

Structural and functional connectivity phenotype in the neocortex of Fmr1−/y mice supports a prominent hypothesis of autism.
Keywords: Fragile X Syndrome, Autism, connectopathy, fMRI, DTI, retrograde tracing, neocortex, structural connectivity, functional connectivity, biomarker
Abstract
Fragile X syndrome (FXS), the most common inherited form of intellectual disability disorder and a frequent cause of autism spectrum disorder (ASD), is characterized by a high prevalence of sensory symptoms. Perturbations in the anatomical connectivity of neocortical circuits resulting in their functional defects have been hypothesized to contribute to the underlying etiology of these disorders. We tested this idea by probing alterations in the functional and structural connectivity of both local and long-ranging neocortical circuits in the Fmr1−/y mouse model of FXS. To achieve this, we combined in vivo ultrahigh-field diffusion tensor magnetic resonance imaging (MRI), functional MRI, and viral tracing approaches in adult mice. Our results show an anatomical hyperconnectivity phenotype for the primary visual cortex (V1), but a disproportional low connectivity of V1 with other neocortical regions. These structural data are supported by defects in the structural integrity of the subcortical white matter in the anterior and posterior forebrain. These anatomical alterations might contribute to the observed functional decoupling across neocortical regions. We therefore identify FXS as a “connectopathy,” providing a translational model for understanding sensory processing defects and functional decoupling of neocortical areas in FXS and ASD.
INTRODUCTION
One of the prevailing theories for explaining the neurobiological basis of autism is an alteration in the connectivity patterns present in the autistic brain. In particular, it is suggested that the long-range connectivity of the neocortex is reduced and that local connectivity is enhanced (1–4). Despite the appeal of this theory, which conceptually explains a number of behavioral symptoms of this disorder (2, 5), hard proof has been difficult to establish, in part because of the range of spatial and temporal scales needed to address this question. Recent studies suggest that although connectivity is largely atypic in autistic patients, the exact pattern of connectivity changes depends, to a large extent, on the age of the subjects and is complicated by the substantial heterogeneity present within the spectrum (4, 6, 7). Given these confounding factors, there is a need for refined models to understand how connectivity might be targeted for therapeutic intervention.
In the context of neurodevelopmental disorders such as autism spectrum disorder (ASD), fragile X syndrome (FXS) presents an attractive model for investigating changes in structural-functional connectivity. FXS is the most prevalent inherited form of intellectual disability disorder and the best-characterized cause of ASD, with at least 5% of ASD cases being attributed to FXS [reviewed by Budimirovic and Kaufmann (8)]. FXS and ASD are intertwined at the molecular level (9, 10). Moreover, FXS is a well-characterized neurobiological disorder of known etiology: In almost all cases, FXS is caused by the transcriptional silencing of the FMR1 gene. Mutational inactivation of the murine homolog of this gene (Fmr1) has led to the generation of a well-validated mouse model, the Fmr1−/y mouse (11, 12), which recapitulates many behavioral and physiological aspects of the disorder (13). Studies in this mouse model have led to a profound understanding of the neurobiological basis of this disorder. For example, early changes in structural connectivity features (14, 15), as well as changes in the strength and nature of synaptic connections (16–18), are suggested to play a fundamental role in the pathophysiology of FXS. However, although changes in local connectivity of neocortical circuits have been revealed during the early developmental stages of the Fmr1−/y mouse (14, 19), long-range connectivity is largely unexplored in this experimental model.
Here, we asked whether structural and functional connectivity is altered at the mesoscopic level in the Fmr1−/y mouse. In particular, we examined the balance between long- and short-range connectivity, placing particular emphasis on the anatomical pathways governing the flow of information to, from, and within the neocortex. The neocortex plays an essential role in the processing and integration of sensory information (among other things). Defects in sensory information processing are a common feature of both FXS (20, 21) and ASD (22–24) and have been suggested to play a role in the expression of a range of behavioral disturbances, such as restricted or stereotyped behavior (23, 25, 26). The Fmr1−/y mouse recapitulates a number of these sensory phenotypes. For example, hyperresponsiveness to acoustic stimuli (27, 28), and the sensory neocortex is hyperexcited in response to tactile and auditory stimuli (28–30), as reviewed by Contractor et al. (31). Here, we used a range of complementary methods to examine differences in the structural organization of white matter and anatomical wiring as well as differences in functional connectivity with the goal of identifying convergent patterns of abnormality in the neocortex of Fmr1−/y mice.
RESULTS
Reduced structural integrity of the corpus callosum
To provide an initial characterization of the anatomical connectivity of the whole brain of adult Fmr1−/y mice and wild-type littermate controls, we performed high-resolution (11.7 T) diffusion tensor imaging (DTI) permitting the identification of the corpus callosum—the major fiber bundle serving the neocortex. We calculated the fractional anisotropy (FA) values, permitting an exploration of the orientation coherence of axons in this fiber bundle (32). We found that the FA values are reduced in several regions of the corpus callosum of Fmr1−/y mice (cf. wild-type controls), suggesting diminished structural integrity of this fiber tract (Fig. 1). Specifically, FA values were decreased in the posterior part of the brain, the splenium/forceps major (FMJ) of the corpus callosum [FA(wild type)FMJ = 0.427 ± 0.005; FA(Fmr1−/y)FMJ = 0.391 ± 0.009; P = 0.0069], and in the anterior part of the brain, the forceps minor (FMI) of the corpus callosum [FA(wild type)FMI = 0.386 ± 0.007; FA(Fmr1−/y)FMI = 0.329 ± 0.019; P = 3.5 × 10−5]. No changes were seen in the external capsule (EC) [FA(wild type)EC = 0.347 ± 0.011; FA(Fmr1−/y)EC = 0.321 ± 0.008; not significant (n.s.)] and the central part of the corpus callosum, Genu/Body (G/B) [FA(wild type)G/B = 0.354 ± 0.003; FA(Fmr1−/y)G/B = 0.345 ± 0.007; n.s.] (Fig. 1). No significant changes were found in the measurements of other diffusion-related parameters, such as average diffusivity and first eigenvalue, either in the corpus callosum or in the neocortical areas (fig. S1). Together, these findings point to an alteration in the orientation of fibers within the corpus callosum rather than to changes in their absolute number. For example, a relative decrease of the overall long-range connectivity compared to the local/short-range connectivity could reduce the FA value.
Fig. 1. Reduced structural integrity of the corpus callosum in Fmr1−/y mice.
