

Abstract
The transporter associated with antigen processing (TAP) translocates peptides from the cytosol to the endoplasmic reticulum (ER) lumen to enable immune surveillance by CD8+ T cells. Peptide transport is preceded by peptide binding to a cytosol-accessible surface of TAP1/TAP2 complexes, but the location of the TAP peptide-binding pocket remains unknown. Guided by the known contributions of polymorphic TAP variants to peptide selection, we combined homology modeling of TAP with experimental measurements to identify several TAP residues that interact with peptides. Models for peptide-TAP complexes were generated, which indicate bent conformation for peptides. The peptide-binding site of TAP is located at the hydrophobic boundary of the cytosolic membrane leaflet, with striking parallels to the glutathione binding site of NaAtm1, a transporter that functions in bacterial heavy metal detoxification. These studies illustrate the conservation of the ligand recognition modes of bacterial and mammalians transporters involved in peptide-guided cellular surveillance.
Introduction
Major histocompatibility complex (MHC) class I molecules play a central role in the adaptive immune response to viruses and cancers by presenting antigenic peptides to CD8+ CTL (1, 2). TAP is localized on the ER membrane and required for the transport of cytosolic peptides into ER lumen for assembly with newly synthesized MHC class I molecules (3). Recent studies have suggested that TAP can also be localized on phagosomal membranes under certain conditions, to transport cytosolic antigens into phagosomes (4). Due to its essential role, TAP deficiency results in human immunodeficiency (5), and inhibition of TAP expression and function are common in viral infections and cancers (6).
TAP is a member of the superfamily of ATP binding cassette (ABC) transporters that translocate a wide range of solutes across membranes using ATP hydrolysis as an energy source (7). TAP is composed of two homologous half-transporters, TAP1 (ABCB2) and TAP2 (ABCB3). TAP shares a common architecture with other ABC transporters, with two transmembrane domains (TMDs) formed by long TM α-helices that are extended into the cytosol and two cytosolic nucleotide-binding domains (NBDs) (Figure 1). The TMD of each subunit has a core domain (core TMD) and an N-terminal accessory domain (TMD0) (8, 9). Two 6-helical core TMDs contain the peptide-binding pocket and translocation channel and, together with NBDs, are essential and sufficient for peptide transport (8, 9).
Figure 1. Localization of polymorphic residues in models of rat and chicken TAP1/TAP2 complexes.
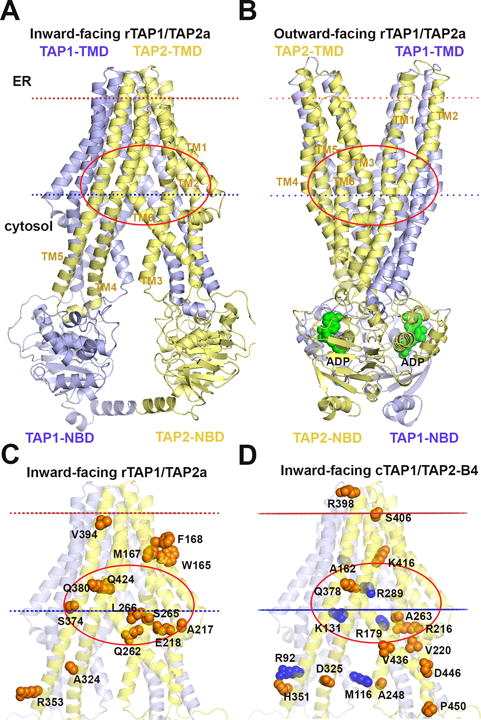
Homology models of the core transport domains of rat TAP complexes in the inward-facing (A, C, D) or the outward-facing (B) conformations were built using crystal structures of bacterial ABC transporters, NaAtm1 (PDB ID: 4mrs), and Sav1866 (PDB ID: 2hyd) respectively. Models are colored blue (for TAP1) and yellow (for TAP2). Membrane boundaries for template structures are taken from the OPM database (http://opm.phar.umich.edu) and shown by red and blue dotted lines. Cartoon representations show the architecture of TAP composed of 2 TMDs and 2 NBDs. TM helices of rTAP2a are indicated by numbers. ADP molecules bound to NBDs in the outward-facing conformation (B) are shown by spheres colored green. All polymorphic residues from TMDs of rat TAP (rTAP1/TAP2a) (C) and chicken TAP from haplotype B4 (cTAP1/TAP2-B4) (D) are shown by spheres colored blue for residues from TAP1, and orange for residues from TAP2. Red ovals show plausible peptide binding sites.
Structural requirements and binding specificities for peptide substrates have been studied for TAP proteins from a number of different species (10–14). In general, the first three N-terminal and the last C-terminal residues of translocated peptides are most critical for peptide-binding specificity (12, 13, 15). Free N- and C-termini are also required for efficient peptide binding (10, 13, 16). The residues in the center of the peptide have small or no effects on the substrate specificity and transport efficiency of TAP (12, 13).
Despite some progress in identification of possible determinants of TAP peptide binding specificity (14, 17–21), the exact location of the peptide binding pocket within the TAP transport core and the preferred configurations of TAP-bound peptides are yet to be identified. Here we present homology models of TAP translocation complexes (excluding TMD0 domains) of rat, human, and two chicken TAPs of the B4 or B15 haplotypes (rTAP1/TAP2a, hTAP1/TAP2, cTAP1/TAP2-B4, cTAP1/TAP2-B15) bound to their peptide substrates. Site-directed mutagenesis and chemical crosslinking were used to assess the proximity of peptides to TAP residues lining the internal cavity and to test the involvement of selected TAP residues upon peptide binding and transport. We used the current results together with previously published data to predict peptide docking modes within the ligand-binding site of TAP transporters.
Materials and Methods
Molecular modeling
TAP sequences
Models of rat, human, and chicken TAP1/TAP2 heterodimers were built using sequences of rTAP1 (residues 147–725) and rTAP2a (residues 135–703) (UniProtKB access codes P36370 and P36372, respectively), hTAP1 (residues 170–748) and hTAP2 (residues 135–686) (UniProtKB access codes Q03518 and Q03519, respectively), and chicken TAP (residues 4–575 for cTAP1 and 133–701 for cTAP2) from alleles B4 and B15 (GenBank protein IDs AEE25613.1 and AEE25616.1 for TAP1 and AEE25620.1 and AEE25623.1 for TAP2).
Homology models were generated for the 12-helical core domain of TAP1/TAP2 heterodimers that represents the core peptide translocation unit shared with other ABC transporters. The N-terminal accessory domain (TMD0), which is not required for peptide translocation but which participates in binding MHC class I molecules via the assembly factor tapasin (8, 9), was not modeled, as it lacks a structural template from a homologous protein domain. The sequence alignments of the core TMDs of TAPs modeled in this work are present on Figure S1. Sequence alignments of NBD domains have been previously published (22).
Structural templates
Models of rat TAP core domains were constructed in both the inward-facing and the outward-facing conformations and carefully analyzed and refined in the context of experimental data. Other models were built only in the inward-facing conformation. The structural templates used for modeling represent crystal structures of bacterial ABC exporters with relatively low homology to TAP (25–28% residue identity) that were crystallized in different conformational states: structure of putative drug exporter Sav1886 (PDB ID: 2hyd) in outward-facing conformation (23) and structure of the heavy metal exporter NaAtm1 in inward-facing conformation (PDB ID: 4mrs) (24). Among numerous available crystal structures in inward-facing conformations (24–28), we chose the structural template of NaAtm1 transporter, because it was co-crystallized with hexapeptide ligands, and the NBDs did not display wide separations. Recent electron single-particle cryomicroscopy study of a bacterial TmrAB exporter in β-DDM micelles supported the functional relevance of C-terminally connected NBDs in the nucleotide-free inward-facing state (29).
Homology modeling
Homology models of rTAP1 and rTAP2a based on Sav1886 or NaAtm1 template were generated by Phyre2-server (30) using the sequence alignments mentioned above. Backbone corrections were introduced in some loops and helices to provide consistency between models generated from different templates. The structure of each half-transporter was refined by energy minimization (100 steps) with the CHARMm force field implemented in QUANTA (Accerlys Inc.) using a dielectric constant (ε) of 10 and the adopted-basis Newton-Raphson method. Models of other half-transporters were generated from NaAtm1-based models of rTAP1 and rTAP2a using residue substitutions and placement of side chains from the rotamer library using in-house software. To obtain 3D structures of different TAP1/TAP2 heterodimers, models of TAP1 and TAP2 were superposed with subunits A and B, respectively, from structures of bacterial exporter dimers. Structures of different TAP heterodimers were further refined by loop correction and side-chain adjustment, mainly at dimerization interfaces.
