

Abstract
Medulloblastoma accounts for 20 % of all primary pediatric intracranial tumors. Current treatment cures 50–80 % of patients but is associated with significant long-term morbidity and thus new therapeutic targets are needed. One such target is cyclin-dependent kinase 6 (CDK6), a serine/threonine kinase that plays a vital role in cell cycle progression and differentiation. CDK6 is overexpressed in medulloblastoma patients and is associated with an adverse prognosis. To investigate the role of CDK6 in medulloblastoma, we assayed the effect of CDK6 inhibition on proliferation by depleting expression with RNA interference (RNAi) or by inhibiting kinase function with a small molecule inhibitor, PD0332991. Cell proliferation was assessed by colony focus assay or by the xCELLigence system. We then investigated the impact of CDK6 inhibition on differentiation of murine neural stem cells by immunofluorescence of relevant markers. Finally we evaluated the effects of PD0332991 treatment on medulloblastoma cell cycle and radiosensitivity using colony focus assays. Gene expression analysis revealed that CDK6 mRNA expression is higher than normal cerebellum in fifteen out of sixteen medulloblastoma patient samples. Inhibition of CDK6 by RNAi significantly decreased medulloblastoma cell proliferation and colony forming potential. Interestingly, CDK6 inhibition by RNAi increased differentiation in murine neural stem cells. PD0332991 treatment significantly decreased medulloblastoma cell proliferation and led to a G0/G1 cell cycle arrest. Furthermore, PD0332991 pretreatment sensitized medulloblastoma cells to ionizing radiation. Our findings suggest that targeting CDK6 with small molecule inhibitors may prove beneficial in the treatment of medulloblastoma, especially when combined with radiation.
Keywords: CDK6, PD0332991, Medulloblastoma
Introduction
Medulloblastoma is the most common primary pediatric brain tumor, accounting for 20 % of all primary pediatric intracranial tumors [1]. Current treatment provides a cure in 50–80 % of patients but is associated with significant long-term morbidity [2, 3]. Currently, patients with medulloblastoma are stratified into “standard” and “high” risk categories for treatment purposes based on features such as age at diagnosis, presence of metastasis, degree of surgical resection, and histology [4]. However, recent genome wide analyses have allowed for subdivision of medulloblastoma into those with Wnt pathway mutations, Sonic Hedgehog pathway mutations, and those termed Group 3 and Group 4, which include intermediate to high risk tumors but whose biology is less well understood [5]. Research within these subgroups has resulted in the development of novel therapeutic strategies [6, 7].
Of the subgroups, Group 4 tumors account for 35 % of childhood medulloblastoma diagnoses and carry an intermediate prognosis [8]. One feature of Group 4 tumors is amplification of cyclin-dependent kinase 6 (CDK6) [5]. CDK6 is a serine/threonine kinase that plays a vital role in cell cycle progression. CDK6 partners with cyclin D to phosphorylate the retinoblastoma (Rb) protein during the G1/S cell cycle transition [9]. CDK6 also plays a role in differentiation. Forced CDK6 expression in mouse astrocytes will alter morphology and expression markers to those of glial progenitor cells and will block differentiation of a mouse erythroid leukemia line [10, 11].
Deregulation of CDK6 is seen in a wide set of tumors, including sarcomas and glioblastoma multiforme (GBM) [12]. CDK6 amplification has been previously described in medulloblastoma patients and is associated with an adverse prognosis [13]. CDK6 is also a target of multiple microRNAs including miR-129 and miR-124 and re-expression of these miRNAs results in a decrease in CDK6 expression and subsequent decrease in cell proliferation [14, 15].
Given the functional importance of CDK6 and known over-expression in medulloblastoma, we hypothesized that direct inhibition of CDK6 would suppress medulloblastoma cell proliferation. In this study we evaluated the effect of CDK6 inhibition by RNA interference (RNAi) and by inhibiting kinase function with a small molecule inhibitor, PD0332991. PD0332991 is an orally available small molecule inhibitor that targets the kinase activity of CDK4 and CDK6 [9]. PD0332291 has shown promising results in Phase I/II studies in adult malignancies [[16, 17], NCT01227434].
We show here that CDK6 is significantly overexpressed in medulloblastoma and inhibition by RNAi and PD0332991 suppresses cell proliferation. In addition, we found treatment with PD0332991 significantly enhances medulloblastoma radiation sensitivity.
Methods
Cell lines and primary patient samples
The Daoy medulloblastoma cell line was purchased from American Type Cell Culture (Rockville, MD). The ONS-76 cells was provided by Dr. James T. Rutka (University of Toronto, Canada) and D425 and D458 by Dr. Darell D. Bigner (Duke University Medical Center, NC). Cell lines were cultured in DMEM (Gibco, Carlsbad, CA) supplemented with 10 % fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA). Daoy and ONS-76 cells are well characterized and thus were selected for use in experiments [18].
