

Abstract
Hapalindoles are bioactive indole alkaloids with fascinating polycyclic ring systems whose biosynthetic assembly mechanism has remained unknown since their initial discovery in the 1980s. In this study, we describe the fam gene cluster from the cyanobacterium Fischerella ambigua UTEX 1903 encoding hapalindole and ambiguine biosynthesis along with the characterization of two aromatic prenyltransferases, FamD1 and FamD2, and a previously undescribed cyclase, FamC1. These studies demonstrate that FamD2 and FamC1 act in concert to form the tetracyclic core ring system of the hapalindoles from cis-indole isonitrile and geranyl pyrophosphate through a presumed biosynthetic Cope rearrangement and subsequent 6-exo-trig cyclization/electrophilic aromatic substitution reaction.
Hapalindoles are a large group of structurally fascinating indole alkaloids from the cyanobacterial order Stigonematales with diverse biological activity profiles,1 including antibacterial,2 antimycotic,3,4 insecticidal,5,6 and anticancer properties.7 This family of metabolites is comprised of at least 81 members isolated from over 18 documented cyanobacterial strains,1,8 and is classified into four subgroups. These subgroups include the hapalindoles (tri-/tetracyclic), ambiguines, fischerindoles, and welwitindolinones, as distinguished by their variant polycyclic ring systems (Figure 1A). Because of their noteworthy biological properties, as well as daunting structural features, several groups have initiated research programs devoted to the total synthesis of these molecules.1 However, the challenge of obtaining the highly functionalized ring systems in a regio- and stereospecific manner has impeded these efforts with some notable exceptions.9 In contrast to this large body of work, limited research concerning the biogenesis10–13 of these structurally diverse terpenoids motivated us to initiate biosynthetic studies.
Figure 1.
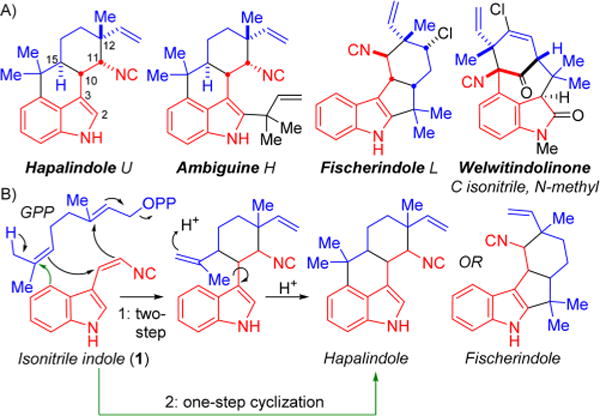
(A) Examples of the four hapalindole alkaloid subgroups with the indole core (red) and monoterpenoid group (blue) highlighted. (B) Current proposed hypotheses for biosynthetic formation of the tri- and tetracyclic cores: Moore proposed a proton-promoted two-step cyclization,15 whereas Carmeli proposed a concerted enzymatic cyclization.18
Although each subgroup has a distinct ring system, all hapalindole compounds share the common features of an indole isonitrile core (1) and a geranyl monoterpene unit (Figure 1B), with many also containing a chlorine substituent. While the biosynthesis of these subunits has been recently identified by Liu and co-workers,10,12 the key fusion step to generate the regio- and stereospecific ring systems from 1 and geranyl pyrophosphate (GPP) remained unknown. Current hypotheses include a proton-promoted two-step polyolefin cyclization to first furnish tricyclic hapalindoles and subsequently tetracyclic hapalindoles and fischerindoles via electrophilic aromatic substitution,14–17 or a one-step enzymatic cyclization to directly form tetracyclic hapalindoles and fischerindoles (Figure 1B).10,18 In this work, we disclose a surprising intermediate generated by the aromatic prenyltransferases and the allied cyclase responsible for forming the tetracyclic core structure of this family, which provides key insights into a new mechanistic proposal.