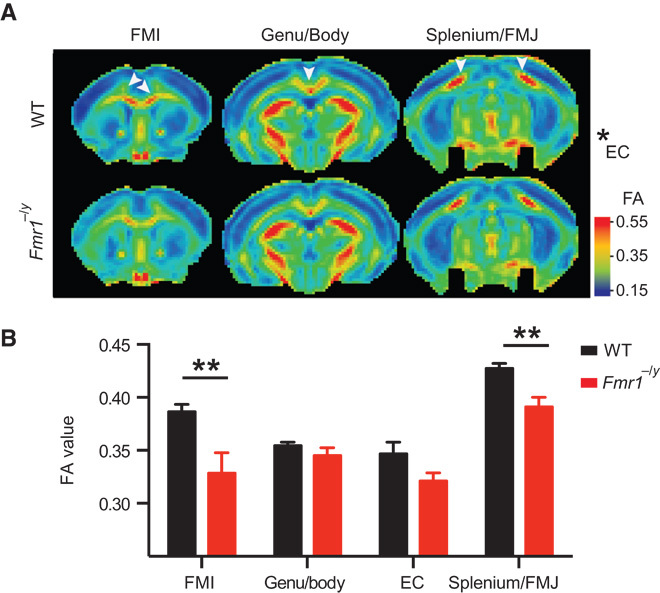
DTI was performed on adult Fmr1−/y and wild type (WT) mice to measure the FA of the corpus callosum. Tensor images were collectively acquired in several horizontal planes from +2.0 to −4.0 mm from the bregma, with an interplane distance of 0.5 mm (WT, n = 12; Fmr1−/y, n = 7). FA values were measured on individual planes and grouped into the splenium/FMJ of the corpus callosum, EC, the Genu/Body of the corpus callosum, and the FMI of the corpus callosum. (A) Color-coded heat maps of the FA values showing the average (of all WT and Fmr1−/y animals) of one plane from each group (from anterior to posterior). Warm colors indicate fiber tracts with strong diffusion coherence. (B) The FA values were significantly reduced in Fmr1−/y mice in the FMI (P = 3.5 × 10−5) and the splenium/FMJ (P = 0.0069). Data are means ± SEM. **P < 0.01 (Fmr1−/y compared with WT). Statistical significance was determined using multiple t tests corrected for multiple comparisons using the Holm-Sidak method with α = 0.05.
Alterations in the structural connectivity of the primary visual cortex
The aforementioned results, showing an altered structural organization of the white matter in Fmr1−/y mice, suggest that the adjacent neocortical areas—in particular the frontal cortex and the primary visual cortex (V1)—may be strongly affected by these changes. Sensory information processing defects are a common feature of both FXS and ASD [for example, Lane et al. (22) and Marco et al. (33)], and functional imaging studies suggest increased activation of the neocortex of ASD subjects in response to sensory stimuli of a range of modalities (24). In addition, alterations in visual perception and fine-scale neuroanatomical defects in the visual cortex (V1) have been reported for FXS patients (34–37). Indeed, rescue of an aberrant spine phenotype in V1 of Fmr1−/y mice has been used as an end point for pharmacological rescue approaches (38). In light of these findings and our own aforementioned results, we probed the reorganization of the anatomical connectivity of V1 by mapping and quantifying neurons projecting to this region. To permit this analysis, we injected a retrograde viral tracer into V1 and subsequently quantified local and long-ranging projections [as the fraction of labeled neurons (FLN)]. To determine the precise location of the injection site, we simultaneously co-injected an anterograde variant of the same virus (39). The cell body locations of all labeled cells were then introduced as fiducial markers into a three-dimensional (3D) brain model (40) to compute the Euclidian distance in the whole brain from the injection site (Fig. 2; representative images of tracing are shown in fig. S2; schematic representation of the experimental procedure is shown in fig. S3; for movies showing the totality of retrogradely labeled cells in the 3D brain model of wild-type and Fmr1−/y mice, see movies S1 and S2, respectively). The largest proportion of inputs to V1 stemmed from a limited number of brain areas, namely, the primary and secondary visual cortices, the thalamic nuclei, and the auditory and motor cortices. Additional projecting cells were detected in the orbitofrontal cortex, the cingulate cortex, the somatosensory cortex, the retrosplenial cortex, the preoptic area of the hypothalamus, and the contralateral visual cortex. We also consistently found cells in the basal telencephalon, most likely belonging to the nucleus basalis of Meynert.
Fig. 2. Reorganization of inputs to V1 of Fmr1−/y mice.

(A) Summary image showing the localization of all retrogradely (input; green) and anterogradely (local; red) labeled neurons of all WT (upper panel; n = 3) and Fmr1−/y (lower panel; n = 3) mice within a 3D model. Scale bar, 1 mm. (B) Average distance of the retrogradely labeled cells from the injection site in Fmr1−/y and WT mice (***P < 0.0001, Mann-Whitney test). (C) The relative FLN (in percentage of total) plotted as a function of distance to the injection site indicates a significant change in the distribution (***P < 0.0001, Kolmogorov-Smirnov test). (C′) In particular, the FLN with a distance of less than 0.5 mm is increased in Fmr1−/y mice (P = 0.0242, unpaired t test). (D) FLN expressed as a function of brain area showing an increased number of local inputs (from V1) in Fmr1−/y mice compared to WT mice (P = 1.57 × 10−5, multiple t tests corrected for multiple comparisons using the Holm-Sidak method with α = 0.05). Ctx, cortical areas; Vi, visual cortex; V1, primary Vi; V2, secondary Vi; V2L, lateral V2; V2MM, mediomedial V2; V2ML, mediolateral V2; Motor, motor cortex; Som, somatosensory cortex; RS, retrosplenial cortex; CG, cingulate cortex; Au, auditory cortex; Thal, thalamus; ON, other nuclei. (E) The average distance of the local input (within V1) is decreased in the Fmr1−/y mice (P < 0.0001, Mann-Whitney test). (F) The distribution of the local input (within V1) shows that the fraction 0 to 400 μm is increased, whereas the fraction above 600-μm distance is decreased (***P < 0.0001, Kolmogorov-Smirnov test). All data are means ± SEM.