Model assessment
Developed models of TAP transport domains closely reproduced structures of their templates. In particular, root-mean-square deviations (r.m.s.d.) between model of rTAP1/TAP2a and 4mrs and 2hyd structures were 1.08 Å (for 684 Cα-atoms) and 0.29 Å (for 752 Cα-atoms), respectively. Model assessment by MolProbity (31) indicated the satisfactory quality of the TAP models. For example, Ramachandran maps of 2hyd- and 4mrs-based models of rTAP1/TAP2a in complex with the FITC-labeled TR peptide showed that 90.7% (1038/1145) and 95.7% (1102/1151), respectively, of all residues were in favored regions, 98.3% of all residues of both models were in allowed regions with 19 outliers. The MolProbity score of 4mrs-based model was 1.94 (79th percentile among 27675 X-ray structures) based on deviations in main chain geometry (<0.3%) and Cβ geometry (<0.6%) and the clash score of 4.24 (96th percentile). To additionally verify correctness of the models of core TMDs, we analyzed locations of evolutionarily conserved residues in transporter sequences and structures (Figure S1). Residues that are conserved in TAP sequences are located exclusively at the interhelical interfaces of each half-transporter in either inward-facing or outward-facing conformation (residues shown by sticks on panels B and C). These residues likely participate in structure stabilization of each half-transporter. Eleven residues are highly conserved in sequences of not only TAPs but also other ABC-transporters (indicated by asterisk in Figure S1A). Five of these residues form TM1/TM6 contacts within each subunit at both membrane sides, which may be essential for structural stabilization of moving elements of these helices. Six highly conserved residues are located between TAP1 and TAP2: between TM2 and CL-2 and between CL-2 and NBD (shown by sticks-and-spheres and marked by circles/ovals on Figure S1 B and C). These residues likely play an essential role in stimulation of the ATPase activity of NBDs by peptides bound to TMDs.
Peptide docking
Structures of peptide ligands were based on conformations of fragments with extended or β-hairpin conformations that were taken from the crystal structure of the membrane β-barrel, VDAC1 (PDB ID: 3emn) (32). Specifically, to model the bent conformation of 8-mers and 9-mers we used β-hairpin fragment 172–180 of the 3emn structure, and for modeling 10-mers we used β-hairpin 224–233, which has a β-bulge. The peptide modeling included residue substitutions followed by energy minimization with QUANTA/CHARMm (100 steps, using ε of 10 and the adopted-basis Newton-Raphson method).
Initial docking of low-energy conformations of the TR nonapeptide in the bent conformation into rTAP1/TAP2a complex was performed manually to satisfy four major structural constraints from this study: (1–2) Bismaleimidoethane (BMOE)-mediated crosslinking between C2 of peptide and TAP1-E436C and between C6 of peptide and TAP1-C273; (3) ionic interactions between R9 of peptide and TAP2-E218; and (4) hydrophobic interactions between TAP1-Y385 and R9 side chain of the peptide. The side chains rotamers of the TR peptide were adjusted to avoid steric clashes with the transporter and to maximize formation of possible hydrogen bonds between ligand and transporter residues. The main chain angles of the second peptide residue (V2) were adjusted to provide favorable ionic interactions between free N-terminal amine with TAP1-E436 residue. Other peptides were similarly positioned inside the human, rat and chicken TAP complexes by overlapping their backbones with the structure of TR docked into rat TAP complex. To avoid peptide-protein hindrances, the manual peptide docking was followed by automated solid docking procedure implemented in QUANTA and short CHARMm minimization (20 steps with the adopted-basis Newton-Raphson method). After minimization of the peptide-TAP complex the main-chain geometry of 9-mer and 10-mer peptides did not significantly differ from the geometry of parent β-hairpins from the VDAC1 structure: r.m.s.d between Cα-atoms of residues 2–9 of 9-mers (TVDNKTAYR, RRYQKSTEL, and their derivatives) and residues 173–180 of the 3emn structure were less than 0.6 Å, while r.m.s.d. between Cα-atoms of residues 2–10 of the 10-meric RYWANATKSR and residues 225–233 of the 3emn structure was 0.33 Å.
The coordinate files (in PDB format) of the 2hyd- and 4mrs-based models of rTAP1/TAP2a in complex with FITC-labeled TVDNKTAYR peptide can be downloaded from the web site: http://mosberglab.phar.umich.edu/resources/. Other developed models can be obtained upon request.
Baculovirus constructs
pFastBac Dual vector encoding both wild type rTAP1 and rTAP2a was kindly gifted by Dr. Gaudet (33). Mutations were made using QuikChange II Site-Directed Mutagenesis Kits (Agilent Technologies). All the primers were purchased from Invitrogen. All sequences were confirmed by DNA sequencing at the University of Michigan Sequencing Core. Baculovirus stocks were prepared according to the Bac-to-Bac manual (Invitrogen).
Insect cell infections, microsome preparations, and peptide translocation
Sf21 cells were cultured in Grace’s insect medium (Invitrogen), supplemented with 10% fetal bovine serum. The cells were grown to confluence and infected with the appropriate baculoviruses at a multiplicity of infections of 5–20, depending on the protein expression level of individual baculoviruses. Following these infections, the cells were harvested after 72 h and microsomal membranes were generated as described (34). Protein expression was analyzed by immunoblotting assays with anti-His or anti-Flag antibodies for rTAP1 and rTAP2 respectively. Peptide transport assays were performed by incubating microsomes with 1 μM of fluorescein isothiocyanate (FITC) labeled peptide (from Peptide 2.0 Inc.) at 37 °C for 5 min in the presence or absence of 5 mM ATP. Microsomes were lysed in 1% NP40 and the fraction of transported and glycosylated peptide was recovered with Con A-Sepharose (Pharmacia, Freiburg, Germany). Fluorescence signals obtained in the presence and absence of ATP were measured in triplicate using a plate reader, and the +ATP/-ATP ratios were obtained. To compute transport efficiencies, +ATP/-ATP ratios from multiple independent experiments were normalized, setting values from microsomes expressing wild type rTAP at 100% and control microsomes lacking TAP at 0%, and averaged. Data are plotted as the mean ± SEM of measurements of indicated replicates. Statistical analyses are based on paired two-tailed t-tests using Prizm 6 (GraphPad, La Jolla, CA) software. For competition assays, peptide libraries XXXXXXXXR (X8R) and XXXXXXXXF (X8F) (where X indicates 19 randomized amino acids excluding cysteine) were synthesized by Sigma-Aldrich. Unlabeled competitor peptide in the appropriate concentration, or buffer for the controls, was added to fluorescent peptide and microsome suspensions to a final volume of 100 μl in the presence or absence of ATP. The concentration needed for 50% inhibition of fluorescent peptide transport was determined using Prizm 6 (GraphPad, La Jolla, CA) software.
Crosslinking assays
Microsomes were incubated with cysteine-containing fluorescently labeled peptides on ice for 10 minutes in PBS (pH 7.4), in a 100 μl reaction volume. BMOE (Pierce) was added to a final concentration of 0.2 mM. After a 10 min reaction at room temperature, crosslinking was quenched by adding 10 mM DTT. Products were further purified by Ni-NTA beads or directly used for SDS-PAGE. For purification on nickel beads, microsomes were lysed in 1% NP40 and TAP complexes were bound to Ni-NTA beads (Qiagen), beads were washed with lysis buffer, and proteins were eluted by SDS sample buffer. 10 μl of each sample was loaded and resolved by SDS/PAGE and visualized on a Typhoon scanner (at 520 nm). The relative fluorescence intensity B was quantified from the fluorimages and plotted as a function of peptide concentration [P]. Data were fitted using Equation: B=Bmax[P]/(KD+[P]), and KD values were determined by nonlinear least squares analysis. It is possible that the use of the cross-linker in these assays interferes with the derivation of true KD values reflective of steady state binding. We thus use the term apparent KD (KD,app) throughout the study.
Results
Homology modeling of the core transport unit of TAP complexes and mapping of polymorphic residues containing determinants of peptide selectivity
TAP from different species and their allelic variants differ in their peptide transport specificities. rTAP2 has two allelic variants, a and u, whereas hTAP and murine TAP (mTAP) have no functional polymorphism. rTAP1/TAP2a complexes are rather promiscuous and allow the transport of peptides with basic C-terminal amino acid (“Arg-permissive”), as also seen with hTAP (hTAP1/TAP2) (12, 13, 16). On the other hand, rTAP1/TAP2u complexes and mTAP1/TAP2 complexes are restrictive for the transport of peptides with charged residues, but permissive to bulky aliphatic and aromatic residues (“Arg-restrictive”) (10, 16). Additionally, cTAP1 and cTAP2 sequences exhibit high sequence polymorphisms and distinct preferences for transport of certain peptide sequence motifs (14). These polymorphic residues are expected to be important in peptide recognition.
In the absence of crystal structures of full-length TAP complexes with and without substrate, homology models of TAP in different functional states are useful for predicting peptide binding modes and uncovering molecular determinants of the selective peptide binding and transport. We thus constructed homology models of the TAP core transport units of rTAP1/TAP2a (Figures 1A–1C) and cTAP1/TAP2-B4 (Figure 1D) (validations of model quality are described in the Methods section). All models were built using the crystal structure of the homodimeric bacterial exporter NaAtm1 which represents the inward-facing conformation with two separated NBD domains that contact only through their C-terminal helices (24) (Figures 1A, 1C and 1D). In addition, rTAP1/TAP2a was modeled in the outward-facing conformation (Figure 1B), using structural template of the ADP-bound bacterial drug exporter Sav1866 (23). In the latter structure, the NBDs dimerize forming one consensus and one degenerate ATPase site at their dimerization interfaces (33), which in the models are occupied by ADP molecules (Figure 1B).