Primary patient samples were obtained from Children’s Hospital Colorado in accordance with local and federal human research protection guidelines and Institutional Review Board (IRB) regulations. Informed consent was obtained for collected specimens. Normal brain tissue was collected from autopsy and purchased from Ambion (Austin, TX) and Clontech Laboratories, Inc (Mountain View, CA). Normal cerebellar samples in Fig. 1 were obtained from nonmalignant brain biopsies at the Children’s Hospital Colorado under IRB guidelines. Normal cerebellar samples UPN 514 and UPN 605 are from 4- and 5-year old patients, respectively.
Fig. 1.
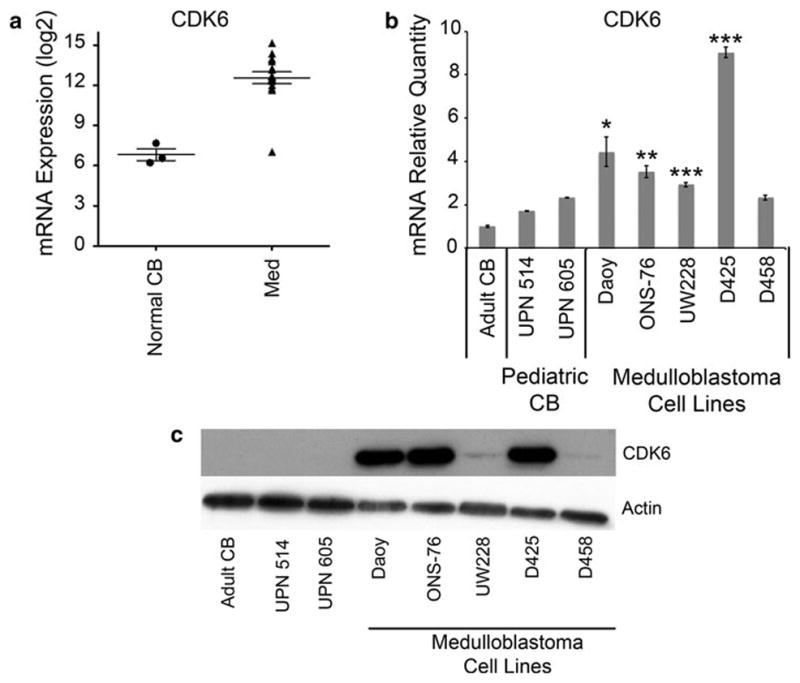
a CDK6 mRNA levels by microarray show increased expression in 15/16 primary medulloblastoma (Med) patient samples as compared to three normal cerebellum samples (Normal CB). Error bars represent standard error of mean (SEM). b Using qRT-PCR, CDK6 mRNA expression in medulloblastoma cell lines is significantly higher than normal adult cerebellum (Clontech) and pediatric cerebellum (UPN 514 and UPN 605). Error bars represent (SEM). (*P <0.05, **P <0.01,***P <0.001). c Western blot of CDK6 protein expression in normal cerebellum and medulloblastoma cell lines
Gene expression microarray analysis
Sixteen patient tumor samples were evaluated for gene expression using Affymetrix U133 Plus 2.0 GeneChip microarrays as previously described [19]. Briefly, samples were collected at the time of surgery and snap-frozen in liquid nitrogen. Ribonucleic acid was extracted from each sample using an RNeasy kit (Qiagen, Valencia, CA) and hybridized to HG-U133 Plus 2.0 GeneChips (Affymetrix, Santa Clara, CA). Microarray data from the samples was background corrected using the gcRMA algorithm. Differential expression of genes was determined using Student’s t test.
Transfection of RNAi vectors
A CDK6 shRNA (shCDK6) and a non-targeting shRNA (shNTC) were purchased from Functional Genomics Facility at University of Colorado, Boulder and transfected into medulloblastoma cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The shCDK6 was used in experiments given the significant decrease in CDK6 mRNA (Supplementary Fig. 1). The ratio used for the forward transfection in a 6-well plate was 1 μg of shRNA DNA:2 μL of Lipofectamine 2000 Transfection Reagent.
Quantitative real-time polymerase chain reaction
Ribonucleic acid was isolated 48 h after transfection using a Qiagen RNeasy kit (Valencia, CA). TaqMan gene expression primers and probes for CDK6 (Hs01026371_m1) and GAPDH (Hs99999905_m1) were purchased from Applied Biosystems (Carlsbad, CA). Assays were performed in triplicate. GAPDH served as endogenous control and gene expression relative quantity was calculated using the delta Ct method. Gene expression assays were performed on ABI StepOnePlus Real-Time PCR system.