We found that the average distance from the injection site of all labeled neurons projecting to V1 was reduced in the Fmr1−/y mice (Fig. 2, A and B; P < 0.0001, Mann-Whitney test), which supports the “long-range hypoconnectivity versus local hyperconnectivity hypothesis of autism” (2). However, the concept of local connectivity is, in general, poorly defined in the literature and can be expressed (i) as a factor of distance from the injection site or (ii) with respect to a functionally defined brain area. We found that the number of local connections was increased in Fmr1−/y mice according to both definitions (Fig. 2, C to F). For example, the FLN for proximal locations is significantly greater for Fmr1−/y cf. wild-type subjects [Fig. 2C′; FLN(wild type)proximal = 28.52 ± 6.17%; FLN(Fmr1−/y)proximal = 53.63 ± 3.52%; P = 0.0242 using unpaired t test]. Likewise, quantifying the FLN as a function of brain area shows a sig????nificant bias toward intrinsic inputs from within V1 [Fig. 2, D to F; FLN(wild type)intrinsic = 45.79 ± 4.29%; FLN(Fmr1−/y)intrinsic = 62.28 ± 5.96%; P = 1.57 × 10−5 using multiple t tests corrected for multiple comparisons using the Holm-Sidak method with α = 0.05]. When considering only the intrinsic input, we additionally found an increased local “input clustering” in Fmr1−/y mice. Specifically, more incoming projections arrive from cells in proximity to the target zone (Fig. 2E; P < 0.0001, Mann-Whitney test) with a peak of input density at ~220 to 260 μm (Fig. 2F).
Functional decoupling of neocortical regions
Increased clustering has previously been shown to alter the dynamics of network activity (41) and may also serve to amplify the intrinsic signal at the expense of more distant inputs. An overrepresentation of the local information and a reduction of input from other regions would cause the target region to be more isolated and functionally decoupled from other brain areas. To test this possibility, we measured spontaneous activity using functional magnetic resonance imaging (fMRI) in head-restrained Fmr1−/y (n = 7) and wild-type (n = 10) mice under light isoflurane anesthesia. In particular, we measured the functional connectivity of the somatosensory, auditory, visual, and motor cortices; the dorsal and ventral hippocampi; and the subcortical areas (Fig. 3 and fig. S4).
Fig. 3. Functional decoupling of neocortical brain areas in Fmr1−/y mice.
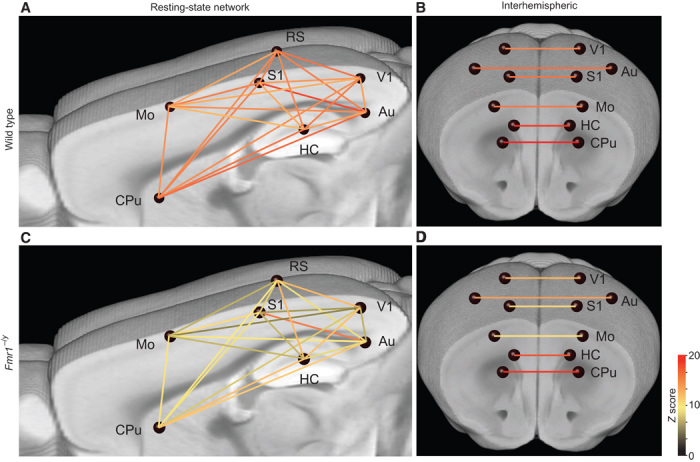
Brain graph showing the network connectivity of nodes (blue spheres) via edges (lines, color-coded for their Z score), indicating the strength of the connections. (A to D) fMRI measurements under light isoflurane anesthesia revealed a reduced functional connectivity between a number of neocortical brain areas in the Fmr1−/y (A and B; n = 7) compared to WT (C and D; n = 10) mice. In particular, the intrahemispheric connections are strongly affected (A and C), whereas the homotopic interhemispheric connectivity is only partially affected (for example, Mo-Mo; B and D). HC, hippocampus; Mo, motor cortex; S1, primary somatosensory cortex; V1, primary visual cortex; CPu, caudate putamen.
We found a functional decoupling that largely affected connections between the hippocampus and neocortical areas, as well as intracortical connectivity. For example, the visual cortex was less linked to the auditory, motor, and somatosensory cortices. Similarly, the connectivity between the auditory cortex and the dorsal and ventral hippocampi as well as the retrosplenial cortex was also deficient. Overall, all examined cortical areas appear to be strongly affected, and none of the probed connections showed an increase in functional connectivity. The predominant pattern of spontaneous activity (under light anesthesia) observed within the network supports the notion of a functional decoupling of numerous brain areas (Fig. 3). The lateral (intrahemispheric) network connectivity (Fig. 3, A and C, and fig. S4) was more strongly affected than the homotopic interhemispheric connectivity (Fig. 3, B and D). However, we noted a general trend for reduced homotypic interhemispheric connectivity in the neocortical areas, with significant differences noted for the motor cortex [fig. S4; P = 0.0034 using multivariate analysis of variance (ANOVA), corrected for multiple comparisons]. Together, we provide evidence for global changes in functional connectivity in the forebrain of adult Fmr1−/y mice.
DISCUSSION
Here, we present converging evidence pointing to both large-scale and local connectivity changes impinging on the sensory circuits in Fmr1−/y mice. In particular, we found a localized reduction in FA values, which points to white matter defects suggestive of alterations in the organization of the corpus callosum. We demonstrate, at the anatomical level, a local hyperconnectivity in V1 versus a long-range hypoconnectivity affecting global inputs into V1. These structural findings are corroborated by evidence of widespread cross-area functional hypoconnectivity, affecting the sensory circuits (among others) of Fmr1−/y mice. Together, these findings lend support to the theory of “long-range hypoconnectivity versus local hyperconnectivity” in ASDs.
The local hyperconnectivity phenotype reported here for the visual cortex is consistent with recent findings from resting-state fMRI studies of adolescents with ASD (42). Although the methods used in the current study are not completely equivalent to those used by Keown et al. (42)—because of the requirement for light anesthesia and the use of invasive tracers in the current study—they nonetheless point to a similar pattern of connectivity changes. Together, these findings support the notion that increased local connectivity might contribute to “islets of superior functioning” in the sensory neocortex (7). They could equally provide an explanation for the heightened sensitivity to visual stimuli (gaze aversion) (43) or the increased amplitude of visually evoked potentials (44) observed in FXS patients.