Both TAP conformations showed spacious internal cavities near the cytosolic membrane leaflet that are lined mostly by polar and charged residues, which form extensive hydrogen-bonding networks and could also interact with polar groups of bound peptides (Figures 1A and 1B; red ovals). Attempts to identify determinants of TAP selectivity have suggested that polymorphic rat TAP2 residues at positions 217/218, 262, 265 and 266, and 374/380 might determine the differential transport of peptides with C-terminal arginine (18, 20). In homology models of rTAP1/TAP2a complexes, residues 218, 262, 265 and 266 of rTAP2a face the internal cavity of TAP1/TAP2 heterodimer (Figure 1C). Several cTAP polymorphic residues (131, 179, 289 of cTAP1 and 216, 263, 378, and 416 of cTAP2 that correspond to positions 278, 326, 436 of rTAP1 and 218, 265, 380, and 418 of rTAP2) also face the internal cavity (Figure 1D). Almost half of all polymorphic rat and chicken residues are located near the cytosolic membrane boundary (Figures 1C and 1D), where the size and the shape of the internal cavity are rather similar in both inward-facing and outward-facing conformations of the transporter (Figures 1A and 1B, red ovals). We hypothesized that this region may represent the binding pocket for peptides, which preferentially interact with the inward-facing conformation of TAP in the first step of the translocation process in a nucleotide-independent manner.
Glu218 of rTAP2a controls specific translocation of Arg-terminated peptides
To elucidate the functional role of polymorphic rTAP residues located near the internal cavity, we generated single mutants of rTAP2a at positions 218, 262, 265 and 266, in which the residues were mutated to the corresponding residues of rTAP2u and co-expressed the mutants with rTAP1. A pair of peptides labeled by FITC that differ only at their C-termini, TVDNK*TAYR (TR) and TVDNK*TAYV (TV), were used in peptide transport assays.
As shown in the peptide transport analysis (Figures 2A and 2B), rTAP1/TAP2a(Q262R), rTAP1/TAP2a(S265P) and rTAP1/TAP2a(L266F) displayed transport efficiencies quite similar to the wild type rTAP1/TAP2a complex for both peptides. In contrast, the E218M mutation in TAP2a that eliminates the local negative charge in the TM2 strongly decreased the transport efficiency of the TR peptide, which has a positively charged arginine at the C-terminus (Figure 2A), but did not significantly affect the translocation of the TV peptide, which has a hydrophobic C-terminal valine (Figure 2B). The selectivity differences of this single mutant resemble the selectivity differences between rTAP1/TAP2a and rTAP1/TAP2u complexes. These results indicated that E218 of rTAP2a is a key residue involved in determining the substrate transport specificity at the peptide C-terminus. To address whether other polymorphic rTAP2 residues have synergistic effects, we introduced a combination of four mutations within TM2 and TM3, which resulted in the quadruple mutant construct rTAP1/TAP2a(E218M/Q262R/S265P/L266F). In the peptide transport assay, the quadruple mutant showed a similar functional fingerprint as the single rTAP1/TAP2a(E218M) mutant (Figures 2A and 2B). Additionally, a stronger effect of the quadruple mutant on reducing transport efficiency of the TR peptide compared to the single rTAP1/TAP2a(E218M) mutant was noted, indicating some contribution of TM3 residues in recognition of peptide C-terminus. Similar results were obtained for the effects of the single (E218M) and quadruple (E218M/Q262R/S265P/L266F) mutations in rTAP2a upon transport of a pair of decapeptides, RYWANATK*SR (RR) and RYWANATK*SF (RF). The effects of the mutations were significant for the RR peptide but not the RF peptide (Figures 2C and 2D). These results confirmed the critical role of the single E218 residue in selective translocation of multiple peptides bearing C-terminal basic residues. All mutants were expressed at similar levels as their wild type rTAP counterparts (Figure 2E), suggesting that the mutations do not compromise TAP folding, as also indicated by full competence for the transport of the TV and RF peptides by the mutants.
Figure 2. Key role for rTAP2a-E218 in the transport of peptides with C-terminal basic residues.
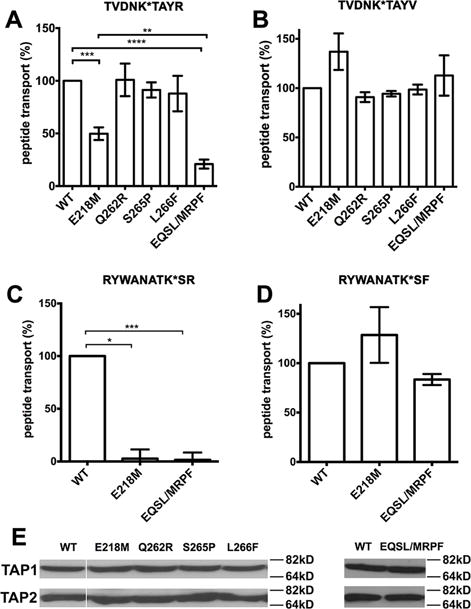
Transport of indicated peptides by the rTAP complexes of wild type (shown as WT) or the indicated rTAP1/TAP2a mutations. The quadruple mutant of rat TAP, TAP1/TAP2a(E218M/Q262R/S265P/L266F), is indicated as EQSL/MPRF. The peptides were fluorescently labeled at the indicated lysine residues (K*) and peptide transport assays were performed as described in Methods. (A–D) Averaged transport efficiencies for indicated nonapeptides and decapeptides. Data are averaged from 6–13 independent transport assays with three different microsome preparations (A), 2 independent transport assays with a single microsome preparation (B) and 3 independent transport assays with a single microsome preparation (C and D). Statistical analyses are based on paired two-tailed t-tests. *, P ≤ 0.05, **, P ≤ 0.01, ***, P ≤ 0.001, ****, P ≤ 0.0001. Error bars represent SEM values. (E) Expression levels of rTAP1 and rTAP2a in a representative microsome preparation were assessed by immunoblotting. In the left panel, between the WT and E218M lanes, the white line indicates irrelevant lanes that were removed.
Cys273 of rTAP1 crosslinks to residue 6 of a nonapeptide
ATP-independent peptide binding to TAP precedes the ATP-dependent peptide translocation step. Peptide transport efficiency usually correlates with peptide binding affinity (35). However, impaired transport might also relate to the obstruction of TMD conformational changes (36, 37). Therefore, a direct peptide binding study is essential to determine whether E218 of rTAP2a is a component of the peptide-binding pocket. We thus developed a fluorescent peptide-based crosslinking assay to examine peptide binding.
The transport unit of the rTAP contains 14 cysteine residues, ten of which are located within the core TMDs (Figure 3A). Two of these cysteines line the internal cavity in the modeled inward-facing TAP conformation: C273 of rTAP1 and C362 of rTAP2a. We predicted that sulhydryl groups of cysteine-containing peptides could be conjugated with free thiol groups of one of these two cysteine residues via a chemical crosslinker. For assessing crosslinking to cysteine-containing peptides, we used two FITC-labeled TR peptides with cysteine substitutions at either the N-terminal P2 position (peptide TC2R), or central P6 position (peptide TC6R) and the homobifunctional crosslinker BMOE with spacer arm length of 8 Å, which can irreversibly crosslink two cysteine residues. After crosslinking, rTAP complexes were recovered from microsome lysates by Ni-NTA beads and loaded on a SDS-PAGE gel. Crosslinked TAP was visualized by fluorimaging of gels. We found that, of the two FITC-labeled peptides, only TC6R was crosslinked to rTAP1/TAP2a (Figure 3B). Notably, an excess of the competitor TR peptide blocked the crosslinking, confirming the specificity of the reaction (Figure 3C). Based on the TAP-specificity of the reaction (Figures 3B and 3C), in subsequent experiments, we did not purify TAP from microsomes, and instead directly loaded the total microsome lysates on gels for fluorimaging.
Figure 3. Proximity of P6 of a TAP-bound nonapeptide to rTAP1-C273.
