 g, 0.0035 mmol) were dissolved in dry toluene (2 mL) over 3Å molecular sieves. A solution of 1M Na2CO3 (0.5 mL) was added and the mixture was stirred at reflux for 15 h. The reaction was quenched with water and extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and the solvent was evaporated. The resulting residue was purified by preparative thin layer chromatography using 5:1 hexanes/ethyl acetate as eluent to obtain an orange solid (0.008 g) in 47% yield. 1H-NMR (400 MHz, CDCl3) δ 7.35-7.33 (d, J = 7.8, 2H), 7.31-7.30 (d, J = 7.8, 2H) 5.99 (s, 2H), 3.55 (s, 2H), 3.0-1.0 (br m, 10H), 2.56 (s, 6H), 2.20 (s, 3H), 1.40 (s, 6H); 13C-NMR (100 MHz, CDCl3) δ 155.75, 142.98, 140.73, 135.84, 131.27, 128.41, 123.20, 121.38, 95.34, 82.90, 71.40, 41.07, 37.50, 14.57, 13.93; MS (ESI-TOF) m/z 495.3772 [M+H]+, calcd for C23H34B11F2N2: 495.3786.
2.1.4. BODIPY 6
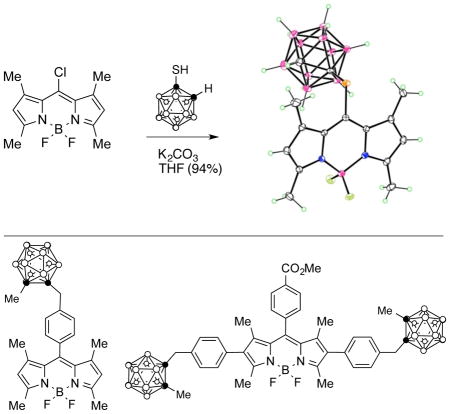
A solution of BODIPY 4 (56.4 mg, 0.2 mmol), 1-mercapto-1,2-carborane (45.6 mg, 0.24 mmol) and K2CO3 (41.5mg, 3.0 mmol) in THF (30 ml) was stirred at room temperature for 12 h or until the starting material was consumed. The solvent was removed in vacuo and the residue was taken up in CHCl3 and washed with water, brine and then dried over anhydrous Na2SO4. The product was purified by silica gel column chromatography using CH2Cl2/hexane as eluent and was obtained as a red solid in 94% yield. 1H NMR (400 MHz; CDCl3) δ 6.15 (s, 2H), 2.57 (s, 6H), 2.49 (s, 6H), 2.20-1.70 (m, 11H); 13C NMR (CDCl3) δ 158.91, 143.66, 129.61, 123.83, 135.66, 60.07, 17.15, 15.10; MS (ESI-TOF): m/z 423.2871 [M+H]+, calcd for C15H24B11F2N2S: 423.2882.
2.1.5. Crystal data
Diffraction data were collected at low temperature on a Bruker Kappa Apex-II DUO diffractometer with MoKα radiation (λ = 0.71073 Å). Refinement was by full-matrix least squares using SHELXL, with H atoms in idealized positions. Crystal data for BODIPY 6: C15H25B11F2N2S, triclinic P-1, a=10.903(2), b=13.495(3), c=16.700(3) Å, α =113.823(9), β =103.287(9), γ =94.166(10)°, Z=4, T=100K, θmax=36.7°, R=0.038 for 17341 data with I>2σ(I) (of 21133 unique), 567 parameters, two independent molecules, CCDC 1056580.
2.1.6. Absorption and Fluorescence Spectroscopy
The spectroscopic properties of BODIPYs 3 and 5–7 were determined on solutions prepared by dissolving crystalline compound in DMSO. Stock solutions (5 × 10−5 M) were prepared and diluted to appropriate concentrations for collection of absorbance and emission spectra. Absorption spectra were obtained on a Varian Cary 50 Bio UV-Vis spectrophotometer. Measurements obtained for determining optical density were taken from solutions with concentrations between 1.2 × 10−5 and 5 × 10−5 M so that λmax was between 0.5 and 1.0. Fluorescence measurements were recorded on solutions with concentrations between 1.2 × 10−6 and 2.5 × 10−5 M to attain an optical density at the excitation wavelength between 0.04 and 0.06. Compounds were excited at 500 nm for 3, 480 nm for 5, 480 nm for 6, and 490 nm for 7. Emission spectra were acquired on a PTI QuantaMaster4/2006SE spectrofluorimeter with the slit width set at 3 nm. Rhodamine 6G was used as a standard in calculating the fluorescence quantum yields (Φf = 0.95 in ethanol) [16]. All measurements, both absorbance and emission, were acquired within 4 h of solution preparation at room temperature (23 – 25 °C), using a 10 mm path length quartz spectrophotometric cell.