Small molecule inhibitor PD0332991
The small molecule inhibitor PD0332991 was purchased from Axon MedChem (Gronigen, The Netherlands), reconstituted in water and aliquots stored at –20 °C.
Cell proliferation assay
Cell proliferation was measured using the xCELLigence system and E-Plate 96 well gold-coated plates (Roche). This system gives real time measurement of cell proliferation [20]. Cell growth is measured by evaluating electrical impedance and converted to Cell Index which is a measure of the number of cells in a given well. Cells were initially transfected with shNTC or shCDK6. After 48 h, cells were then trypsinized and plated 2,000 cells/well on to an E-plate for use in the system.
Colony focus assay
Cells were initially transfected with shCDK6 or shNTC. After 48 h, cells were trypsinized and replated in triplicates of 500 cells/well in a 6-well plate. For drug treatment, 500 cells were plated in triplicate 24 h before addition of PD0332991. Wells were treated for 48 h and then allowed to grow in normal culture medium. After seven days of growth, medium was aspirated and colonies were stained with 0.5 % crystal violet, 25 % methanol solution. Colonies per well were counted using a dissecting microscope with a threshold of 50 cells necessary to constitute a colony.
Differentiation assay
C17.2 murine neural stem cells were kindly provided by Dr. E.Y. Snyder (Burnham Institute, La Jolla, CA) and cells were established in DMEM medium supplemented with 10 % FBS, 5 % Equine horse serum, L-Glutamine and Pen/Strep. C17.2 cells were transfected with either shNTC or shCDK6. After 48 h of transfection, cells were trypsinized and seeded (3000 cells/well) on poly-D Lysine coated chamber slides. To evaluate the effect of CDK6 inhibition on differentiation, C17.2 cells were grown in differentiation medium (DM) [21]. After 5 days in DM, cells were processed for immunofluorescence. Briefly, cells were fixed with 4 % paraformaldehyde and permeabilized with 0.05 % Triton X100. Cells were then blocked at room temperature with blocking buffer (0.05 % Triton X100 containing 5 % Milk) for 30 min. Slides were incubated with anti-IgG as control and anti-Tuj1 (1:200 to label immature neurons), (Cell Signaling Technologies) overnight at 4 °C. Antibodies were diluted with 0.05 % Triton X. Slides were washed with PBS and incubated with Alexafluor 488 conjugated goat anti-mouse (1:500) (Molecular Probes) antibody for 1 h at RT. Slides were washed thrice with PBS and coated with anti-fading reagent containing DAPI. Coverslips were mounted and the cells were visualized using confocal microscopy with 40× oil objective. Confocal images were taken using 3I Marianas inverted spinning disk imaging system built on a Zeiss Axio Observer Z1 microscope equipped with Yokogawa CSU-X1 and Photometrics Evolve 16-bit EM-CCD camera. The images were viewed and quantified using 3I Slidebook 5.0 software.
Cell cycle assay
Cells were seeded in 6 well plates (1×105 cells/well) and 24 h later were treated with PD0332991. After 48 h of drug treatment, cells were harvested and fixed with 70 % ethanol overnight. Collected cells were treated with 250 μl cell cycle reagent (Millipore) and evaluated by flow cytometric analysis per the manufacturer’s recommendations.
Western blotting
Protein lysates were obtained from samples using RIPA buffer (Thermo Scientific, Rockford, IL) with protease inhibitors added. Western blotting was performed per standard methods. Antibodies for CDK6 (#3136) and Actin (MAB1501) were purchased from Cell Signaling Technology (Danvers, MA) and Millipore, respectively. Antibodies for RB and pRB were purchased from Cell Signaling Technology (Danvers, MA). Secondary antibodies conjugated to horseradish-peroxidase were used in conjunction with a chemiluminescent reagent to visualize protein bands.
Combination of PD0332991 and ionizing radiation
Six well plates were set up as previously described. After 48 h of drug treatment, media was replaced and the cells were immediately irradiated. After seven days of additional growth, wells were stained with crystal violet solution and colonies were counted. Survival curves were generated after normalizing for the amount of PD0332991 induced death. Non-linear regressions were calculated for each line. The radiation dose intersecting the non-linear regression for a 10 % (SF0.1) and 50 % (SF0.5) surviving fraction was calculated for each drug dose. The sensitizer enhancement ratio (SER) was then calculated as follows:
Calculation of SER allows for direct comparison of the effect of the potential sensitizing agent relative to control. SER ratio >1 indicates sensitization.
Statistical analysis
Statistical significance was determined using a Student’s t test. Error bars represent the standard error of the mean (n ≥ 3). GraphPad Prism 5 was used to calculate IC50 values and to compute the non-linear regression equations.