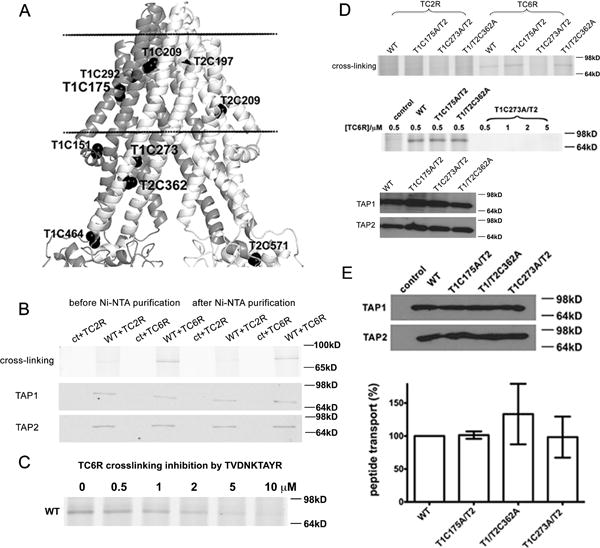
(A) Locations of cysteine residues (shown by spheres) of rTAP1 (colored dark gray) and rTAP2a (colored light gray) subunits of the NaAtm1-based homology model. Three cysteine residues mutated in this work are indicated in larger size. Positions of membrane boundaries are as defined in Figure 1. (B) Top panel: Prominent BMOE-mediated crosslinking of wild type (shown as WT) rTAP1/TAP2a to a fluorescent peptide containing a cysteine at position 6 (TVDNK*CAYR (TC6R)) compared to that observed with peptide bearing a cysteine at position 2 (TCDNK*TAYR (TC2R)) at peptide concentration of 1 μM (K* represents lysine-FITC). Crosslinked products were loaded directly on SDS-PAGE gels (left lanes) or further purified using Ni-NTA beads before loading (right lanes). Data are representative of two independent experiments, each in duplicate. TAP expression was confirmed by immunoblotting (middle and lower panels). (C) Peptide TC6R (1 μM) crosslinking with wild type TAP was blocked by peptide TVDNKTAYR. Representative data of two independent experiments with two microsome preparations are shown. (D) Mutation at TAP1-C273 disrupted TC6R peptide crosslinking. Top panel: Crosslinking between TC2R or TC6R (1 μM) and wild type or mutants, rTAP1(C175A)/TAP2a (T1C175A/T2), rTAP1/TAP2a(C362A) (T1/T2C362A) and rTAP1(C273A)/TAP2a (T1C273A/T2). Data are representative of four independent experiments. Middle panel: T1C273A/T2 does not crosslink with TC6R even at higher peptide concentrations. Data are representative of two independent experiments. Bottom panel: protein expression levels by immunoblots. (E) Similar expression levels and transport activities of mutants and wild type rTAP1/TAP2a were confirmed by immunoblotting (top panel) and peptide transport assays (bottom panel, average of 2 independent transport assays with 2 different microsome preparations). Error bars represent SEM values. Differences were not statistically significant, based on paired two-tailed t-tests.
To further investigate which cysteine residue within the internal cavity of the wild type rTAP becomes conjugated with TC6R, two mutant constructs (rTAP1(C273A) and rTAP2a(C362A)) were generated and tested for crosslinking to the TC6R peptide. Additionally, as a negative control, we generated the rTAP1(C175A) mutant with alanine-substituted cysteine in a region outside of the predicted peptide-binding pocket (Figure 3A). We found that only the rTAP1(C273A) mutant completely abolished the ability of the rat TAP complex to be linked to TC6R via BMOE in the absence of ATP, even under conditions of slightly higher expression compared to wild type TAP complexes (Figure 3D). In contrast, the rTAP1(C175A)/TAP2a and the rTAP1/TAP2a(C362A) mutants retained the ability to crosslink with the TC6R peptide (Figure 3D). The peptide transport efficiency of rTAP1(C273A)/TAP2a was similar to that of wild type TAP complex (Figure 3E), indicating that impaired crosslinking to this mutant complex is not related to protein misfolding. These results demonstrated that the central region (P6) of the nonameric peptide is located close to C273 of rTAP1.
Glu218 of rTAP2a determines the specific binding of Arg-terminated peptides
We used the crosslinking assay described above to further evaluate the peptide binding by different TAP constructs (in the absence of ATP). By quantifying the fluorescence intensities, the averaged KD,app value of the TC6R peptide for rTAP1/TAP2a was calculated to be 186.6±23.2 nM. Crosslinking of the TC6R peptide was largely impaired for the E218M and the quadruple mutants of rTAP2a (Figure 4A), indicating decreased binding efficiencies of the TC6R peptide to both mutants (KD,app could not be accurately determined). These results are in line with the significant impairment in TR transport in both cases (Figure 2). In contrast, the binding efficiencies of the TC6V peptide to rTAP1/TAP2a(E218M) (KD,app = 339.2±50.6 nM) or the quadruple mutant complexes (KD,app = 107.3±5.8 nM) were not reduced compared with wild type rTAP (KD,app = 416.7±115.0 nM) (Figure 4B). These experiments provide direct evidence that E218 of rTAP2a is a key residue that determines ATP-independent binding of peptides with basic C-terminal residues.
Figure 4. Key role of rTAP2a-E218 in the selective binding of peptides with basic C-terminal residues.
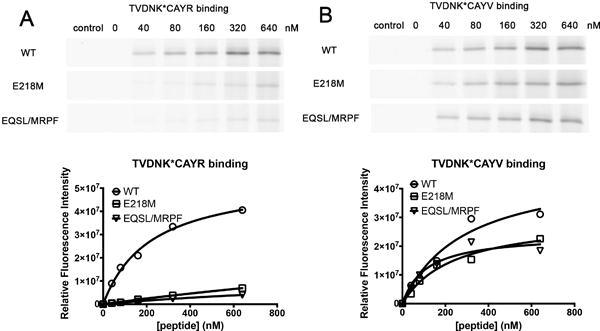
Binding of indicated peptides (K* represents lysine-FITC) to wild type (shown as WT), single mutant rTAP1/TAP2a(E218M) and quadruple mutant rTAP1/TAP2a(E218M/ Q262R/S265P/L266F) (shown as EQSL/MRPF) was assessed using BMOE-mediated crosslinking assays performed as described in Figure 3. The same microsome preparation was used in the TC6R and TC6V binding assays. Data are representative of 2 independent experiments with a single microsome preparation.
Tyr385 of rTAP1 is a general determinant of peptide binding that favors interactions with peptides containing hydrophobic C-termini
TAP transporters across many species are permissive for the transport of peptides with C-terminal hydrophobic residues (16). We found that TAP1-Y385 in TM5, which is spatially proximal to TAP2a-E218 from TM2 (Figure 5A), is conserved across TAP1 from many species. To investigate whether Y385 of rTAP1 determines TAP preference for peptides with hydrophobic C-termini, we examined peptide transport by the rTAP1(Y385A)/TAP2a mutant complex. As shown in Figure 5B, there was a significant reduction in the transport efficiency of the TV peptide, and a less significant reduction in transport of the TR peptide by rTAP1(Y385A)/TAP2a, compared to the wild type TAP. We also found that the binding to rTAP1(Y385A)/TAP2a of TC6V peptide was decreased relative to those observed with the rTAP1/TAP2a complexes, again to a greater extent compared to that observed with the TC6R peptide (Figures 5C and 5D; KD,app values for rTAP1(Y385A)/TAP2a could not be accurately assessed).
Figure 5. Key role of rTAP1-Y385 in the transport and specific recognition of peptides with hydrophobic C-termini.
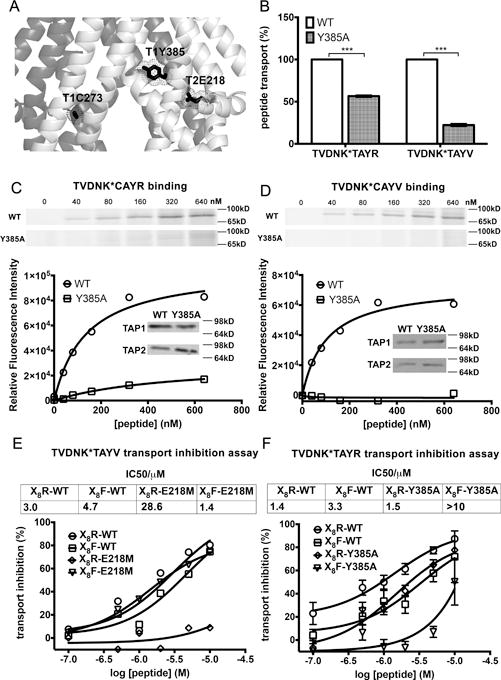
(A) Location of rTAP1-Y385 (T1Y385) in the inward-facing rat TAP model. rTAP1-C273 (T1C273) and rTAP2a-E218 (T2E218) are indicated as reference points. Residues are shown as sticks. (B) Transport of peptides TR (TVDNK*TAYR) and TV (TVDNK*TAYV) (K* represents lysine-FITC) by wild type rTAP1/TAP2a and rTAP1(Y385A)/TAP2a complexes. Data are averages of three independent experiments, each in triplicate, with 2 different microsome preparations. Statistical analyses are based on paired two-tailed t-tests. ***, P ≤ 0.001. Error bars represent SEM values. (C-D) Impaired binding of peptides TVDNK*CAYR (TC6R) (C) and TVDNK*CAYV (TC6V) (D) to rTAP1(Y385A)/TAP2a complexes, using BMOE-based crosslinking as described in Figures 3 and 4. Insets show representative immunoblots. Data are from five or two independent analyses for TC6R and TC6V, respectively. (E–F) Effects of rTAP2a(E218M) (E) and rTAP1(Y385A) (F) mutations on peptide transport inhibition by the indicated peptide libraries X8R and X8F, where X stands for 19 randomized amino acids excluding cysteine. Data are representative of four (rTAP1/TAP2a(E218M)) or two (rTAP1(Y385A)/TAP2a) independent experiments each performed in duplicate with a single microsome preparation.