Altered sensory function has been consistently shown to be a feature of both FXS syndrome and ASD (20–22, 33). Although an altered balance of excitation/inhibition [for example, Gibson et al. (16)] and increased neocortical/neuronal excitability (28, 45) clearly play a role in this phenomenon, structural connectivity changes resulting in, for example, the enhanced local hyperconnectivity demonstrated here could likely also exacerbate local network excitability. Alterations in visual perception, in particular an enhanced processing of fine detail or local structure, have consistently been shown to be a feature of ASD (46). Such perceptual alterations could likely contribute to some of the core behavioral features of the disorder (46). A better mechanistic understanding of how neuronal circuitry contributes to such phenomena could thus provide a platform for understanding some of the behavioral alterations observed in ASD (31, 47).
The development of quantitative biomarkers either for the evaluation of treatment response or for patient stratification is seen as a critical step toward the development of improved clinical trials for both ASD and FXS (48–50). Such markers could be used to document the effect of a clinical treatment or provide an advanced means of selecting patients who are likely to respond to a particular treatment especially in a multifaceted disorder like ASD (22, 47, 51, 52). Neuroimaging approaches are suggested to be particularly promising because they are objectively measurable and thought to reflect the underlying neurobiology of the disorder (53). The convergence of ASD susceptibility genes and FMRP (fragile X mental retardation protein) targets on pathways and processes governing connectivity (9, 10) further suggest that these molecules might also be targeted for pharmacological rescue. The next step in the validation of our findings would be to test whether candidate drugs can influence the aforementioned quantifiable parameters of structural or functional connectivity.
Although short-range connectivity changes have been described in Fmr1−/y mice during early postnatal development (that is, 2 to 4 weeks postnatal), these alterations are generally thought to be transient in nature (14, 16, 19, 45, 54–56). Such changes have led to a theory of a critical window for cellular and structural plasticity, suggesting that intervention during this developmentally sensitive time window is crucial for this neurodevelopmental disorder (56, 57). Here, we show a connectivity phenotype present in adult Fmr1−/y mice that challenges the notion that alterations in connectivity are restricted to this early developmental window. Largely in agreement with these findings are recent studies in humans, which identified large-scale network deficits in adolescent and adult FXS patients (58, 59). Particularly compelling are the findings that functional connectivity affecting a number of neocortical networks was reduced (58). In addition, the integrity of certain white matter structures was also reduced based on DTI in adolescence/adulthood and childhood (60, 61). Although it is clearly beyond the scope of the current study, it is important to note that rewiring of cortical circuits is possible, even during adulthood in healthy animals (62). Moreover, therapeutic rescue of connectivity features has previously been demonstrated in adolescence/adulthood in murine models of both FXS and ASD (38, 63).
Although we have chosen to focus on connectivity changes affecting the sensory regions of the neocortex in adulthood, this is by no means the only region implicated in the pathophysiology of FXS or ASD. Indeed, even within the context of sensory information processing, a variety of brain structures may play a role in the refinement of the neocortical circuits required for the integration of sensory information (64). Previous neuroimaging studies in individuals with FXS have identified a range of brain structures with abnormal volume, including caudate nucleus, amygdala, hippocampus, and specific regions of the neocortex and cerebellum [reviewed by Lightbody and Reiss (53)]. More diverse changes have been more recently reported in both gray and white matter volumes in younger children with FXS (65), possibly reflecting refinements in imaging technology and developmental stage. For ASD in general, the findings are more diverse. Indeed, the overall pattern is one of heterogeneity, rather than defects in specific structures, reflecting perhaps differences in image analysis, varying criteria for subject inclusion, and the heterogeneity inherent within this population (22, 47, 66). With relevance to the current work, a meta-analysis of the existing structural MRI data suggests both an alteration of cortical thickness in the parietal lobes and reduced structural integrity of the corpus callosum (66). A recent study using MRI-based neuroanatomical characterization of diverse mouse models of ASD suggests that a mathematical clustering approach might be used to identify mouse models likely to respond to the same therapeutic rescue approach (67). In light of these findings, it would be interesting to determine whether mouse models co-clustering with the Fmr1−/y model exhibit similar patterns of long- and short-range connectivity.
MATERIALS AND METHODS
Mice
Adult (9 to 12 weeks old) male Fmr1−/y mice (12) in a C57BL/6 background and their male wild-type littermates [as described by Zhang et al. (28)] were used in all experiments. Animals were bred at two sites, namely, the SPF (specific pathogen–free) animal facility of the Neurocentre Magendie (Bordeaux, France) and the animal facility of the Radboud University (Nijmegen, Netherlands). All experiments were performed according to the European Directive governing the use of experimental animals (2010/63/EU) and local institutional guidelines; all experiments received previous ethical approval from the Ethics Committee of Bordeaux (CE2A50; approval #5012024-A). Genotypes were determined by a polymerase chain reaction analysis of DNA extracted from tail samples. All experiments and analysis were performed with the experimenter being blind to the genotype.
Virus production
SAD ΔG-eGFP (RG), a retrograde variant of glycoprotein-deleted recombinant rabies virus, was produced as previously described by Haberl et al. (39). VSV GRtmC-pseudotyped SAD ΔG-mCherry, an anterograde variant of the same virus, was produced as previously described by Haberl et al. using BSR T7/5 cells (39).
Stereotactic injections
Stereotactic injections were performed in Fmr1−/y and wild-type mice at 10 to 12 weeks of age. The stereotactic injections of viral vectors were performed in isoflurane-anesthetized and head-fixed mice using a 10-μl glass syringe fitted with a 34-gauge needle or a pulled glass pipette. Injection volume and speed were controlled using a WPI Ultra Micro Pump. Viral injections were performed using a 9:1 mixture of retrograde RABV ΔG (expressing enhanced green fluorescent protein) and anterograde RABV ΔG (expressing mCherry) as described by Haberl et al. (68). mCherry expression at the site of injection was used to verify injection coordinates and determine the precise location of the injection. Injection coordinates for upper layer 5 of V1 were at anterior/posterior (A/P) 2.8 mm, lateral (L) 2.25 mm, and dorsal/ventral (D/V) 0.5 mm. A/P and L coordinates are given with respect to the bregma, whereas D/V coordinates are given with respect to the brain surface. Precise coordinates were later validated for each injection by calculating the center of mass of all anterogradely [SAD ΔG-mCherry (VSV GRtmC)] labeled neurons in the respective brain (COMi). To verify that all injections are comparable (mapping the same area), we calculated the SD from the injection coordinates using the COMi of all analyzed injections: A/P, 2.8 ± 0.175 mm; L, 2.25 ± 0.126 mm; and D/V, 0.5 ± 0.06 mm. All distance calculations were performed for every cell to the COMi of the respective brain.