Results
CDK6 is overexpressed in medulloblastoma
To verify previously published data, we first examined the expression of CDK6 in a cohort of sixteen medulloblastoma patients. We found that CDK6 mRNA expression was higher than normal cerebellum in fifteen of sixteen samples (Fig. 1a). We then examined whether CDK6 mRNA expression was sub-group specific. We analyzed publically available data as published by Northcott et al. [22]. We found CDK6 mRNA expression is highest in the Wnt subgroup (Supplementary Fig. 2). We next evaluated CDK6 mRNA expression in a panel medulloblastoma cell lines. Consistent with our patient tissue data, all medulloblastoma cell lines, except for D458, were significantly higher than pediatric normal cerebellum (UPN 514 and UPN 605) (Fig. 1b). CDK6 protein expression is also increased in the medulloblastoma lines as compared to normal pediatric and adult cerebellum (Fig. 1c).
Inhibition of CDK6 suppresses medulloblastoma cell proliferation and colony forming ability
Next we examined the functional significance of CDK6 expression in medulloblastoma cell lines. We decreased expression of CDK6 mRNA by shRNA in Daoy and ONS-76 cells to evaluate the impact on cell proliferation. Using the xCELLigence system, we found a significant decrease in cell proliferation over time upon RNAi mediated inhibition of CDK6 as compared to control vector transfected cells (Fig. 2a). To evaluate the long-term effect of CDK6 inhibition, we then performed colony focus assays on transfected cells. Again, inhibition of CDK6 significantly decreased cell proliferation as measured by their ability to form colonies (Fig. 2b, c). CDK6 knockdown was verified by qRT-PCR (Supplementary Fig. 1) and by western blot analysis (Fig. 2d).
Fig. 2.
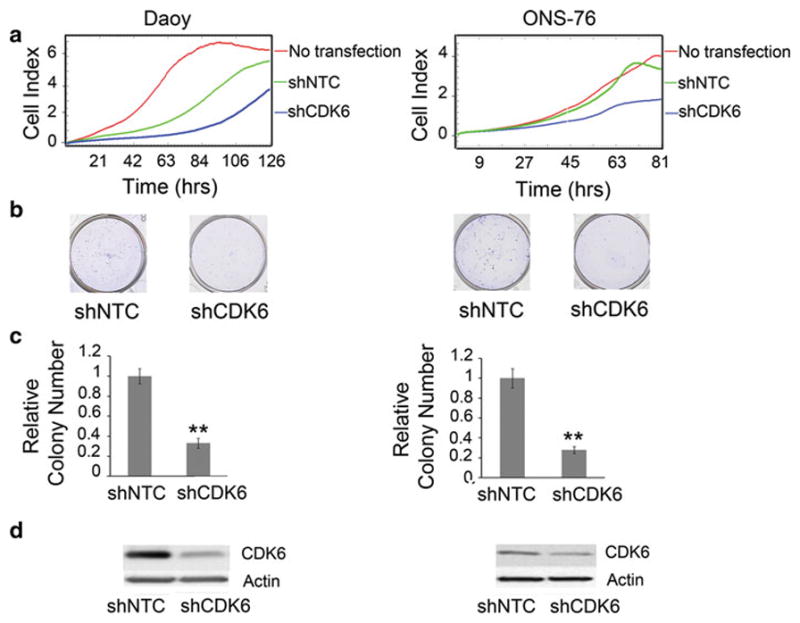
a Using the xCELLigence system for assessing cell proliferation, inhibition of CDK6 by shRNA decreases cell proliferation in Daoy and ONS-76 medulloblastoma cell lines. b Representative pictures of wells for each cell line for colony focus assay. c CDK6 inhibition by shRNA decreases long-term growth as seen with colony focus assay. Error bars represent SEM. (**P <0.01) d Western blot showing a decrease in CDK6 protein after transfection with shRNA
CDK6 inhibition induces differentiation in C17.2 murine neural stem cells
In searching for the etiology of decreased medulloblastoma cell proliferation, we found that Daoy and ONS-76 cells do not undergo increased apoptosis or cell cycle arrest with CDK6 inhibition (data not shown). We then examined the role of CDK6 in differentiation as a possible mechanism. We chose to use C17.2 murine neural stem cells because they are well characterized and can be induced to differentiate in vitro [21]. C17.2 cells were transfected with either shNTC or shCDK6. After 5 days of growth in differentiation media, cells were stained using antibodies to IgG as control and Tuj1 the marker of immaturity in neurons. Cells transfected with shCDK6 had expressed Tuj1 in 62 % of cells (DAPI positive) compared to 27 % of cells transfected with shNTC, illustrating that with CDK6 inhibition, differentiation was increased (Fig. 3a, b).