To assess peptide translocation by the rTAP1(Y385A)/TAP2a and the rTAP1/TAP2a(E218M) mutant complexes for a wider range of peptides, we examined peptide transport inhibition, using permissive or partially permissive peptides as reporters: TV for rTAP1/TAP2a(E218M) and TR for rTAP1(Y385A)/TAP2a (Figures 5E and 5F), and two libraries of nonapeptides (X8R and X8F), which had random amino acids (Cys excluded) at positions 1–8 and a fixed basic (Arg) or hydrophobic (Phe) C-terminal residue. We found that transport of TV by the rTAP1/TAP2a(E218M) complexes was more efficiently inhibited by peptide libraries with a C-terminal Phe than with C-terminal Arg (IC50 values were 1.4 and 28.6 μM, respectively) (Figure 5E). Transport of TR by rTAP1(Y385A)/TAP2a complexes was better inhibited by a peptide library with C-terminal Arg than Phe (IC50 were 1.5 and >10 μM, respectively) (Figure 5F). However, both X8R and X8F peptide libraries inhibited binding of TR and TV peptides to rTAP1/TAP2a to more similar extents (IC50 between 1.4 and 4.7 μM).
These results imply that both E218 of TAP2a and Y385 of TAP1 are functionally important in determining the transport specificities of a range of peptides. TAP2a-E218 specifically favors transport of peptides with basic C-terminal residues, while Y385 of TAP1 favors transport of peptides with hydrophobic C-termini. We also suggest that TAP1-Y385 is more important for TAP function than TAP2a-E218, as mutations of TAP1-Y385 resulted in reduced binding and transport of peptides with both basic and hydrophobic C-terminal residues.
Proximity of rTAP1-E436 to the peptide N-terminus supports a model for a bent conformation of TAP-bound peptides
The experimental results thus far provided three anchor points for a peptide substrate inside the rTAP internal cavity: direct contacts of peptide C-terminal side chains with rTAP2a-E218 and rTAP1-Y385 and covalent linking of peptide P6 with rTAP1-C273. Based on this information, we docked the FITC- and BMOE-labeled TC6R peptide into the TAP complex. The presence of two bulky covalently bound probes, FITC linked to K5 and BMOE linked to C6 of the peptide, significantly increases the peptide volume and restricts its orientation inside the internal cavity. These spatial restrictions and three identified anchoring points allowed us to unambiguously define the position of the peptide C-terminus but not of the N-terminus. The N-terminal part of a peptide may adopt quite different conformations and orientations within the large peptide translocation channel, either pointing toward cytosol in an extended, quasi-linear conformation or being directed toward the luminal side in a bent configuration (Figure 6A).
Figure 6. Proximity of rTAP1-E436 to the peptide N-terminus supports a model for a bent peptide conformation.
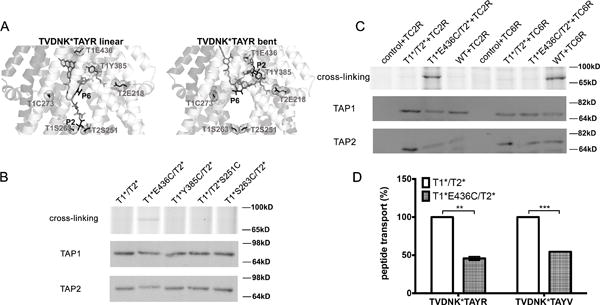
(A) Models for linear (left) and bent (right) conformations of the TVDNK*TAYR (TR) peptide in the translocation channel of the rTAP1/TAP2a model in an inward-facing conformation. TM helices are shown as cartoons. Residues that could anchor the C-terminus, P2, or P6 of the peptide are shown by sticks. The TR peptide is also shown by sticks. (B) Crosslinking between C2 of TCDNK*TAYR (TC2R) peptide (at 1 μM; K* represents lysine-FITC) and indicated TAP complexes (top panel). Data are representative of four experiments with 2 different microsome preparations. Lower panels show representative immunoblots. (C) Crosslinking occurred between rT1*(E436C)/T2* and C2 of TC2R peptide but not C6 of TVDNK*CAYR (TC6R) (at 1 μM; K* represents lysine-FITC) (top panel). Data are representative of four experiments with a single microsome preparation. Middle and Lower panels show representative immunoblots. (D) Impaired transport of TR and TV peptides by the rT1*(E436C)/T2* mutant. Data are averages of three experiments each in triplicate, with 2 different microsome preparations. All constucts shown in the figure are on the rTAP1(C273A)/TAP2a(C362A) background (labeled as T1*/T2*). Statistical analyses are based on paired two-tailed t-tests. **, P ≤ 0.01, ***, P ≤ 0.001. Error bars represent SEM values.
To localize the N-terminus of the TAP-bound peptide inside the ligand-binding pocket and examine the preferred (bent vs linear) conformation of the TAP-bound peptides, we performed additional crosslinking assays with the TC2R peptide containing a cysteine at the peptide residue 2 (C2). We designed rTAP mutants by introducing cysteines into positions 385 or 436 of rTAP1, which are predicted to be close to the C2 of TC2R in a bent conformation (Cβ-Cβ distances in the inward-facing state rTAP model are ~7 Å and ~9 Å, respectively). We also designed mutants with cysteines at positions 263 of rTAP1 and 251 of rTAP2a, predicted to be close to C2 of TC2R peptide in a linear conformation (Cβ-Cβ distances are ~4 and ~5 Å, respectively). We expected that crosslinking of TC2R with either C436 or C385 of rTAP1 would support a bent conformation for the peptide, while crosslinking with either C263 of rTAP1 or C251 of rTAP2a would point to a linear peptide conformation. To avoid crosslinking to other cysteine residues in the vicinity of the cavity, the cysteine mutants were generated on the rTAP1(C273A)/TAP2a(C362A) background (named rTAP1*/TAP2a*) to remove the other cysteines that face the internal cavity of the TAP heterodimer (Figure 3A).
We found that the TC2R peptide could be conjugated via BMOE only to rTAP1*(E436C)/TAP2a* but not to rTAP1*(S263C)/TAP2a* or rTAP1*/TAP2a* (S251C) (Figure 6B). This result provided direct evidence supporting a bent model of the TAP-bound peptide. There was no significant crosslinking between TC2R and TAP1-C385 of rTAP1*(Y385C)/TAP2a*, which is also predicted to be in crosslinking proximity to peptide residue 2 in a bent conformation. The significant impairment in peptide binding that results from rTAP1(Y385) mutations (Figure 5) could explain this result. On the other hand, the lack of crosslinking of rTAP1*/TAP2a*(S251C) or rTAP1*(S263C)/TAP2a* mutants to TC2R could not be explained by the impaired binding of these mutants, as transport and binding of the TR peptide by rTAP1/TAP2(S251A) and rTAP1(S263A)/TAP2 complexes were not reduced relative to the wild type TAP (Figure S2). Crosslinking of rTAP1*(E436C)/TAP2a* was selective towards peptide TC2R but not TC6R (Figure 6C) verifying the closer proximity of rTAP1–436 to the peptide N-terminus compared to the central residues of the peptide.
The rTAP1*(E436C)/TAP2a* complex, which was efficiently crosslinked to TC2R, demonstrated partial reduction in transport of both TR and TV peptides as compared to the rTAP1*/TAP2a* complex (Figure 6D). rTAP1-E436 is a site of polymorphism in cTAP (R289; Figure 1D). To further explore the role of acidic TAP1-E436 residue in plausible interactions with peptides, we designed the rTAP1(E436R) mutant with a charge inversion and assessed binding and transport of four FITC-labeled peptides that carried different net charges (TV: −1, TR: 0, RF: +1, RR: +2). We found a 2-fold increased translocation efficiency of the negatively charged TV peptide and reduced transport of neutral TR peptide (by ~28%) (Figure 7A). The rTAP1(E436R)/TAP2a complexes also showed reductions in transport efficiencies for the basic RR and RF peptides compared to the wild type TAP complex (by ~43% and ~45%, respectively) (Figure 7B). In the peptide binding assessments, although the differences between wild type TAP and rTAP1(E436R)/TAP2a appeared to be small (Figures 7C and 7D), in pairwise comparisons within experiments, both TC6R and TC6V displayed higher binding affinities to rTAP1(E436R)/TAP2a complexes compared to wild type complexes (KD,app values derived from individual experiments are listed in the legends to Figures 7C and 7D). In parallel experiments, we found that the RR and RF peptides inhibit crosslinking of peptide TC6R to rTAP1(E436R)/TAP2a with higher IC50 values compared to those for the wild type TAP complex (Figures 7E and 7F), indicating reduced binding affinities of both RR and RF peptides to rTAP1(E436R)/TAP2a.
Figure 7. Effects of the rTAP1(E436R) mutation on transport (A–B) and binding (C–F) of peptides with different net charges (q).