Mouse brain slice preparation
Mice were perfused and brains were sectioned as previously described by Haberl et al. (39). Briefly, mice were administered a lethal dose of sodium pentobarbital and then transcardially perfused with 30 ml of normal Ringer’s solution followed by 100 ml of a 4% paraformaldehyde (PFA) solution in 1× phosphate-buffered saline. The mouse brains were dissected and postfixated in 4% PFA solution and slices were cut using a vibratome (Leica). For whole-forebrain sectioning, brains were immersed in 10% gelatin, postfixed 2 hours in 4% PFA, sectioned in 50-μm slices, and mounted using Prolong Gold Antifade Reagent.
Fluorescence microscopy and analysis
Images of the entire forebrain were acquired using a scanning mosaic wide-field fluorescence acquisition system (NanoZoomer, Hamamatsu) equipped with a 20× 0.75 numerical aperture objective. Images were acquired by scanning each section at multiple (about five to six) z-positions with an 8-μm step size. Analysis of retrogradely and locally labeled cells was performed blind to the genotype. Fiducial markers were placed manually on the cell position, and images were segmented using the mouse brain atlas (The Mouse Brain in Stereotaxic Coordinates by K. B. J. Franklin and G. Paxinos, 1997). Fiducial points transformed in a 3D average brain atlas in Vaa3D software (69) were used to compute the Euclidian distance of each marker in the whole brain in xyz directions from the injection site. The brain model was previously generated from anatomical MRI scans of adult C57BL/6 mice at 16-μm spatial resolution (40). The precise coordinates of each injection site were confirmed by calculating the center of mass of cells infected by the local tracer SAD ΔG-mCherry (VSV-G). We counted a total of 7209 retrogradely labeled neurons and calculated an average distance of 973 μm to its respective COMi (1183 μm in the wild-type mice and 810 μm in the Fmr1−/y mice) equaling a total minimum wiring length of 6.94 m (1.22 ± 0.09 m in wild type versus 1.09 ± 0.07 m in Fmr1−/y).
Definition
To test the hypothesis of short-ranging hyperconnectivity, we determined the intrinsic and the proximal FLN. Retrogradely labeled neurons within V1 are intrinsic. Proximity was set as distance below 500 μm.
Magnetic resonance imaging
MRI measurements were performed with an 11.7-T BioSpec Avance III small animal MR system (Bruker BioSpin) equipped with an actively shielded gradient set of 600 mT/m and operated by Paravision 5.1 software. We used a circular polarized volume resonator for signal transmission and an actively decoupled mouse brain quadrature surface coil for signal reception (Bruker BioSpin). The levels of anesthesia and mouse physiological parameters were monitored following an established protocol to obtain a reliable measurement of functional connectivity (70). Briefly, during the MR experiments, low-dose isoflurane was used (3.5% for induction and ~1.5% for maintenance), slightly adjusted throughout the experiment to maintain a fast and stable breathing frequency (>130 beats/min). The mice were placed in a stereotactic device to immobilize the head. Body temperature was measured with a rectal thermometer and maintained at 37°C by a heated airflow device.
After standard adjustments and shimming, fMRI data sets were acquired using a single-shot spin-echo sequence combined with echo-planar imaging (SE-EPI) sequence. Six hundred repetitions with a repetition time (TR) of 1.8 s and an echo time (TE) of 16.9 ms were recorded for a total acquisition time of 18 min. Other imaging parameters were as follows: field of view, 25 × 25 mm; image matrix, 96 × 96; spatial resolution, 260 × 260 × 500 μm; number of slices, 9.
Diffusion of water was imaged as previously described by Zerbi et al. (32) and Harsan et al. (71). In short, 22 axial slices covering the whole brain were acquired with a four-shot SE-EPI protocol. B0 shift compensation, navigator echoes, and an automatic correction algorithm to limit the occurrence of ghosts and artifacts were implemented. Encoding b factors of 0 s/mm2 (b0 images; 5×) and 1000 s/mm2 were used and diffusion-sensitizing gradients were applied along 30 noncollinear directions in 3D space. Other imaging parameters were as follows: TR, 7.55 s; TE, 20 ms; field of view, 20 × 20 mm; image matrix, 128 × 128; spatial resolution, 156 × 156 × 500 μm; total acquisition time, 18 min.
Functional connectivity measurements
The fMRI data sets were processed as previously described by Zerbi et al. (70). Briefly, the data were first realigned using a least-squares method and rigid-body transformation with Statistical Parametric Mapping (SPM) mouse toolbox [SPM5, University College London (72)]. The mean SE-EPI images of each mouse were then used to generate and normalize the data into a study-specific template through linear affine and nonlinear diffeomorphic transformation [ANTs (Advanced Normalization Tools) V1.9, http://picsl.upenn.edu/ANTS/]. On the template, 17 areas were selected in the left and right hemispheres and back-transformed in each subject space using the inverse of the affine and diffeomorphic transformations. Brain regions were segmented based on an MRI atlas (40) and include the following: dorsal hippocampus, ventral hippocampus, auditory cortex, primary motor cortex, somatosensory cortex, primary visual cortex, retrosplenial cortex, piriform cortex, amygdala, pretectal area, caudate putamen, lateral geniculate nucleus, globus pallidus, parafascicular nucleus, ventral posterolateral nucleus, and ventral posteromedial nucleus. In-plane spatial smoothing (0.4 × 0.4 mm) and temporal high-pass filtering (cutoff at 0.01 Hz) were applied to compen??????sate for small across-mouse misregistration and temporal low-frequency noise using the FEAT tool of FSL [FSL 5.0 (73)]. Functional connectivity between regions of interest (ROIs) was calculated from the blood oxygen level–dependent time series after movement regression using total correlation analyses implemented in FSLNets (FSLNets, V0.3, www.fmrib.ox.ac.uk/fsl). Pearson correlation values were Fisher-transformed to Z scores for group comparisons and statistical analysis.