2.2. BBB Permeability
The hCMEC/D3 cells were incubated in a 6-well, 0.4 μm porosity PET transwell plate (Corning) and allowed to grow for 48 h. EBM-2 medium containing 5% FBS (fetal bovine serum), 1% penicillin-streptomycin, hydrocortisone, ascorbic acid, chemically defined lipid concentrate (1/100), HEPES, and bFGF was used. Samples of 0.5 mL for each BODIPY and standard lucifer yellow at a concentration of 1 mg/ml were used. To determine the BODIPY and standard concentrations in the lower chamber that mimicks the cerebral compartment, fluorescence emission was read at 520/590 and 420/540 nm (excitation/emission), respectively, using a BMG FLUOstar plate reader. The permeability coefficients (Pe, expressed in cm/s) were determined following the clearance principle, as previously reported [12,21]. A value of Pe = 25.23 ± 0.88 × 10−6 cm/s was determined under the above conditions for the standard lucifer yellow.
2.3. Octanol-HEPES buffer partition coefficients
The partition coefficients (log P) were measured at room temperature by adding 0.5 mL of a BODIPY stock solution in DMSO (50 μM) to a 15 mL volumetric tube containing 4.0 mL of HEPES buffer (1 M, pH 7.4), followed by addition of 4.0 mL of 1-octanol. After vortexing for 5 min, the phases were allowed to separate completely. An aliquot of 0.5 mL from each layer was diluted with 0.5 mL of methanol and the absorbance was measured on a Varian Cary 50 Bio UV-Vis spectrophotometer with a 10 mm path length quartz cuvette.
3. Results and Discussion
3.1. Syntheses and structural characterization
Halogenated BODIPYs are versatile starting materials for functionalization via palladium(0)-catalyzed cross-coupling reactions, such as Stille, Suzuki and Sonogashira, at the α-(3,5)- or β-(2,6)-pyrrolic carbons, and at the meso-8-position [19,22–26]. In particular, para-carborane clusters have been previously introduced at the 2,6-positions via Sonogashira cross-coupling of a 2,6-diiodo-BODIPY with 1-ethynyl-p-carborane [26]. Based on these studies, we decided to explore another palladium(0)-catalyzed cross-coupling, the Sukuki reaction, for the preparation of carborane-containing BODIPYs.
Symmetric 2,6-diiodo-BODIPYs, such as 1, can be easily obtained in four steps by condensation of 2,4-dimethylpyrrole with the corresponding aryl aldehyde, followed by oxidation, boron complexation, and iodination reactions [18]. We have previously reported that BODIPY 1 shows very low cytotoxicity (IC50 > 200 μM) in human HEp2 cells [18], an important property of a potential BNCT agent, and its methyl ester group allows the introduction of further functionality via hydrolysis and conjugation with alcohols or amines [27]. The Suzuki coupling reaction of BODIPY 1 with o-carborane-containg boronic acid 2 [20], under similar conditions to those previously reported for bromo-BODIPYs [25], produced BODIPY 3 in 89% yield after purification, as shown in Scheme 1. The ortho-carborane clusters were stable under the basic conditions used in the reaction, and no deboronated byproducts were observed.
Scheme 1.

Synthesis of BODIPY 3 via Suzuki coupling of BODIPY 1 and o-carborane-substituted boronic acid 2.
Under similar reaction conditions, chloro-BODIPY 4 [19] reacted with boronic acid 2 [20] producing BODIPY 5 in 47% yield, as shown in Scheme 2. The lower yield obtained for the cross-coupling reaction at the 8- versus the 2,6-positions is likely a result of the lower reactivity of chlorine versus iodine under the cross-coupling conditions. We have recently reported that in polychlorinated BODIPYs, palladium(0)-catalyzed Stille cross-coupling reactions occur regioselectively first at the 8-position, followed by the 3,5- and the 2,6-positions [28].