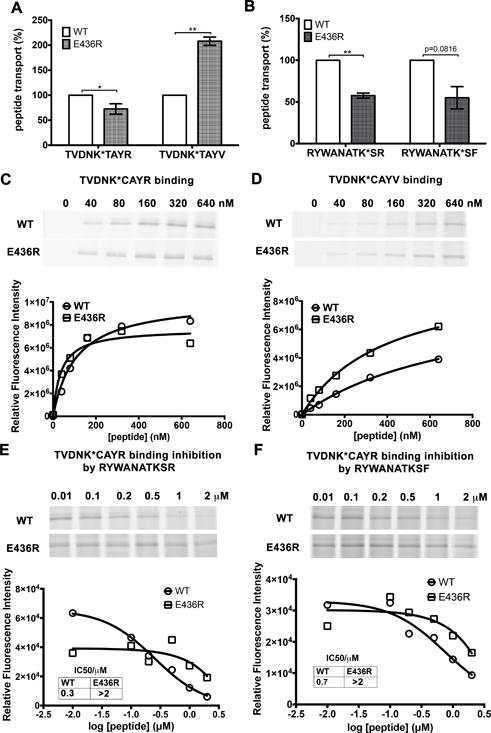
(A) The rTAP1(E436R) mutation reduces transport efficiency for the neutral peptide TVDNK*TAYR (q = 0) but increases transport efficiency for the negatively charged peptide TVDNK*TAYV (q = −1). (B) The rTAP1(E436R) mutation reduces the efficiency of transport of both basic peptides RYWANATK*SF (q = +1) and RYWANATK*SR (q = +2). Peptide transport data are averages of three independent experiments each in triplicate with 2 (A) or a single microsome preparation (B). (C–F) The rTAP1(E436R) mutation increases binding affinities of the negatively charged TVDNK*CAYV (TC6V) (D) nonapeptides, but decreases the binding of two basic decapeptides, RYWANATKSR (RR) (E) and RYWANATKSF (RF) (F). Peptide binding of nonapeptides (C–D) was assessed by direct BMOE-mediated crosslinking assays as described in the Method section. Derived apparent peptide binding affinities from independent experiments for rTAP1(E436R)/TAP2a and wild type TAP complexes were respectively: TC6R, 14.8±9.8 nM vs 90.6±25.6 nM, 36.6±13.4 nM vs 106.9±25.5 nM, 18.9±11.1 nM vs 104.4±11.1 nM, 23.7±8.1 nM vs 43.9±10.9 nM, 90.0±27.9 nM vs 93.9±13.7, p=0.0345. TC6V, 263.2±55.7 nM vs 410.1±143.1 nM, 397.4±50.6 nM vs 790.4±121.9 nM, 345.5±65.1 nM vs 894.0±505.2 nM, 318.0±105.1 nM vs 649.3±248.6 nM, p=0.0067). Peptide binding of decapeptides (E-F) was measured by their competitive inhibition of binding of TC6R (1 μM). K* represents lysine-FITC. For C-F, representative data of two independent experiments with a single microsome preparation are shown. Statistical analyses for A-D are based on paired two-tailed t-tests. *, P ≤ 0.05, **, P ≤ 0.01. Error bars represent SEM values.
Thus, the wild type TAP complex with negatively charged rTAP1-E436 preferentially binds the positively charged peptides (RR and RF). A TAP mutant with a basic R436 at the same position favors the binding and transport of the negatively charged TV peptide. These results indicate that the TAP-E436 residue is a determinant for recognition of peptide charge, further discussed below based on modeling.
Modeling of peptide-TAP complexes
The 9-mer peptide could be unambiguously modeled inside the ligand pocket in a bent configuration based on identification of key anchor points for the TAP-bound nonapeptide, including (i) crosslinking between TC2R and the rTAP1(E436C) mutant (Figure 6B), (ii) crosslinking between TC6R and rTAP1-C273 (Figure 3D), and (iii) demonstration of contributions of rTAP2a-E218 and rTAP1-Y385 to binding of peptide C-terminus (Figures 4 and 5). To model bent peptides inside the translocation channel, we chose β-hairpin-like peptide conformations stabilized by internal hydrogen bonds as an energetically preferred structure in an environment with the lowered dielectric constant (ε~10) that is expected inside the translocation channel. Validations of model qualities are described in the Methods section.
The TR peptide docked into the inward-facing rTAP1/TAP2a complex in a β-hairpin-like conformation is surrounded by thirteen TAP residues (Figure 8A). The involvement of seven of these residues (C273, Y385, E436 of rTAP1, and E218, Q262, S265, L266 of rTAP2a) in the proposed peptide binding pocket is supported by our current experiments. Four residues (D278 and Y326 of rTAP1 and E418 and Q424 of rTAP2a) are variable in rat or chicken TAP1 and TAP2 sequences, which suggests their role in selective recognition of different peptide ligands. Three residues, E381, Y385 of rTAP1 and Y428 of rTAP2a, are conserved across species and may be essential for the peptide translocation along the channel. Interactions of peptide with these three conserved residues are different in models of TR-TAP complexes in the inward-facing and the outward-facing states (Figure S3). For example, rTAP1-Y385 forms hydrophobic interactions with side chains of V2 and R9 in the inward-facing state model, but becomes more distant from V2 in the outward-facing state model. Similarly, only in the inward-facing state model, π-stacking interactions are formed between aromatic rings of rTAP2a-Y428 and peptide Y8, and rTAP1-E381 forms H-bonds with peptide R9. Alterations in the interactions of peptide with these residues of TAP in the outward-facing state compared to the inward-facing state could reduce the binding affinity, and initiate the release of peptide into the ER lumen.
Figure 8. Peptide-TAP complexes of rat and chicken TAPs in inward-facing conformation.
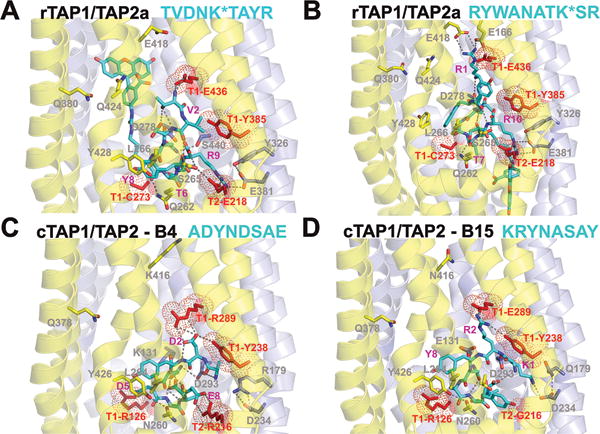
Structural models of rTAP1/TAP2a (A, B) and chicken TAP complexes from B4 (C) and B15 (D) haplotypes were built using crystal structures of NaAtm1 (PDB ID: 4mrs). The following transporter-specific peptides are shown inside the ligand binding pockets: FITC-labeled 9-meric TVDNK*TAYR (A) and 10-meric RYWANATK*SR (B), and 8-meric ADYNASAY (C) and KRYNASAY (D). TM helices are presented as cartoons colored blue (for TAP1) and yellow (for TAP2). Peptides ligands are shown by sticks with C atoms colored cyan; surrounding TAP residues are shown by sticks with C atoms colored blue (for TAP1) and yellow (for TAP2); O atoms are colored red, N atoms are colored blue. Residues mutated in this work are colored red and marked by dots. Hydrogen bonds formed between main-chain atoms of ligands and between ligands and transporter side chains are shown by gray dashes.
In the models of rTAP1/TAP2a with docked FITC-labeled TR and RR peptides (Figures 8A and 8B), electrostatic (H-bonding and ionic) interactions are formed between peptide R9/R10 and TAP2a-E218 and between the peptide amino-terminus (NH3+) and rTAP1-E436, while hydrophobic interactions are formed between peptide R9/R10 and rTAP1-Y385. Additionally, in the RR model, H-bonding and ionic contacts are formed between peptide R1 and rTAP1-E436, and with rTAP2a-E166 and rTAP2a-E418. Thus, the RR decapeptide has more contacts within the modeled ligand-binding pockets than the TR nonapeptide, which could explain the higher binding affinity of RR compared to TR observed in inhibition experiments (Figures 7E, IC50 values of 0.3 μM for RR but > 2 μM for TR (data not shown)). Based on models of mutant rTAP1(E436R)/TAP2a complex, RR and RF peptides likely encounter ionic repulsions due to close proximity between R1 and TAP1-R436 (Figure 8B), which could explain the reduced binding and transport activities of RR and RF peptides by rTAP1(E436R)/TAP2a relative to wild type TAP (Figures 7B, 7E and 7F). On the other hand, TR and TV might form favorable ionic interactions between their D3 and rTAP1-R436 in rTAP1(E436R)/TAP2a mutant (Figure 8A), which could explain the tendency towards improved binding affinities of nonapeptides to the mutant TAP, as discussed above (Figures 7C and 7D). During transition of TAP to the outward-facing conformation, all electrostatic and hydrophobic interactions of TR and TV seem to be weakened, based on the models (Figure S3). Thus, TV, with fewer ionic interactions near the C-terminus compared to TR (Figure 8A), may dissociate more readily than TR, thereby increasing transport efficiency in the context of rTAP1(E436R)/TAP2a complexes (Figure 7A).