Fig. 3.
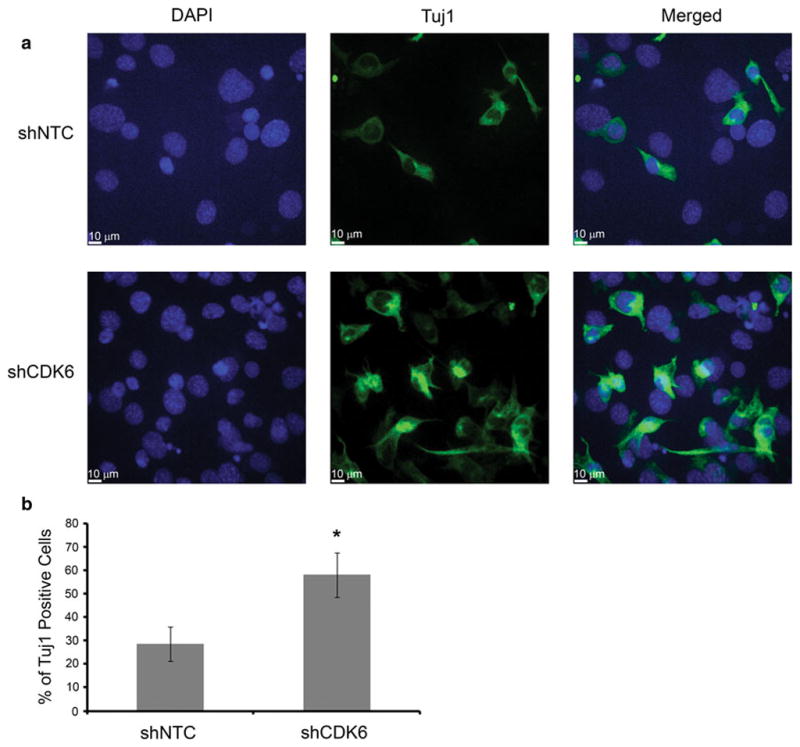
a Inhibition of CDK6 by shRNA leads to increased differentiation of murine neural stem cells (C17.2) as seen by increased expression of Tuj1, a marker of immature neurons. Images captured at 40× magnification. b Quantitation of immunofluorescent images for the percentage of DAPI positive cells that were also Tuj1 positive. Error bars represent SEM. (*P <0.05)
Small molecule inhibitor PD0332991 decreases medulloblastoma cell proliferation by inducing cell cycle arrest with time dependent decrease in phosphorylated Rb
Early evidence of aberrant increased expression of CDKs in cancers led to the development small molecule CDK inhibitors as therapeutics [23]. One such inhibitor, PD0332991, is of the pyridopyrimidine class that potently inhibits the kinase activity of CDK4 and CDK6 [24]. Daoy and ONS-76 cells were treated with varying concentrations of PD0332991 for 48 h, after which the media was replaced with normal growth media. After seven days, effect on long-term growth was evaluated by colony focus assay. Treatment with PD0332991 inhibited the ability of medulloblastoma cells to form colonies with an IC50 of 1.5 μM for Daoy and 790 nM for ONS-76 cells (Fig. 4a, b).
Fig. 4.
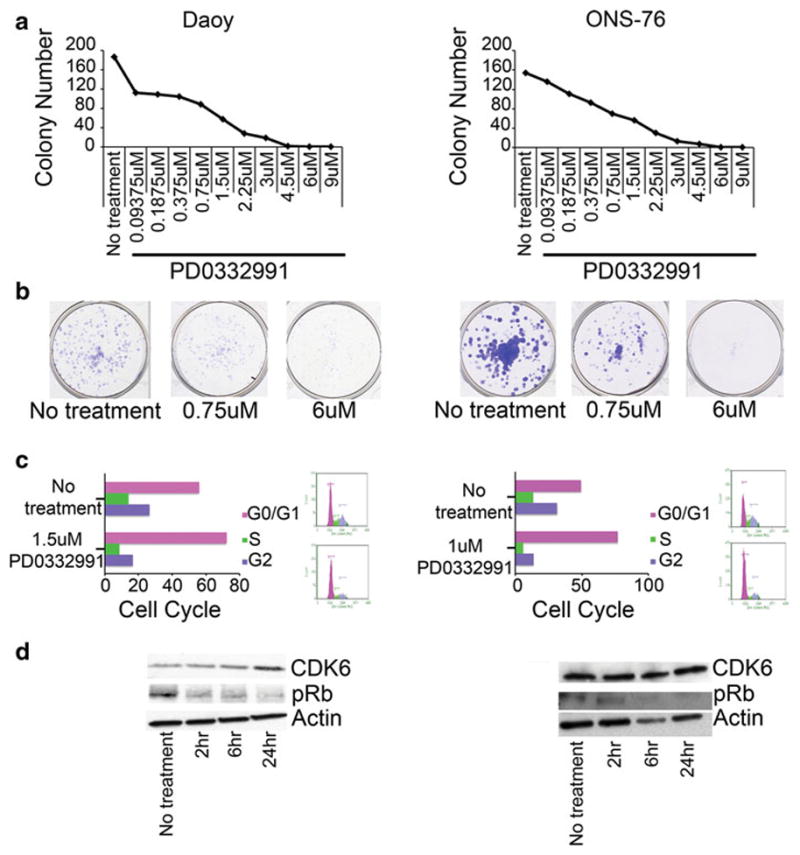
a Decrease in long-term growth in Daoy and ONS-76 cells as measured by colony focus assay after 48 h of treatment with varying concentrations of PD0332991. b Representative pictures of no treatment, 0.75 μM PD0332991 and 6 μM PD0332991 wells for each cell line. c Treatment with PD0332991 results in G0/G1 cell cycle arrest. d Western blot showing a time dependent decrease in the amount of phosphorylated Rb protein
Treatment with PD0332991 resulted in G0/G1 arrest as compared to untreated cells (Fig. 4c). It is important to note that PD0332991 inhibits both CDK6 and CDK4 and likely inhibition of both is required to inhibit medulloblastoma cell growth and induce the G1 arrest. Western blot analysis shows a corresponding time dependent decrease in the amount of phosphorylated Rb, indicating an inhibition of the Cyclin D-CDK6-Rb pathway (Fig. 4d). Interestingly while phosphorylated Rb decreased in response to PD0332991, CDK6 protein increased, perhaps as a feedback mechanism in response to decreased activity.
PD0332991 enhances radiation sensitivity of medulloblastoma cells
To evaluate for potential radiosensitivity, Daoy and ONS-76 were pretreated with PD0332991. After 48 h, they were exposed to different doses of radiation and effects measured using the colony focus assay. The results show that the surviving fractions (SF) were reduced in Daoy and ONS-76 cells treated with PD0332991 (Fig. 5a). Surviving fractions of PD0332991-pretreated cells after 2 Gray (Gy) irradiation were significantly lower than