Scheme 2.

Synthesis of BODIPYs 5 – 7 from 8-chloro-BODIPY 4 via Suzuki coupling with 2, or by nucleophilic substitution using 1-mercapto-ortho-carborane or 1-mercaptomethyl-para-carborane.
In addition to cross-coupling reactions, 8-halogenated-BODIPYs, such as 4, can also undergo nucleophilic substitutions in the presence of S-, N- and O-centered nucleophiles [19,29]. Using this strategy, BODIPY 4 reacted with commercially available 1-mercapto-ortho-carborane in the presence of K2CO3 in THF, to give BODIPY 6 in 94% yield (Scheme 2). Unlike the Suzuki cross-coupling reaction, the nucleophilic substitution at the most electrophilic 8-position afforded the target BODIPY in high yield. We have previously reported the synthesis of BODIPY 7 from reaction of 4 with 1-mercaptomethyl-1,12-carborane in high yield [19]. Under these conditions boron substitution was not observed, however, we have previously substituted the para-fluoro phenyl groups of meso-tetra(pentafluorophenyl)porphyrin using 1-mercapto-ortho-carborane [30] and 1-mercaptomethyl-1,12-carborane [12,31].
The structures of the new BODIPYs 3, 5, and 6 were confirmed by 1H- and 13C-NMR, mass spectrometry, UV-Vis and fluorescence spectroscopies. We have previously reported the X-ray structure and spectroscopic properties of BODIPY 7 [19]. A single crystal of 6 suitable for X-ray analysis (see Figure 1) was grown from slow evaporation of hexane. The X-ray crystal structure revealed the expected approximate planarity of the C3BN2 ring of the BODIPY core, with mean deviation of 0.040 Å (average of two independent molecules). The other six pyrrole carbon atoms tip out of this plane by up to 0.246 Å, and the S atom of the 8-thiocarborane group lies out of plane on the opposite side by 0.335 Å (average of two). The boron atom possesses tetrahedral geometry with the BF2 groups lying nearly perpendicular to the BODIPY core, forming a dihedral angle of 89.1° (average of two) with the C3BN2 ring. In the 1H-NMR spectra in CDCl3, significant difference was observed in the chemical shifts of the 1,7-methyl groups of 6 compared with those of 3 and 5. The presence of the electron-withdrawing sulfur at the 8-position caused a downfield shift in the chemical shift to 2.5 ppm, compared to the shielding effect of the phenyl ring in 3 and 5, causing the 1,7-methyl groups to appear between 1.3–1.4 ppm. On the other hand, the BH protons were observed as a broad signal between 1–3 ppm for all carboranylBODIPYs. BODIPYs 3, 5, 6 and 7 allowed us to investigate the effects of the position (8 versus 2,6) and the nature of the carborane cages (ortho versus para), on the spectroscopic properties and BBB permeability of this type of compounds (vide infra).
Figure 1.

Molecular structure of 6 from X-ray crystal structure determination. Ellipsoids are drawn at the 50% probability level. Selected distances are S1-C5 1.7797(8), S1-C14 1.7949(8), B1-F1 1.3994(10), B1-F2 1.3875(10), B1-N1 1.5438(11), B1-N2 1.5405(11), C14-C15 1.6618(11) Å.
3.2. Spectroscopic properties
The spectroscopic properties of BODIPYs 3, and 5–7 were evaluated in DMSO, and the results are summarized in Table 1. The fluorescence quantum yields were calculated using rhodamine 6G in ethanol (ΦF = 0.95) as the standard. The carboranyl-BODIPYs displayed spectra characteristic of boron dipyrromethene dyes with strong absorption bands (log ε = 4.4 – 4.8) corresponding to the S0-S1 (π-π*) transitions, accompanied by a shoulder at slightly lower wavelength attributed to vibrational transitions. The absorption λmax for BODIPYs 3, 6 and 7 were red-shifted by about 30 nm compared to BODIPY 5 due to 8-sulfur substitution [19] or 2,6-aryl substitution [25], respectively. The Stokes’ shifts varied between 7 – 27 nm, the larger value observed for BODIPY 3, as a result of 2,6-aryl substitution, as previously observed [25]. BODIPYs 6 and 7 were poorly emissive in DMSO, as previously reported for 8-sulfur substituted BODIPYs [19,32], while in contrast BODIPYs 3 and 5 showed high fluorescence quantum yields, demonstrating that the ortho-carborane clusters do not quench the fluorescence of boron dipyrromethene dyes. This result is in agreement with previous reports showing that closo-carboranes generally enhance the fluorescence of chromophores [33,34].