To identify additional determinants of TAP translocation specificity, we constructed models of peptide-transporter complexes for two cTAPs from haplotypes B4 and B15, which have been shown to be selective for acidic (ADYNDSAE) and basic (KRYNASAY) peptides, respectively (14). As expected, substitutions in polymorphic residues of TAP1 and TAP2 sequences correlate with distinct sequence preferences for transported peptides. In particular, the exclusive acidic residues at positions 2, 5, and 8 of ADYNDSAE form specific ionic interactions with basic residues of TAP from B4 haplotype, cTAP1-R289 (rTAP1-E436), cTAP1-K131 (rTAP1-D278), and cTAP2-R216 (rTAP2a-E218), respectively (Figure 8C). On the other hand, the essential basic and aromatic residues at positions 1, 2, and 8 of KRYNASAY interact with residues of TAP from B15 haplotype, cTAP1-D234 (rTAP1-E381), cTAP1-E289 (rTAP1-E436), and cTAP2-G216 (rTAP2a-E218) of B15 haplotype, respectively (Figure 8D). These results indicate that several residues from TM3, TM5, and TM6 of TAP1 (positions 278, 381, 436 in rTAP1) could represent important determinants of selective recognition of peptide motifs.
It has been previously reported that the nonapeptide RRYQKCTEL with a cysteine at the P6 position crosslinks with human TAP1(V288C) using BM[PEO]3 as crosslinker, and that the same peptide with an iron-dependent chemical protease BABE at P6 can cleave TAP1 near residue 290 (36). To examine these previous data in the context of the proposed binding model (Figure 8), we constructed models of hTAP1/TAP2-peptide complexes (Figure S4). RRYQKCTEL with BM[PEO]3 or BABE incorporated into P6 was docked onto hTAP1/TAP2 complexes in silico, based on the models described in Figure 8. In the resulting models, BM[PEO]3 is in sufficient proximity to V288C to form a covalent linkage with this cysteine in TM3 of hTAP1 (Figure S4A), in agreement with the previous experimental data (36). Additionally, BABE is positioned near residue 290 of hTAP1 (Figure S4B), consistent with the previous experimental identification of the site of polypeptide cleavage by the chemical protease (36). Recent electron paramagnetic resonance (EPR) studies showed that interspin distance between spin probes at positions 2 and 8 of the double-labeled nonapeptide was around 22 Å (38). We analyzed orientations and distances between nitroxyl radicals of proxyl-labeled side chains at positions 2 and 8 of the TAP-bound spin-labeled RCYQKSTCL peptide by docking the peptide into the TAP model, based on the docking mode discussed in Figure 8A. We found that the conformation and placement of the spin-labeled nonapeptide and its long proxyl-modified side chains are restricted inside the peptide binding pocket. Therefore, the extended configuration of proxyl spin probes, their opposite orientations, and rather large distance between nitroxyl groups (22.6 Å) are unequivocally defined (Figure S4C). The obtained distance is in agreement with the EPR data, although the bent conformation in our model contradicts the author’s conclusions about plausible extended conformation of the TAP-bound peptide (38).
Discussion
An understanding of the peptide translocation specificity of TAP is largely impeded by the absence of experimental structures of the full-length transporters in complexes with peptide substrates. Based on the results described in this study, we constructed three-dimensional models of peptide-TAP complexes for rat, chicken and human TAPs (Figures 8 and S3–S4), which were extensively validated in the context of known experimental data and functional polymorphism of rat and chicken TAPs. Our model of rat TAP in the outward-facing state is similar to previously published Sav1866-based models of the human TAP (22, 37). In contrast, the inward-facing conformation of our TAP complexes significantly differ from previously published human TAP models that were produced based on P-glycoprotein, MsbA, and ABCB10 crystal structures (22).
Our models of peptide-TAP complexes for different species are mainly based on four interaction sites for TAP-bound peptides elucidated from this work (residues C273, Y385, E436 of rTAP1 and E218 of rTAP2a) (Figures 8 and S3). In all the models, TAP-bound peptides are located at ~7 Å distances from both sides of the cytosolic membrane boundary (Figure 9A). In the NaAtm1 structural template, two molecules of co-crystallized GSSG occupy a similar position (Figure 9B). The location of TAP-bound peptide also overlaps with the binding site of an unknown molecule that was co-purified with the TmrAB dimer (29), but differs from positions of P-glycoprotein ligands that are inserted deeper into membrane-embedded TMD domains, 5–10 Å away from the cytoplasmic surface (39).
Figure 9. Similar locations of peptides in the bacterial NaAtm1-based model of the rat TAP1/TAP2a complex (A) and in the crystal structure of the NaAtm1 transporter (B).
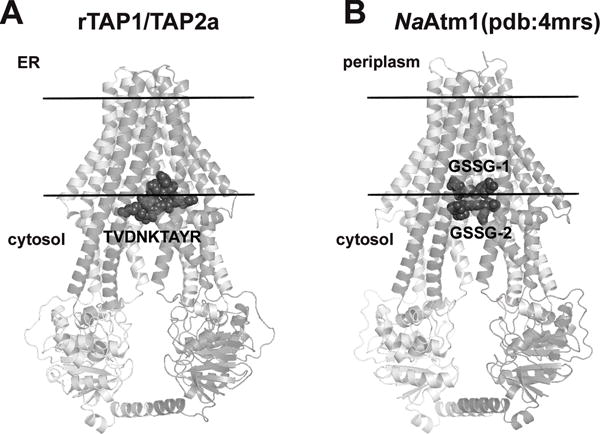
Two molecules of oxidized glutathione (GSSG) co-crystallized with bacterial transporter (PDB ID: 4mrs) occupy similar space near the cytosolic membrane boundary as the predicted site for the TVDNKTAYR (TR) peptide in the rTAP-peptide complex. TM helices of transporters are shown by cartoons colored light gray (for subunit A and TAP1) and dark gray (for subunit B and TAP2a). GSSG molecules and the TR peptide are shown by spheres.
We found strong inhibition of binding and translocation of peptides with C-terminal arginines by rTAP complexes with either a single TAP2a(E218M) or the quadruple mutant (Figures 2 and 4). It is known that the presence of Met at TAP2-218 position coincides with “Arg-restrictive” phenotypes of mTAP1/TAP2 and rTAP1/TAP2u complexes (20). Comparison of models of chicken TAPs from haplotypes B4 and B15, which significantly differ in peptide translocation specificity (14), also emphasized a role for residue 216 from cTAP2 (position 218 in rTAP2) in the specific recognition of peptide C-terminal residues. In particular, it became clear from the models that basic R216 forms an ionic pair with acidic E8 residue of octapeptides, while G216 favorably interacts with peptide with a bulky aromatic Phe residue at peptide C-terminus (Figures 8C and 8D).
Further analyses of peptide docking in models of chicken and rat TAPs indicate that several residues from the TM3, TM5, and TM6 of TAP1 (cTAP1-K/E131 or rTAP1-D278, cTAP1-D234 or rTAP1-E381, cTAP1-R/E289 or rTAP1-E436) and TM2 and TM6 of TAP2 (cTAP2-R/G216 or rTAP2-E/M218, cTAP2-K/N416 or rTAP2-E418, cTAP2-Q422 or rTAP2-Q424) are likely to function as determinants of selective peptide recognition (Figures 8 and S3–S4). In particular, we found that a charged residue from the TM6 of TAP1 (rTAP1-E436 or cTAP1-R/E289) participates in charge recognition at the peptide N-terminus, and therefore plays an important role for TAP translocation selectivity.
Notably, the localization of bound RRYQKSTEL in our model of human TAP-peptide complex (Figure S4) significantly differs from the peptide positioning proposed by Corradi et al. (22), using homology modeling and replica exchange simulations in the absence of experimental data. The Corradi model, based on the structure of the ABCB10 in the inward-facing conformation, places the peptide in an extended conformation in the large cytoplasmic opening between both TAP subunits, ~10–15 Å below the cytosolic membrane border. The latter model, which was not based on experimental constraints, is inconsistent with several components of the experimental data described here, and does not explain the peptide repositioning in the outward-facing state of the transporter, when a large part of the proposed peptide binding site becomes occluded by moving TM helices.
Based on the current modeling studies, we propose that the peptide, in a β-hairpin-like conformation, binds to the ligand binding site of the nucleotide-free inward-facing conformation of TAP, which is located between TM helices at the cytoplasmic membrane boundary (Figure 9). Peptide binding to TMDs facilitates the transition of the transporter to the outward-facing state (40). In that state, the peptide likely maintains a bent conformation as well as its overall position within the translocation channel but may have altered interactions with TAP residues (Figure S3), which would facilitate peptide release from the TMD translocation channel to the ER lumen.
In conclusion, this study provides structural models for the TAP-peptide complexes in different functional states of the transporters that were extensively validated using current and previous experimental data. These models can be used to guide future discovery of TAP inhibitors for treatment of autoimmune diseases.
Supplementary Material
Acknowledgments
This work utilized the DNA sequencing and Hybridoma Cores of the University of Michigan. The authors thank Dr. Rachelle Gaudet for the pFastBac Dual vector encoding wild type rTAP1 and rTAP2a and Dr. Andrew Tai for the use of the plate reader.
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI044115 (to MR). Additional financial support was from the Division of Biological Infrastructure of the National Science Foundation under award number 1145367 (to IDP) and from the National Institute of Drug Abuse of the National Institute of Health under award number R01DA003910 (to HIM and IDP).