We initiated our investigation using the hapalindole/ambiguine producing strain Fischerella ambigua UTEX 19033,4,19 as a model. The whole genome was independently sequenced and mined for the ambiguine biosynthetic gene cluster using prenyltransferases as an in silico probe, revealing a 41 kb region encoding 32 proteins that was putatively annotated as the ambiguine biosynthetic gene cluster (fam) (Figure 2A). This sequence is 100% identical to the recently published amb gene cluster10 (Figure 2B), and now includes the complete protein functional annotation for FamC1–4 based on our findings detailed below.
Figure 2.
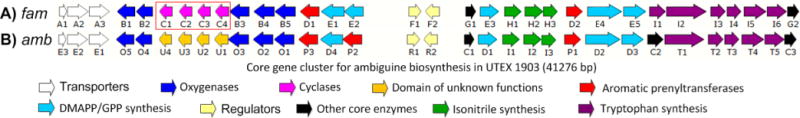
Comparison of the gene cluster from Fischerella ambigua UTEX 1903: (A) fam gene cluster identified in this study; (B) amb gene cluster assigned previously.10
FamD1 and FamD2 align with ABBA aromatic prenyltransferases that catalyze prenyl transfer to aromatic groups.20–22 Thus, the presence of a monoterpene unit and a dimethylallyl group in the ambiguine natural products indicate that FamD1 and FamD2 may play critical roles in fusing 1 with GPP to generate tri- or tetracyclic hapalindoles (Figure 1B), and prenylating tetracyclic hapalindoles with dimethylallyl pyrophosphate (DMAPP) to form the ambiguines (Figure 1A). Hence, we cloned, overexpressed, and purified FamD1 and FamD2 in Escherichia coli as 6× His-tagged proteins to assess their biochemical function in vitro. Contrary to previous reports,10,13 we observed that FamD2 prenylates 1 with GPP to give two distinct products that we have isolated and identified as novel geranylated C-2 and C-3 isocyano-indoles 2 and 3 (Figure 3A; SI Tables 1 and 2). Their production is pH dependent: a slightly acidic pH (6.0) gives C-2 prenylation (2) as the major product, and a high pH (10.5) gives C-3 prenylation (3) as the major product (Figure 3C). Compound 3 rearranges to 2 in the presence of FamD2 at pH 6.0 and 7.8 but not pH 10.5, while 2 did not convert to 3 at any pH tested (SI Figure 1). FamD2 can also accept DMAPP and react with 1 in a pH dependent manner to give dimethylallylated isocyano-indoles 4 and presumably 5 (Figure 3B; SI Table 3), the latter structure being determined by comparing its 1H/COSY NMR and MS to 4 and 3. We also examined the activity of FamD1 toward substrate 1. Surprisingly, FamD1 was able to prenylate 1 with GPP and DMAPP in an analogous manner to FamD2, albeit with much lower efficiency. Compared to DMAPP, FamD1 shows diminished conversion with GPP and produced only 2 (Figure 3D-ii). FamD1 also preferentially accepted DMAPP when incubated with 1:DMAPP:GPP (1:1:1) (SI Figure 2), indicating that DMAPP is the natural substrate for FamD1. We also investigated whether FamD1 or FamD2 could tailor tetracyclic hapalindole U to ambiguine H (SI Figure 3) and confirmed10 that only FamD1 was capable of this transformation. Neither enzyme prenylated hapalindole U with GPP.
Figure 3.
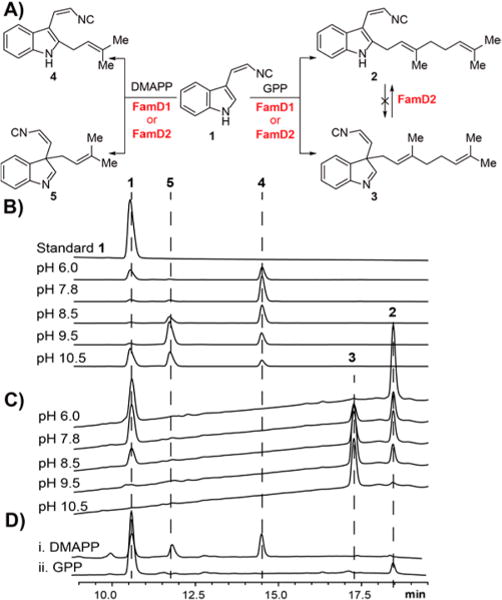
In vitro assay of aromatic prenyltransferases FamD1 and FamD2. (A) The four products generated by FamD1 and FamD2 in reaction with GPP or DMAPP. (B) pH effects of 1 and DMAPP treated with FamD2. (C) pH effects of 1 and GPP treated with FamD2. (D) An assessment of FamD1 reactivity showing production of (i) 4 and 5 from 1 and DMAPP and (ii) 2 from 1 and GPP.