those of untreated cells (Fig. 5b). Non-linear regressions were fit to the curves to calculate the sensitizer enhancement ratio (SER) (Supplementary Fig. 3). The SERs were 1.6 for Daoy at 10 % cell survival (SF0.1) and 1.5 at 50 % cell survival (SF0.5) with 750 nM PD0332991 pretreatment. For ONS-76 cells pretreated with 750 nM PD0332991, the SERs were 2.3 for both SF0.1 and SF0.5. Thus, with SER ratios greater than 1, the radiation survival curves obtained by the colony focus assay show that treatment with PD0332991 sensitized human medulloblastoma cells to ionizing radiation.
Fig. 5.

a Line graph plotting the surviving fraction of Daoy and ONS-76 cells given different doses of radiation following 48 h of treatment with PD0332991. b Bar graph showing the surviving fraction following irradiation with a dose of 2 Gray (Gy). Error bars represent SEM. (**P <0.01, ***P <0.001)
Discussion
Current medulloblastoma therapy using a combination of surgery, chemotherapy, and radiation has improved overall survival; however this comes at a cost with the potential for significant long-term cognitive and endocrine dysfunction [4]. As with other tumors, this treatment strategy likely results in the overtreatment of some low risk patients and the need for intensification and novel therapeutics in high-risk patients. Recent genomic studies are beginning to illustrate a more subdivided molecular makeup of medulloblastoma and this information will be important to identify novel therapeutic strategies [8].
Alterations of the cell cycle kinases are associated with aberrant division and uncontrolled proliferation of cancer cells [23]. The expression of one such kinase, CDK6, is increased in many malignancies. CDK6 partners with cyclin D to play an important role in cell cycle progression at the G1-S-phase boundary [9]. The G1-S checkpoint is a critical control mechanism in medulloblastoma. Additionally CDK6 plays an important role in differentiation and regulation of neurogenesis [25].
In this study we demonstrated that CDK6 mRNA expression is higher in our patient cohort than normal cerebellum controls. Our cohort of patients does not have enough numbers to power comparisons within molecular subgroup. However, our findings are in line with other published data showing that CDK6 is amplified in medulloblastoma [13]. Decreasing the expression of CDK6 mRNA by RNAi resulted in significant long-term growth suppression and a reduction in the ability to form colonies by inducing differentiation thereby establishing CDK6 as a potential therapeutic target. Inhibition of CDK6 by PD0332991 significantly reduced proliferation over time with IC50 values in therapeutically achievable doses. Treatment with PD0332991 causes G0/G1 cell cycle arrest secondary to decreased phosphorylated Rb protein.