Table 1.
Photophysical properties of BODIPYs 3 and 5–7 in DMSO.
| Compound | λmax abs (nm) | λmax em (nm) | log ε | ΦF | Stokes Shift (nm) |
|---|---|---|---|---|---|
| 3 | 534 | 561 | 4.38 | 0.99 | 27 |
| 5 | 501 | 508 | 4.81 | 0.99 | 7 |
| 6 | 538 | n/a | 4.37 | <0.01 | n/a |
| 7 | 531 | 545 | 4.68 | 0.03 | 14 |
3.3. In vitro BBB Permeability
The BBB permeability of BODIPYs 3 and 5–7 was investigated using the human brain endothelial cell line hCMEC/D3 as BBB model [7–9]. The permeabilities were evaluated by determining the permeability coefficients (Pe), as previously described [12] and the results are shown in Table 2. Only one carboranyl-BODIPY, 6, showed higher permeability than the standard lucifer yellow (determined Pe = 25.23 ± 0.88 × 10−6 cm/s), probably as a result of its lower molecular weight and lower hydrophobic character, as measured by the partition coefficients in 1-octanol-HEPES, in comparison with the other BODIPYs. BODIPYs 3 and 5 in which the carborane moiety is conjugated to the BODIPY core via a phenyl spacer, were more hydrophobic and showed decreased solubility in the cellular medium compared with 6 and 7; this property, in addition to their higher molecular weight compared with 6 and 7, resulted in lower permeabilities. The permeability of molecules across the hCMEC/D3 monolayer is known to depend on molecular weight and hydrophobic character, and we have recently shown that a series of carboranylporphyrins of larger molecular weights and hydrophobic characters compared with the BODIPYs, also show relatively small Pe values in the order of 0.8–3.3 × 10−6 cm/s [12]. It is however remarkable that BODIPY 6 shows about 10-fold higher permeability than the other BODIPYs tested, and we are currently investigating other cellular properties of carboranyl-BODIPYs, including cytotoxicity and uptake.
Table 2.
Permeabilty coefficients (Pe) using human endothelium hCMEC/D3 cells, and partition coefficients (log P) in 1-octanol-HEPES buffer for BODIPYs.
| BODIPY | Pe x 10−6 (cm/s) | log P |
|---|---|---|
| 3 | 0.05±0.02 | 1.41 |
| 5 | 0.10±0.003 | 1.35 |
| 6 | 38.75±1.12 | 0.85 |
| 7 | 0.38±0.006 | 0.89 |
4. Conclusions
The synthesis, spectroscopic properties and BBB permeability, using the human brain endothelial cell line hCMEC/D3 as BBB model, of four BODIPYs were investigated. Two different synthetic routes, Pd(0)-catalyzed Suzuki cross-coupling and nucleophilic substitution reactions on halogenated BODIPYs, were employed for attachment of the carborane moieties to the BODIPY scaffold. Studies of the spectroscopic properties of this series of BODIPYs revealed strong absorptions at 501–538 nm and emissions at 508–561 nm, with high fluorescence quantum yields (Φf = 0.99) for the 8-aryl-BODIPYs and very low (Φf < 0.04) for the 8-sulfo-BODIPYs. The BBB permeability assessment of the four carboranyl-BODIPYs revealed that only BODIPY 6 shows higher permeability (Pe =3.9 × 10−5 cm/s) than lucifer yellow, probably as a result of its lower molecular weight and lower hydrophobic character (log P = 0.85) compared with the other BODIPYs.