These results show that FamD1 is responsible for processing tetracyclic hapalindoles into ambiguines and that FamD2 prenylates 1 with GPP, although not to the tri- or tetracyclic hapalindoles. Instead, FamD2 catalyzes the first step of a ring-forming cascade, generating an intermediate that we reasoned subsequently serves as substrate for an additional enzyme responsible for the final ring formations. Thus, as the ultimate goal for this investigation, we set out to determine which gene encodes the hapalindole Fam cyclase.
After the characterization of FamD1 and FamD2, the remaining genes that could play a role in the cyclization included five oxygenases (FamB1–5) and four genes previously annotated as unknown proteins (FamC1–4). Additionally, it was possible that the gene encoding the cyclase was located outside of the defined gene cluster. To identify the gene(s) responsible for cyclization, we employed traditional lysate fractionation techniques to isolate and identify the enzyme responsible.
F. ambigua was cultured and filtered to obtain sufficient cell mass, which was disrupted and extracted to give a crude cell-free lysate.23 We tested the lysate activity by reacting it with 1 and GPP, and two new products (6 and 7) were generated (Figure 4, i). We examined the possible relationship between 2/3 and 6/7 (Figure 4, iv–ix) by treating 2 and 3 with cell-free lysate, observing 2 is unreactive (Figure 4, iv–vi), while 3 converts into a mixture of 6 and 7 (Figure 4, vii and viii). Surprisingly, the negative control where the cell-free lysate was boiled for 5 min (Figure 4, ix) showed the formation of 6 only. Boiling the lysate for an extended period (15 min) showed a decreased conversion to 6 (SI Figure 4). These results suggested that 2 is a shunt metabolite, while 3 is an intermediate between starting materials 1 and GPP on the biosynthetic pathway to 6 and 7, and a thermostable protein in the boiled supernatant cyclizes 3 to 6. The protein responsible for forming 7 from 3 is present in the cell-free lysate but is thermolabile. A large scale reaction enabled us to identify 6 as a previously unreported tetracycle, 12-epi-hapalindole U (SI Table 4); our efforts to identify 7 are ongoing.
Figure 4.
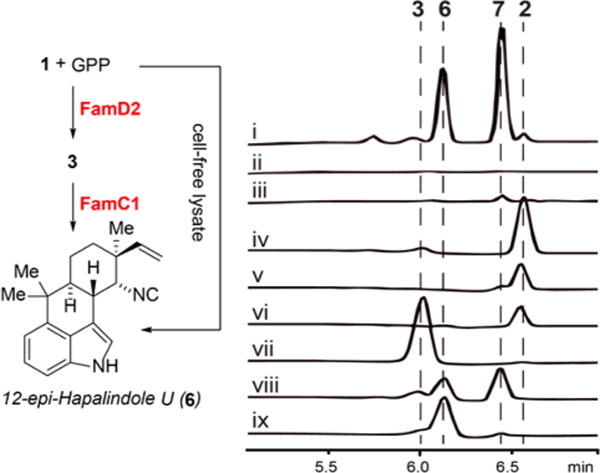
In vitro assays with cell-free lysate: (i) 1 + GPP + cell-free lysate; (ii) 1 + GPP + boiled cell-free lysate; (iii) blank with only cell-free lysate; (iv) standard of 2; (v) 2 + cell-free lysate; (vi) 2 + boiled cell-free lysate; (vii) standard of 3; (viii) 3 + cell-free lysate; (ix) 3 + boiled cell-free lysate.