Diffusion tensor MRI parameter estimation
The calculation of the two commonly used DT-MRI parameters, mean diffusivity (MD) and FA, was performed following a protocol as previously described by Zerbi et al. (32). Briefly, the diffusion images were first realigned with SPM mouse toolbox to compensate for small movement artifacts; thereafter, the data sets were spatially normalized to a study-specific template through linear affine and nonlinear diffeomorphic transformation using ANTs. Following these preprocessing steps, the diffusion tensor was estimated for every voxel using the PATCH algorithm (74). ROIs in several white matter and gray matter areas were drawn on the template image based on an anatomical atlas (The Mouse Brain in Stereotaxic Coordinates), and the resulting FA and MD values were measured for further statistical analyses.
Statistical analysis
Prism 6.0e was used for analysis with statistical tests as described. All values were presented as means ± SEM unless stated otherwise. Two-tailed unpaired t tests were performed to evaluate the difference between two groups of data. Mann-Whitney test was used to compare two groups in the absence of Gaussian distribution. Two-sample Kolmogorov-Smirnov test was used to test for the equality of distributions. Multiple t tests corrected for multiple comparison using the Holm-Sidak method and multivariate ANOVA corrected for multiple comparison using Bonferroni’s method were used for the analysis of more than two parameters. The specific details are provided in the text and in each figure legend.
Acknowledgments
Microscopy was performed using equipment of the Bordeaux Imaging Center. We thank S. Viana Da Silva for feedback and help with experiments and K. Conzelmann for rabies virus starter stocks. Funding: This project was supported by the European Commission FP7 Erasmus Mundus Doctoral Fellowship (M.G.H.) as well as funding from INSERM, Conseil de la Region, d’Aquitaine, LABEX BRAIN ANR-10-LABX-43, and the Euro-BioImaging initiative. The MR instrument was acquired with the (partial) support of Nederlandse Organisatie voor Wetenschappelijk Onderzoek (investment grant no. 91106021). Author contributions: The study was designed by M.G.H., A.H., and A.F. Experiments were conducted and analyzed by M.G.H., V.Z., and A.V. Virus was produced by M.G.H. and M.G. The manuscript was written by M.G.H., M.G., and A.F. with the help of the rest of the authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors at M.G.H. (haberlmatt@gmail.com) or A.F. (andreas.frick@inserm.fr).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/10/e1500775/DC1
Fig. S1. High-field 11.7-T DT-MRI measurements of the white and gray matter of adult Fmr1−/y and wild-type littermate mice.
Fig. S2. Tracing of the input to V1.
Fig. S3. Schematic representation of the experimental strategy for the 3D anatomical registration of projection neurons into V1.
Fig. S4. Functional connectivity matrix of wild-type and Fmr1−/y mice.
Movie S1. Slice view and 3D view, illustrating the 3D mouse brain model with the combined positions of all retrogradely (input; green) and anterogradely (local; red) labeled neurons in wild-type mice.
Movie S2. Slice view and 3D view, illustrating the 3D mouse brain model with the combined positions of all retrogradely (input; green) and anterogradely (local; red) labeled neurons in Fmr1−/y mice.
REFERENCES AND NOTES
- 1.Belmonte M. K., Allen G., Beckel-Mitchener A., Boulanger L. M., Carper R. A., Webb S. J., Autism and abnormal development of brain connectivity. J. Neurosci. 24, 9228–9231 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Courchesne E., Pierce K., Why the frontal cortex in autism might be talking only to itself: Local over-connectivity but long-distance disconnection. Curr. Opin. Neurobiol. 15, 225–230 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Rippon G., Brock J., Brown C., Boucher J., Disordered connectivity in the autistic brain: Challenges for the “new psychophysiology”. Int. J. Psychophysiol. 63, 164–172 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Rane P., Cochran D., Hodge S. M., Haselgrove C., Kennedy D. N., Frazier J. A., Connectivity in autism: A review of MRI connectivity studies. Harv. Rev. Psychiatry 23, 223–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geschwind D. H., Levitt P., Autism spectrum disorders: Developmental disconnection syndromes. Curr. Opin. Neurobiol. 17, 103–111 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Hahamy A., Behrmann M., Malach R., The idiosyncratic brain: Distortion of spontaneous connectivity patterns in autism spectrum disorder. Nat. Neurosci. 18, 302–309 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Rudie J. D., Dapretto M., Convergent evidence of brain overconnectivity in children with autism? Cell Rep. 5, 565–566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Budimirovic D. B., Kaufmann W. E., What can we learn about autism from studying fragile X syndrome? Dev. Neurosci. 33, 379–394 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darnell J. C., Van Driesche S. J., Zhang C., Hung K. Y. S., Mele A., Fraser C. E., Stone E. F., Chen C., Fak J. J., Wook Chi S., Licatalosi D. D., Richter J. D., Darnell R. B., FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parikshak N. N., Luo R., Zhang A., Won H., Lowe J. K., Chandran V., Horvath S., Geschwind D. H., Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The Dutch-Belgian Fragile X Consortium , Fmr1 knockout mice: A model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33 (1994). [PubMed] [Google Scholar]
- 12.Mientjes E. J., Nieuwenhuizen I., Kirkpatrick L., Zu T., Hoogeveen-Westerveld M., Severijnen L., Rifé M., Willemsen R., Nelson D. L., Oostra B. A., The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol. Dis. 21, 549–555 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Kazdoba T. M., Leach P. T., Silverman J. L., Crawley J. N., Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res. 3, 118–133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bureau I., Shepherd G. M. G., Svoboda K., Circuit and plasticity defects in the developing somatosensory cortex of Fmr1 knock-out mice. J. Neurosci. 28, 5178–5188 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nimchinsky E. A., Oberlander A. M., Svoboda K., Abnormal development of dendritic spines in FMR1 knock-out mice. J. Neurosci. 21, 5139–5146 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibson J. R., Bartley A. F., Hays S. A., Huber K. M., Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huber K. M., Gallagher S. M., Warren S. T., Bear M. F., Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J., Pelletier M. R., Perez Velazquez J.-L., Carlen P. L., Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 19, 138–151 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Testa-Silva G., Loebel A., Giugliano M., de Kock C. P. J., Mansvelder H. D., Meredith R. M., Hyperconnectivity and slow synapses during early development of medial prefrontal cortex in a mouse model for mental retardation and autism. Cereb. Cortex 22, 1333–1342 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller L. J., McIntosh D. N., McGrath J., Shyu V., Lampe M., Taylor A. K., Tassone F., Neitzel K., Stackhouse T., Hagerman R. J., Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: A preliminary report. Am. J. Med. Genet. 83, 268–279 (1999). [PubMed] [Google Scholar]