BODIPYs functionalized with closo-carboranes were synthesized via Suzuki cross-coupling or nucleophilic substitution reactions
The structural features of the BODIPYs were studied by NMR, MS, and X-ray crystallography
The in vitro BBB permeability of the BODIPYs was investigated using hCMEC/D3 cells as BBB model.
The closo-carborane cages do not quench the fluorescence of BODIPY dyes.
Acknowledgments
The authors thank the National Institutes of Health, grant number R01 CA179902, and the National Science Foundation, grant number 1362641, for supporting the work described.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hawthorne MF. Angew Chem Int Ed. 1993;32:950–984. [Google Scholar]
- 2.Soloway AH, Tjarks W, Barnum BA, Rong FG, Barth RF, Codogni IM, Wilson JG. Chem Rev. 1998;98:1515–1562. doi: 10.1021/cr941195u. [DOI] [PubMed] [Google Scholar]
- 3.Barth RF, Coderre JA, Vicente MGH, Blue TE. Clin Cancer Res. 2005;11:3987–4002. doi: 10.1158/1078-0432.CCR-05-0035. [DOI] [PubMed] [Google Scholar]
- 4.Barth RF, Vicente MGH, Harling OK, Kiger WS, III, Riley KJ, Binns PJ, Wagner FM, Suzuki M, Aihara T, Kato I, Kawabata S. Rad Oncol. 2012;7:146–167. doi: 10.1186/1748-717X-7-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huber JD, Egleton RD, Davis TP. Trends in Neurosciences. 2001;24:719–725. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- 6.Deeken JF, Löscher W. Clin Cancer Res. 2007;13:1663–1674. doi: 10.1158/1078-0432.CCR-06-2854. [DOI] [PubMed] [Google Scholar]
- 7.Weksler BB, Subileau EA, Perrière N, Charneau P, Holloway K, Leveque M, Tricoire-Leignel H, Nicotra A, Bourdoulous S, Turowski P, Male DK, Roux F, Greenwood J, Romero IA, Couraud PO. FASEB J. 2005;19:1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 8.Poller B, Gutmann H, Krähenbühl S, Weksler B, Romero IA, Couraud PO, Tuffin G, Drewe J, Huwyler J. J Neurochem. 2008;107:1358–1368. doi: 10.1111/j.1471-4159.2008.05730.x. [DOI] [PubMed] [Google Scholar]
- 9.Weksler B, Romero IA, Couraud PO. Fluids Barriers CNS. 2013;10:10, 16–25. doi: 10.1186/2045-8118-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markoutsa E, Pampalakis G, Niarakis A, Romero IA, Weksler B, Couraud PO, Antimisiaris SG. Eur J Pharm Biopharm. 2011;77:265–274. doi: 10.1016/j.ejpb.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Rooy I, Cakir-Tascioglu S, Couraud PO, Romero IA, Weksler B, Storm G, Hennink WE, Schiffelers RM, Mastrobattista E. Pharm Res. 2010;27:673–682. doi: 10.1007/s11095-010-0053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhupathiraju NVSDK, Hu X, Zhou Z, Fronczek FR, Couraud P-O, Romero IA, Weksler B, Vicente MGH. J Med Chem. 2014;57:6718–6728. doi: 10.1021/jm500786c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hopewell JW, Gorlia T, Pellettieri L, Giusti V, Stenstam BH, Skold K. Appl Radiat Isot. 2011;69:1737–1740. doi: 10.1016/j.apradiso.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 14.Kankaanranta L, Seppälä T, Koivunoro H, Saarilahti K, Atula T, Collan J, Salli E, Kortesniemi M, Uusi-Simola J, Välimäki P, Mäkitie A, Seppänen M, Minn H, Revitzer H, Kouri M, Kotiluoto P, Seren T, Auterinen I, Savolainen S, Joensuu H. Int J Rad Oncol Biol Phys. 2012;82:e67–e75. doi: 10.1016/j.ijrobp.2010.09.057. [DOI] [PubMed] [Google Scholar]