PD0332991 competitively targets both CDK4-Cyclin D1 as well as CDK6-cyclin D3 in the nanomolar range [23]. Several adult Phase I and Phase II trials show that PD0332991 is well tolerated with dose limiting toxicity being myelosuppression [17]. PD0332991 also has been shown to effectively cross the blood–brain barrier [26]. More recent studies suggest PD0332991 can act in concert with other therapeutics. For example, PD0332991 can enhance the anti-tumor effects of bortezomib [27]. However there are no current clinical trials with PD0332991 in pediatric patients. Our data show that PD0332991 can inhibit medulloblastoma cell growth, likely by inhibiting both CDK4 and CDK6.
Here we show that pretreatment with biologically achievable doses of PD0332991 significantly decreased the surviving fraction of tumors cells in response to radiation and increased the sensitizer enhancement ratio. These results indicate that PD0332991 can effectively enhance medulloblastoma cell radiosensitivity in vitro, which is in accordance with previous studies showing radiosensitivity in GBM cells treated with PD0332991 [26]. These data are important because radiation is an integral part of medulloblastoma therapy even though the side effects can be deleterious to long term quality of life. Thus any agents that can decrease the dose of radiation required would be of significant benefit.
In total our data strongly suggest that CDK6 is a viable molecular target in medulloblastoma therapy and the use of CDK6 inhibitors in combination with radiation therapy represents a novel therapeutic strategy for patients with medulloblastoma. These data provide the rationale for examining the efficacy of PD0332991 in patients with medulloblastoma and in particular evaluating its combination with radiation and other targeted therapeutics.
Supplementary Material
Acknowledgments
This work was supported by NINDS K08 NS059790 (RV) and the Morgan Adams Foundation (RV, NKF). Imaging experiments were performed in the University of Colorado Anschutz Medical Campus Advance Light Microscopy Core supported in part by NIH/NCRR Colorado CTSI Grant Number UL1 RR025780.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (doi:10.1007/s11060-012-1000-7) contains supplementary material, which is available to authorized users.
Contributor Information
Susan L. Whiteway, Email: susan.whiteway@us.af.mil, Department of Pediatrics, Children’s Hospital Colorado and University of Colorado Denver, Anschutz Medical Campus, Aurora, CO, USA
Peter S. Harris, Email: peter.harris@ucdenver.edu, Department of Pediatrics, Children’s Hospital Colorado and University of Colorado Denver, Anschutz Medical Campus, Aurora, CO, USA
Sujatha Venkataraman, Email: sujatha.venkataraman@ucdenver.edu, Department of Pediatrics, Children’s Hospital Colorado and University of Colorado Denver, Anschutz Medical Campus, Aurora, CO, USA.
Irina Alimova, Email: irina.alimova@ucdenver.edu, Department of Pediatrics, Children’s Hospital Colorado and University of Colorado Denver, Anschutz Medical Campus, Aurora, CO, USA.
Diane K. Birks, Email: diane.birks@ucdenver.edu, Department of Pediatric Neurosurgery, University of Colorado Denver, Anschutz Medical Campus, Aurora, CO, USA
Andrew M. Donson, Email: andrew.donson@ucdenver.edu, Department of Pediatrics, Children’s Hospital Colorado and University of Colorado Denver, Anschutz Medical Campus, Aurora, CO, USA
Nicholas K. Foreman, Email: nicholas.foreman@childrenscolorado.org, Department of Pediatrics, Children’s Hospital Colorado and University of Colorado Denver, Anschutz Medical Campus, Aurora, CO, USA
Rajeev Vibhakar, Email: rajeev.vibhakar@ucdenver.edu, Department of Pediatrics, Children’s Hospital Colorado and University of Colorado Denver, Anschutz Medical Campus, Aurora, CO, USA.