- 21.Baranek G. T., Roberts J. E., David F. J., Sideris J., Mirrett P. L., Hatton D. D., Bailey D. B. Jr, Developmental trajectories and correlates of sensory processing in young boys with fragile X syndrome. Phys. Occup. Ther. Pediatr. 28, 79–98 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Lane A. E., Young R. L., Baker A. E. Z., Angley M. T., Sensory processing subtypes in autism: Association with adaptive behavior. J. Autism Dev. Disord. 40, 112–122 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Boyd B. A., Baranek G. T., Sideris J., Poe M. D., Watson L. R., Patten E., Miller H., Sensory features and repetitive behaviors in children with autism and developmental delays. Autism Res. 3, 78–87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green S. A., Rudie J. D., Colich N. L., Wood J. J., Shirinyan D., Hernandez L., Tottenham N., Dapretto M., Bookheimer S. Y., Overreactive brain responses to sensory stimuli in youth with autism spectrum disorders. J. Am. Acad. Child Adolesc. Psychiatry 52, 1158–1172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baumgardner T. L., Reiss A. L., Freund L. S., Abrams M. T., Specification of the neurobehavioral phenotype in males with fragile X syndrome. Pediatrics 95, 744–752 (1995). [PubMed] [Google Scholar]
- 26.Wigham S., Rodgers J., South M., McConachie H., Freeston M., The interplay between sensory processing abnormalities, intolerance of uncertainty, anxiety and restricted and repetitive behaviours in autism spectrum disorder. J. Autism Dev. Disord. 45, 943–952 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Chen L., Toth M., Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103, 1043–1050 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y., Bonnan A., Bony G., Ferezou I., Pietropaolo S., Ginger M., Sans N., Rossier J., Oostra B., LeMasson G., Frick A., Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1−/y mice. Nat. Neurosci. 17, 1701–1709 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Rotschafer S., Razak K., Altered auditory processing in a mouse model of fragile X syndrome. Brain Res. 1506, 12–24 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Arnett M. T., Herman D. H., McGee A. W., Deficits in tactile learning in a mouse model of fragile X syndrome. PLOS One 9, e109116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Contractor A., Klyachko V. A., Portera-Cailliau C., Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zerbi V., Kleinnijenhuis M., Fang X., Jansen D., Veltien V., Van Asten J., Timmer N., Dederen P. J., Kiliaan A. J., Heerschap A., . Gray and white matter degeneration revealed by diffusion in an Alzheimer mouse model. Neurobiol. Aging 34, 1440–1450 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Marco E. J., Hinkley L. B. N., Hill S. S., Nagarajan S. S., Sensory processing in autism: A review of neurophysiologic findings. Pediatr. Res. 69, 48R–54R (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cornish K. M., Munir F., Cross G., Spatial cognition in males with fragile-X syndrome: Evidence for a neuropsychological phenotype. Cortex 35, 263–271 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Farzin F., Whitney D., Hagerman R. J., Rivera S. M., Contrast detection in infants with fragile X syndrome. Vision Res. 48, 1471–1478 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scerif G., Cornish K., Wilding J., Driver J., Karmiloff-Smith A., Visual search in typically developing toddlers and toddlers with fragile X or Williams syndrome. Dev. Sci. 7, 116–130 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Irwin S. A., Patel B., Idupulapati M., Harris J. B., Crisostomo R. A., Larsen B. P., Kooy F., Willems P. J., Cras P., Kozlowski P. B., Swain R. A., Jeanne Weiler I., Greenough W. T., Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: A quantitative examination. Am. J. Med. Genet. 98, 161–167 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Michalon A., Sidorov M., Ballard T. M., Ozmen L., Spooren W., Wettstein J. G., Jaeschke G., Bear M. F., Lindemann L., Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haberl M. G., Viana da Silva S., Guest J. M., Ginger M., Ghanem A., Mulle C., Oberlaender M., Conzelmann K.-K., Frick A., An anterograde rabies virus vector for high-resolution large-scale reconstruction of 3D neuron morphology. Brain Struct. Funct. 220, 1369–1379 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ullmann J. F. P., Watson C., Janke A. L., Kurniawan N. D., Reutens D. C., A segmentation protocol and MRI atlas of the C57BL/6J mouse neocortex. Neuroimage 78, 196–203 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Litwin-Kumar A., Doiron B., Slow dynamics and high variability in balanced cortical networks with clustered connections. Nat. Neurosci. 15, 1498–1505 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keown C. L., Shih P., Nair A., Peterson N., Mulvey M. E., Müller R.-A., Local functional overconnectivity in posterior brain regions is associated with symptom severity in autism spectrum disorders. Cell Rep. 5, 567–572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merenstein S. A., Sobesky W. E., Taylor A. K., Riddle J. E., Tran H. X., Hagerman R. J., Molecular-clinical correlations in males with an expanded FMR1 mutation. Am. J. Med. Genet. 64, 388–394 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Knoth I. S., Vannasing P., Major P., Michaud J. L., Lippé S., Alterations of visual and auditory evoked potentials in fragile X syndrome. Int. J. Dev. Neurosci. 36, 90–97 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Gonçalves J. T., Anstey J. E., Golshani P., Portera-Cailliau C., Circuit level defects in the developing neocortex of fragile X mice. Nat. Neurosci. 16, 903–909 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dakin S., Frith U., Vagaries of visual perception in autism. Neuron 48, 497–507 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Amaral D. G., Schumann C. M., Nordahl C. W., Neuroanatomy of autism. Trends Neurosci. 31, 137–145 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Ghosh A., Michalon A., Lindemann L., Fontoura P., Santarelli L., Drug discovery for autism spectrum disorder: Challenges and opportunities. Nat. Rev. Drug Discov. 12, 777–790 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Jacquemont S., Berry-Kravis E., Hagerman R., von Raison F., Gasparini F., Apostol G., Ufer M., Des Portes V., Gomez-Mancilla B., The challenges of clinical trials in fragile X syndrome. Psychopharmacology 231, 1237–1250 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berry-Kravis E., Hessl D., Abbeduto L., Reiss A. L., Beckel-Mitchener A., Urv T. K. The Outcome Measures Working Groups , Outcome measures for clinical trials in fragile X syndrome. J. Dev. Behav. Pediatr. 