- 15.Loudet A, Burgess K. Chem Rev. 2007;107:4891–4932. doi: 10.1021/cr078381n. [DOI] [PubMed] [Google Scholar]
- 16.Boens N, Leen V, Dehaen W. Chem Soc Rev. 2012;41:1130–1172. doi: 10.1039/c1cs15132k. [DOI] [PubMed] [Google Scholar]
- 17.Grimes RN. Carboranes. 2. Academic Press; Oxford: 2011. [Google Scholar]
- 18.Gibbs JH, Robins LT, Zhou Z, Bobadova-Parvanova P, Cottam M, McCandless GT, Fronczek FR, Vicente MGH. Bioorg Med Chem. 2013;21:5770–5781. doi: 10.1016/j.bmc.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Vicente MGH, Fronczek FR, Smith KM. Chem Eur J. 2014;20:5064–5074. doi: 10.1002/chem.201304310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hao E, Fabre B, Fronczek FR, Vicente MGH. Chem Mat. 2007;19:6195–6205. [Google Scholar]
- 21.Gaillard PJ, de Boer AG. Eur J Pharmac Sci. 2000;12:95–102. doi: 10.1016/s0928-0987(00)00152-4. [DOI] [PubMed] [Google Scholar]
- 22.Rohand T, Qin W, Boens N, Dehaen W. Eur J Org Chem. 2006:4658–4663. [Google Scholar]
- 23.Bonardi L, Ulrich G, Ziessel R. Org Lett. 2008;10:2183–2186. doi: 10.1021/ol800560b. [DOI] [PubMed] [Google Scholar]
- 24.Jiao L, Yu C, Uppal T, Liu M, Li Y, Zhou Y, Hao E, Hu X, Vicente MGH. Org Biomol Chem. 2010;8:2517–2519. doi: 10.1039/c001068e. [DOI] [PubMed] [Google Scholar]
- 25.Jiao L, Pang W, Zhou J, Wei Y, Mu X, Bai G, Hao E. J Org Chem. 2011;76:9988–9996. doi: 10.1021/jo201754m. [DOI] [PubMed] [Google Scholar]
- 26.Godoy J, Vives G, Tour JM. Org Lett. 2010;12:1464–1467. doi: 10.1021/ol100108r. [DOI] [PubMed] [Google Scholar]
- 27.Niu SL, Ulrich G, Ziessel R, Kiss A, Renard P-Y, Romieu A. Org Lett. 2009;11:2049–2052. doi: 10.1021/ol900302n. [DOI] [PubMed] [Google Scholar]
- 28.Zhao N, Xuan S, Fronczek FR, Smith KM, Vicente MGH. submitted. [Google Scholar]
- 29.Leen V, Yuan P, Wang L, Boens N, Dehaen W. Org Lett. 2012;14:6150–6153. doi: 10.1021/ol3028225. [DOI] [PubMed] [Google Scholar]
- 30.Hao E, Friso E, Miotto G, Jori G, Soncin M, Fabris C, Sibrian-Vazquez M, Vicente MGH. Org Biomol Chem. 2008;6:3732– 3740. doi: 10.1039/b807836j. [DOI] [PubMed] [Google Scholar]
- 31.Bhupathiraju NVSDK, Vicente MGH. Bioorg Med Chem. 2013;21:485–495. doi: 10.1016/j.bmc.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boens N, Wang L, Leen V, Yuan P, Verbelen B, Dehaen W, der Auweraer MV, De Borggraeve WD, Meervelt LV, Jacobs J, Beljonne D, Tonnelé C, Lazzaroni R, Ruedas-Rama MJ, Orte A, Crovetto L, Talavera EM, Alvarez-Pez JM. J Phys Chem A. 2014;118:1576–1594. doi: 10.1021/jp412132y. [DOI] [PubMed] [Google Scholar]
- 33.Ferrer-Ugalde A, Juárez-Pérez EJ, Teixidor F, Viñas C, Sillanpää R, Pérez-Inestrosa E, Núñez R. Chem Eur J. 2012;18:544–553. doi: 10.1002/chem.201101881. [DOI] [PubMed] [Google Scholar]
- 34.Ferrer-Ugalde A, González-Campo A, Viñas C, Rodríguez-Romero J, Santillan R, Farfán N, Sillanpää R, Sousa-Pedrares A, Núñez R, Teixidor F. Chem Eur J. 2014;20:9940–9951. doi: 10.1002/chem.201402396. [DOI] [PubMed] [Google Scholar]