References
- 1.Pizer BL, Clifford SC. The potential impact of tumour biology on improved clinical practice for medulloblastoma: progress towards biologically driven clinical trials. Br J Neurosurg. 2009;23(4):364–375. doi: 10.1080/02688690903121807. [DOI] [PubMed] [Google Scholar]
- 2.Packer RJ, Cogen P, Vezina G, et al. Medulloblastoma: clinical and biologic aspects. Neuro Oncol. 1999;1:232–250. doi: 10.1215/15228517-1-3-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Packer RJ. Progress and challenges in childhood brain tumors. J Neurooncol. 2005;75:239–242. doi: 10.1007/s11060-005-6745-9. [DOI] [PubMed] [Google Scholar]
- 4.Dhall G. Medulloblastoma. J Child Neurol. 2009;24:1418–1430. doi: 10.1177/0883073809341668. [DOI] [PubMed] [Google Scholar]
- 5.Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyers-Needham M, Lewis JA, Gencer S, et al. Off-target function of the sonic hedgehog inhibitor cyclopamine in mediating apoptosis via nitric oxide-dependent neutral sphingomye-linase2/ceramide induction. Mol Cancer Ther. 2012;11(5):1092–1102. doi: 10.1158/1535-7163.MCT-11-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee MJ, Hatton BA, Villavicencio EH, et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in mouse medulloblastoma model. Proc Natl Acad Sci USA. 2012;109(20):7859–7864. doi: 10.1073/pnas.1114718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Northcott PA, Korshunov A, Pfister S, et al. The clinical implications of medulloblastoma subgroups. Nat Rev Neurol. 2012;8(6):340–351. doi: 10.1038/nrneurol.2012.78. [DOI] [PubMed] [Google Scholar]
- 9.Musgrove EA, Caldon CE, Barraclough J, et al. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11(8):558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 10.Ericson KK, Krull D, Slomiany P, et al. Expression of cyclin-dependent kinase 6, but not cyclin-dependent kinase 4, alters morphology of cultured astrocytes. Mol Cancer Res. 2003;1(9):654–664. [PubMed] [Google Scholar]
- 11.Grossel MJ, Hinds PW. Beyond the cell cycle: a new role for CDK6 in differentiation. J Cell Biochem. 2006;97(3):485–493. doi: 10.1002/jcb.20712. [DOI] [PubMed] [Google Scholar]
- 12.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 13.Mendryk F, Radlwimmer B, Joos S, et al. Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J Clin Oncol. 2005;23(34):8853–8862. doi: 10.1200/JCO.2005.02.8589. [DOI] [PubMed] [Google Scholar]
- 14.Wu J, Qian J, Li C, et al. miR-129 regulates cell proliferation by downregulating CDK6 expression. Cell Cycle. 2010;9(9):1809–1818. doi: 10.4161/cc.9.9.11535. [DOI] [PubMed] [Google Scholar]
- 15.Pierson J, Hostager B, Fan R, et al. Regulation of cyclin dependent kinase 6 by microRNA 124 in medulloblastoma. J Neurooncol. 2008;90:1–7. doi: 10.1007/s11060-008-9624-3. [DOI] [PubMed] [Google Scholar]
- 16.Leonard JP, LaCasce AS, Smith MR, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012;119(20):4597–4607. doi: 10.1182/blood-2011-10-388298. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz GK, LoRusso PM, Dickson MA, et al. Phase I study of PD0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1) Br J Cancer. 2011;104:1862–1868. doi: 10.1038/bjc.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venkataraman S, Alimova I, Fan R, et al. MicroRNA 128a increases intracellular ROS level by targeting Bmi-1 and inhibits medulloblastoma cancer cell growth by promoting senescence. PLoS One. 2010;5(6):e10748. doi: 10.1371/journal.pone.0010748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barton VN, Donson AM, Kleinschmidt-DeMasters BK, et al. Unique molecular characteristics of pediatric myxopapillary ependymoma. Brain Pathol. 2010;20(3):560–570. doi: 10.1111/j.1750-3639.2009.00333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xing JZ, Zhu L, Jackson JA, et al. Dynamic monitoring of cytotoxicity on microelectronic sensors. Chem Res Toxicol. 2005;18:154–161. doi: 10.1021/tx049721s. [DOI] [PubMed] [Google Scholar]
- 21.Su X, Gopalakrishnan V, Stearns D, et al. Abnormal expression of REST/NRSF and Myc in neural stem/progenitor cells causes cerebellar tumors by blocking neuronal differentiation. Mol Cell Biol. 2006;26(5):1666–1678. doi: 10.1128/MCB.26.5.1666-1678.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8(7):547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- 24.Toogood P, Harvey PJ, Repine JT, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48(7):2388–2406. doi: 10.1021/jm049354h. [DOI] [PubMed] [Google Scholar]
- 25.Beukelaers P, Vandenbosch R, Caron N, et al. CDK6-dependent regulation of G1 length controls adult neurogenesis. Stem Cells. 2011;29:713–724. doi: 10.1002/stem.616. [DOI] [PubMed] [Google Scholar]
- 26.Michaud K, Solomon DA, Oermann E, et al. Pharmacologic inhibition of CDK4/6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70(8):3228–3238. doi: 10.1158/0008-5472.CAN-09-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menu E, Garcia J, Huang X, et al. A novel therapeutic combination using PD 0332991 and Bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008;68(14):5519–5523. doi: 10.1158/0008-5472.CAN-07-6404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.