34, 508–522 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veenstra-VanderWeele J., Blakely R. D., Networking in autism: Leveraging genetic, biomarker and model system findings in the search for new treatments. Neuropsychopharmacology 37, 196–212 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amaral D. G., The promise and the pitfalls of autism research: An introductory note for new autism researchers. Brain Res. 1380, 3–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lightbody A. A., Reiss A. L., Gene, brain, and behavior relationships in fragile X syndrome: Evidence from neuroimaging studies. Dev. Disabil. Res. Rev. 15, 343–352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel A. B., Loerwald K. W., Huber K. M., Gibson J. R., Postsynaptic FMRP promotes the pruning of cell-to-cell connections among pyramidal neurons in the L5A neocortical network. J. Neurosci. 34, 3413–3418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.La Fata G., Gärtner A., Domínguez-Iturza N., Dresselaers T., Dawitz J., Poorthuis R. B., Averna M., Himmelreich U., Meredith R. M., Achsel T., Dotti C. G., Bagni C., FMRP regulates multipolar to bipolar transition affecting neuronal migration and cortical circuitry. Nat. Neurosci. 17, 1693–1700 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Meredith R. M., Sensitive and critical periods during neurotypical and aberrant neurodevelopment: A framework for neurodevelopmental disorders. Neurosci. Biobehav. Rev. 50, 180–188 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Krueger D. D., Bear M. F., Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu. Rev. Med. 62, 411–429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hall S. S., Jiang H., Reiss A. L., Greicius M. D., Identifying large-scale brain networks in fragile X syndrome. JAMA Psychiatry 70, 1215–1223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van der Molen M. J. W., Stam C. J., van der Molen M. W., Resting-state EEG oscillatory dynamics in fragile X syndrome: Abnormal functional connectivity and brain network organization. PLOS One 9, e88451 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barnea-Goraly N., Eliez S., Hedeus M., Menon V., White C. D., Moseley M., Reiss A. L., White matter tract alterations in fragile X syndrome: Preliminary evidence from diffusion tensor imaging. Am. J. Med. Genet. B Neuropsychiatr. Genet. 118B, 81–88 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Villalon-Reina J., Jahanshad N., Beaton E., Toga A. W., Thompson P. M., Simon T. J., White matter microstructural abnormalities in girls with chromosome 22q11.2 deletion syndrome, fragile X or Turner syndrome as evidenced by diffusion tensor imaging. Neuroimage 81, 441–454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oberlaender M., Ramirez A., Bruno R. M., Sensory experience restructures thalamocortical axons during adulthood. Neuron 74, 648–655 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baudouin S. J., Gaudias J., Gerharz S., Hatstatt L., Zhou K., Punnakkal P., Tanaka K. F., Spooren W., Hen R., De Zeeuw C. I., Vogt K., Scheiffele P., Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science 338, 128–132 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Wang X.-S., Peng C.-Z., Cai W.-J., Xia J., Jin D., Dai Y., Luo X.-G., Klyachko V. A., Deng P.-Y., Activity-dependent regulation of release probability at excitatory hippocampal synapses: A crucial role of fragile X mental retardation protein in neurotransmission. Eur. J. Neurosci. 39, 1602–1612 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoeft F., Walter E., Lightbody A. A., Hazlett H. C., Chang C., Piven J., Reiss A. L., Neuroanatomical differences in toddler boys with fragile X syndrome and idiopathic autism. Arch. Gen. Psychiatry 68, 295–305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen R., Jiao Y., Herskovits E. H., Structural MRI in autism spectrum disorder. Pediatr. Res. 69, 63R–68R (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ellegood J., Anagnostou E., Babineau B. A., Crawley J. N., Lin L., Genestine M., DiCicco-Bloom E., Lai J. K. Y., Foster J. A., Peñagarikano O., Geschwind D. H., Pacey L. K., Hampson D. R., Laliberté C. L., Mills A. A., Tam E., Osborne L. R., Kouser M., Espinosa-Becerra F., Xuan Z., Powell C. M., Raznahan A., Robins D. M., Nakai N., Nakatani J., Takumi T., van Eede M. C., Kerr T. M., Muller C., Blakely R. D., Veenstra-VanderWeele J., Henkelman R. M., Lerch J. P., Clustering autism: Using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol. Psychiatry 20, 118–125 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haberl M. G., Ginger M., Frick A., Dual anterograde and retrograde viral tracing of reciprocal connectivity. Methods Mol. Biol., in press. [DOI] [PubMed] [Google Scholar]
- 69.Peng H., Bria A., Zhou Z., Iannello G., Long F., Extensible visualization and analysis for multidimensional images using Vaa3D. Nat. Protoc. 9, 193–208 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Zerbi V., Wiesmann M., Emmerzaal T. L., Jansen D., Van Beek M., Mutsaers M. P. C., Beckmann C. F., Heerschap A., Kiliaan A. J., Resting-state functional connectivity changes in aging apoE4 and apoE-KO mice. J. Neurosci. 34, 13963–13975 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harsan L.-A., Paul D., Schnell S., Kreher B. W., Hennig J., Staiger J. F., von Elverfeldt D., In vivo diffusion tensor magnetic resonance imaging and fiber tracking of the mouse brain. NMR Biomed. 23, 884–896 (2010). [DOI] [PubMed] [Google Scholar]
- 72.Sawiak S. J., Wood N. I., Williams G. B., Morton A. J., Carpenter T. A., Voxel-based morphometry with templates and validation in a mouse model of Huntington’s disease. Magn. Reson. Imaging 31, 1522–1531 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jenkinson M., Beckmann C. F., Behrens T. E. J., Woolrich M. W., Smith S. M., FSL. Neuroimage 62, 782–790 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Zwiers M. P., Patching cardiac and head motion artefacts in diffusion-weighted images. Neuroimage 53, 565–575 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/10/e1500775/DC1
Fig. S1. High-field 11.7-T DT-MRI measurements of the white and gray matter of adult Fmr1−/y and wild-type littermate mice.
Fig. S2. Tracing of the input to V1.
Fig. S3. Schematic representation of the experimental strategy for the 3D anatomical registration of projection neurons into V1.
Fig. S4. Functional connectivity matrix of wild-type and Fmr1−/y mice.
Movie S1. Slice view and 3D view, illustrating the 3D mouse brain model with the combined positions of all retrogradely (input; green) and anterogradely (local; red) labeled neurons in wild-type mice.
Movie S2. Slice view and 3D view, illustrating the 3D mouse brain model with the combined positions of all retrogradely (input; green) and anterogradely (local; red) labeled neurons in Fmr1−/y mice.