Abbreviations
- (ER)
Endoplasmic reticulum
- (ABC)
ATP binding cassette
- (TMD)
Transmembrane domain
- (NBD)
Nucleotide-binding domain
- (BMOE)
Bismaleimidoethane
- (EPR)
Electron paramagnetic resonance
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Raghavan M, Del Cid N, Rizvi SM, Peters LR. MHC class I assembly: out and about. Trends Immunol. 2008;29:436–443. doi: 10.1016/j.it.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blum JS, Wearschand PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seyffer F, Tampe R. ABC transporters in adaptive immunity. Biochim Biophys Acta. 2015;1850:449–460. doi: 10.1016/j.bbagen.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 4.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 5.Raghavan M. Immunodeficiency due to defective antigen processing: the molecular basis for type 1 bare lymphocyte syndrome. J Clin Invest. 1999;103:595–596. doi: 10.1172/JCI6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lankat-Buttgereit B, Tampe R. The transporter associated with antigen processing: function and implications in human diseases. Physiol Rev. 2002;82:187–204. doi: 10.1152/physrev.00025.2001. [DOI] [PubMed] [Google Scholar]
- 7.Rees DC, Johnsonand E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koch J, Guntrum R, Heintke S, Kyritsisand C, Tampe R. Functional dissection of the transmembrane domains of the transporter associated with antigen processing (TAP) J Biol Chem. 2004;279:10142–10147. doi: 10.1074/jbc.M312816200. [DOI] [PubMed] [Google Scholar]
- 9.Procko E, Raghuraman G, Wiley DC, Raghavanand M, Gaudet R. Identification of domain boundaries within the N-termini of TAP1 and TAP2 and their importance in tapasin binding and tapasin-mediated increase in peptide loading of MHC class I. Immunol Cell Biol. 2005;83:475–482. doi: 10.1111/j.1440-1711.2005.01354.x. [DOI] [PubMed] [Google Scholar]
- 10.Schumacher TN, Kantesaria DV, Heemels MT, Ashton-Rickardt PG, Shepherd JC, Fruh K, Yang Y, Peterson PA, Tonegawa S, Ploegh HL. Peptide length and sequence specificity of the mouse TAP1/TAP2 translocator. J Exp Med. 1994;179:533–540. doi: 10.1084/jem.179.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neefjes J, Gottfried E, Roelse J, Gromme M, Obst R, Hammerlingand GJ, Momburg F. Analysis of the fine specificity of rat, mouse and human TAP peptide transporters. Eur J Immunol. 1995;25:1133–1136. doi: 10.1002/eji.1830250444. [DOI] [PubMed] [Google Scholar]
- 12.van Endert PM, Riganelli D, Greco G, Fleischhauer K, Sidney J, Setteand A, Bach JF. The peptide-binding motif for the human transporter associated with antigen processing. J Exp Med. 1995;182:1883–1895. doi: 10.1084/jem.182.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uebel S, Kraas W, Kienle S, Wiesmuller KH, Jung G, Tampe R. Recognition principle of the TAP transporter disclosed by combinatorial peptide libraries. Proc Natl Acad Sci U S A. 1997;94:8976–8981. doi: 10.1073/pnas.94.17.8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker BA, Hunt LG, Sowa AK, Skjodt K, Gobel TW, Lehnerand PJ, Kaufman J. The dominantly expressed class I molecule of the chicken MHC is explained by coevolution with the polymorphic peptide transporter (TAP) genes. Proc Natl Acad Sci U S A. 2011;108:8396–8401. doi: 10.1073/pnas.1019496108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gubler B, Daniel S, Armandola EA, Hammer J, Caillat-Zucman S, van Endert PM. Substrate selection by transporters associated with antigen processing occurs during peptide binding to TAP. Mol Immunol. 1998;35:427–433. doi: 10.1016/s0161-5890(98)00059-5. [DOI] [PubMed] [Google Scholar]
- 16.Momburg F, Roelse J, Howard JC, Butcher GW, Hammerling GJ, Neefjes JJ. Selectivity of MHC-encoded peptide transporters from human, mouse and rat. Nature. 1994;367:648–651. doi: 10.1038/367648a0. [DOI] [PubMed] [Google Scholar]
- 17.Armandola EA, Momburg F, Nijenhuis M, Bulbuc N, Fruh K, Hammerling GJ. A point mutation in the human transporter associated with antigen processing (TAP2) alters the peptide transport specificity. Eur J Immunol. 1996;26:1748–1755. doi: 10.1002/eji.1830260813. [DOI] [PubMed] [Google Scholar]
- 18.Momburg F, Armandola EA, Post M, Hammerling GJ. Residues in TAP2 peptide transporters controlling substrate specificity. J Immunol. 1996;156:1756–1763. [PubMed] [Google Scholar]
- 19.Nijenhuis M, Schmitt S, Armandola EA, Obst R, Brunner J, Hammerling GJ. Identification of a contact region for peptide on the TAP1 chain of the transporter associated with antigen processing. J Immunol. 1996;156:2186–2195. [PubMed] [Google Scholar]
- 20.Deverson EV, Leong L, Seelig A, Coadwell WJ, Tredgett EM, Butcherand GW, Howard JC. Functional analysis by site-directed mutagenesis of the complex polymorphism in rat transporter associated with antigen processing. J Immunol. 1998;160:2767–2779. [PubMed] [Google Scholar]
- 21.Baldauf C, Schrodt S, Herget M, Kochand J, Tampe R. Single residue within the antigen translocation complex TAP controls the epitope repertoire by stabilizing a receptive conformation. Proc Natl Acad Sci U S A. 2010;107:9135–9140. doi: 10.1073/pnas.1001308107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corradi V, Singhand G, Tieleman DP. The human transporter associated with antigen processing: molecular models to describe peptide binding competent states. J Biol Chem. 2012;287:28099–28111. doi: 10.1074/jbc.M112.381251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 24.Lee JY, Yang JG, Zhitnitsky D, Lewinsonand O, Rees DC. Structural basis for heavy metal detoxification by an Atm1-type ABC exporter. Science. 2014;343:1133–1136. doi: 10.1126/science.1246489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ward A, Reyes CL, Yu J, Rothand CB, Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci U S A. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatschand IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hohl M, Briand C, Grutter MG, Seeger MA. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat Struct Mol Biol. 2012;19:395–402. doi: 10.1038/nsmb.2267. [DOI] [PubMed] [Google Scholar]
- 28.Shintre CA, Pike AC, Li Q, Kim JI, Barr AJ, Goubin S, Shrestha L, Yang J, Berridge G, Ross J, Stansfeld PJ, Sansom MS, Edwards AM, Bountra C, Marsden BD, von Delft F, Bullock AN, Gileadi O, Burgess-Brown NA, Carpenter EP. Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc Natl Acad Sci U S A. 2013;110:9710–9715. doi: 10.1073/pnas.1217042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J, Wu S, Tomasiak TM, Mergel C, Winter MB, Stiller SB, Robles-Colmanares Y, Stroud RM, Tampe R, Craikand CS, Cheng Y. Subnanometre-resolution electron cryomicroscopy structure of a heterodimeric ABC exporter. Nature. 2015;517:396–400. doi: 10.1038/nature13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 31.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ujwal R, Cascio D, Colletier JP, Faham S, Zhang J, Toro L, Ping P, Abramson J. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci U S A. 2008;105:17742–17747. doi: 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Procko E, Ferrin-O’ConnellS I, Ng L, Gaudet R. Distinct structural and functional properties of the ATPase sites in an asymmetric ABC transporter. Mol Cell. 2006;24:51–62. doi: 10.1016/j.molcel.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 34.Lapinski PE, Neubigand RR, Raghavan M. Walker A lysine mutations of TAP1 and TAP2 interfere with peptide translocation but not peptide binding. J Biol Chem. 2001;276:7526–7533. doi: 10.1074/jbc.M009448200. [DOI] [PubMed] [Google Scholar]
- 35.Gorbulev S, Abeleand R, Tampe R. Allosteric crosstalk between peptide-binding, transport, and ATP hydrolysis of the ABC transporter TAP. Proc Natl Acad Sci U S A. 2001;98:3732–3737. doi: 10.1073/pnas.061467898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herget M, Oancea G, Schrodt S, Karas M, Tampe R, Abele R. Mechanism of substrate sensing and signal transmission within an ABC transporter: use of a Trojan horse strategy. J Biol Chem. 2007;282:3871–3880. doi: 10.1074/jbc.M608480200. [DOI] [PubMed] [Google Scholar]
- 37.Oancea G, O’Mara ML, Bennett WF, Tieleman DP, Abele R, Tampe R. Structural arrangement of the transmission interface in the antigen ABC transport complex TAP. Proc Natl Acad Sci U S A. 2009;106:5551–5556. doi: 10.1073/pnas.0811260106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herget M, Baldauf C, Scholz C, Parcej D, Wiesmuller KH, Tampe R, Abele R, Bordignon E. Conformation of peptides bound to the transporter associated with antigen processing (TAP) Proc Natl Acad Sci U S A. 2011;108:1349–1354. doi: 10.1073/pnas.1012355108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Jaimesand KF, Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geng J, Sivaramakrishnanand S, Raghavan M. Analyses of conformational states of the transporter associated with antigen processing (TAP) protein in a native cellular membrane environment. J Biol Chem. 2013;288:37039–37047. doi: 10.1074/jbc.M113.504696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.