We examined the composition of the cell-free lysate and boiled supernatant by protein gel electrophoresis (SI Figure 5) and subsequently purified the active cyclase directly from the supernatant after finding that it contained relatively few proteins. Thus, the lysate was boiled for 5 min prior to fractionation on an anion exchange column, and the resulting fractions were tested for activity with 3. The HPLC traces revealed that two fractions retained the ability to effectively cyclize 3 to 6 (SI Figure 6). Protein gel analysis of the active fractions showed one major band with a second minor component (SI Figure 7). These bands were excised and analyzed at the University of Michigan Proteomics & Peptide Synthesis Core, with the active protein being conclusively identified as FamC1 (Figure 5; SI Figure 8). BLAST analysis of the three additional previously unknown proteins in the fam gene cluster against FamC1 showed high sequence identities of 63%, 63%, and 73%, respectively, thus leading us to propose that the encoding genes represent a new class of cyclases (famC1–4) (SI Figure 9).
Figure 5.

FamC1 protein identification. The highlighted residues were identified as contiguous peptide segments encoded exclusively by the famC1 gene of F. ambigua UTEX 1903.
The results above indicate that an unexpected biosynthetic mechanism is mediating formation of the core common to all hapalindoles, ambiguines, fischerindoles, and welwitindolinones, in contrast to previous proposals (Figure 1B).10,14–18 Our data suggest the mechanism begins with the FamD2 catalyzed prenylation of GPP to the C3 position of 1 (Figure 6), generating a quaternary product (3) that can rearomatize to form C2 prenylated shunt metabolite 2 in the presence of FamD2 at neutral or low pH (SI Figure 1).24–26 To continue along the biosynthetic pathway, prenylated indole 3 must undergo a Cope rearrangement24,25,27 catalyzed by FamC1 to achieve alkene intermediate 8. The stereochemistry of the C11 and C12 centers would be set during this [3,3]-sigmatropic rearrangement, with the 12-epi-hapalindoles being directed through a chair-like transition state by FamC1, and the normal hapalindoles likely the result of another cyclase. FamC1 subsequently catalyzes the formation of the tetracyclic hapalindole core (6), setting the stereochemistry of the C10 and C15 positions in either a concerted (8 to 6) or stepwise manner (8 to 9 to 6).
Figure 6.
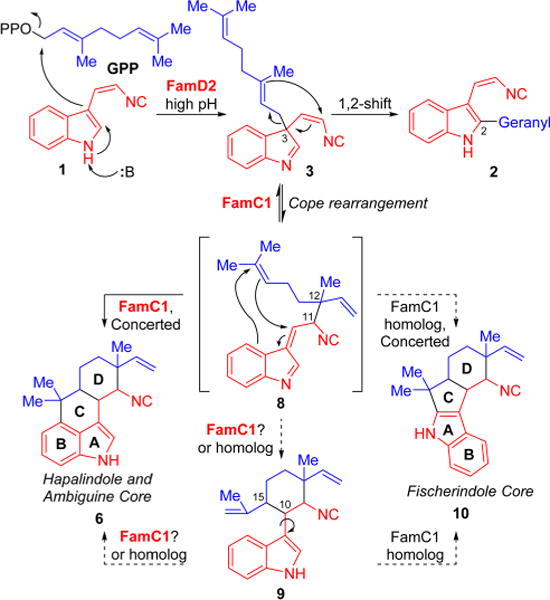
Proposed mechanism for the origin of hapalindole and fischerindole core ring systems.
The modular mechanism where rearomatization first generates tricyclic hapalindoles (9) from 8 and electrophilic aromatic substitution forms 6 as a separate step is particularly intriguing as it retains the possibility for 9 to re-enter the catalytic cycle and diverge to either hapalindoles (6) or fischerindoles (10) as controlled by the directing effect of pathway-specific cyclase enzymes. Further study is necessary to determine if 9 is indeed an active intermediate on the pathway to 6 or 10, as was previously proposed15 and effectively utilized in total syntheses of the tetracyclic hapalindole and ambiguine natural products,9 or a shunt metabolite that is unable to undergo further cyclization.
We identified the functions of aromatic prenyltransferases FamD1 and FamD2, discovering a previously unreported ability to prenylate indole isonitrile 1 with either GPP or DMAPP to give C-2 prenylation (2 and 4) and C-3 prenylation (3 and 5). We also confirmed previous reports10 that FamD1 prenylates tetracyclic hapalindoles with DMAPP into ambiguines while FamD2 does not. The hypothesis that 3 could act as precursor for tri- or tetracyclic hapalindoles led to our identification of the FamC1 cyclase from the cell-free lysate based on its ability to convert 3 into tetracyclic 12-epi-hapalindole U (6). Thus, we propose that the core ring formation is the combined result of a prenyltransferase (FamD2) generating intermediate 3 and a cyclase (FamC1) completing the ring fusion to generate tetracyclic hapalindoles, which represents the first biocatalytic demonstration of such a process.
The studies described herein support a novel mechanism involving the union of indole isonitrile 1 and GPP to generate key biosynthetic intermediate 3, and a subsequent Cope rearrangement and ring-cyclization cascade to generate the tetracyclic hapalindoles. It is especially significant that the current mechanistic hypothesis constitutes perhaps one of the only biosynthetic Cope rearrangements. The rarity of pericyclic reactions in biosynthetic transformations has been the subject of much recent debate,24 and the current mechanistic hypothesis is consistent with all of the available experimental data. The underlying molecular basis for the biocatalytic conversion of 3 to 9 via the proposed [3,3]-sigmatropic rearrangement and subsequent 6-exo-trig ring closure/electrophilic aromatic substitution constitutes a provocative starting point for further studies in this intriguing arena. Moreover, the discovery of 12-epi-hapalindole U (6) indicates a larger metabolite scope produced by UTEX 1903 than previously recognized and suggests that our work characterizing the other famC genes in this biosynthetic pathway will facilitate access to the previously isolated hapalindole natural products and additional structural diversity within this class of secondary metabolites.
Supplementary Material
Acknowledgments
The authors thank the University of Michigan Proteomics & Peptide Synthesis Core for assistance with protein determination. We are grateful to the National Institutes of Health for financial support (CA70375 to R.M.W. and D.H.S.) as well as the National Science Foundation under the CCI Center for Selective C–H Functionalization (CHE-1205646) and the Hans W. Vahlteich Professorship (to D.H.S.). We are grateful to Prof. Phil S. Baran and Prof. Jimmy Orjala for kind gifts of authentic hapalindoles.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b10136.
Full experimental details, NMR data, protein gels, and additional LCMS and HPLC traces (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Bhat V, Dave A, MacKay JA, Rawal VH. In: The Alkaloids: Chemistry and Biology. Hans-Joachim K, editor. Vol. 73. Academic Press; New York: 2014. p. 65. [DOI] [PubMed] [Google Scholar]
- 2.Asthana RK, Srivastava A, Singh AP, Deepali, Singh SP, Nath G, Srivastava R, Srivastava B. J Appl Phycol. 2006;18:33. [Google Scholar]
- 3.Mo S, Krunic A, Santarsiero BD, Franzblau SG, Orjala J. Phytochemistry. 2010;71:2116. doi: 10.1016/j.phytochem.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mo S, Krunic A, Chlipala G, Orjala J. J Nat Prod. 2009;72:894. doi: 10.1021/np800751j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becher PG, Keller S, Jung G, Süssmuth RD, Jüttner F. Phytochemistry. 2007;68:2493. doi: 10.1016/j.phytochem.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 6.Cagide E, Becher PG, Louzao MC, Espiña B, Vieytes MR, Jüttner F, Botana LM. Chem Res Toxicol. 2014;27:1696. doi: 10.1021/tx500188a. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Smith CD. Mol Pharmacol. 1996;49:288. [PubMed] [Google Scholar]
- 8.Walton K, Gantar M, Gibbs PDL, Schmale MC, Berry JP. Toxins. 2014;6:3568. doi: 10.3390/toxins6123568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.References for the accomplished total synthesis of the hapalindole alkaloids are listed in the Supporting Information.
- 10.Hillwig ML, Zhu Q, Liu X. ACS Chem Biol. 2014;9:372. doi: 10.1021/cb400681n. [DOI] [PubMed] [Google Scholar]
- 11.Hillwig ML, Fuhrman HA, Ittiamornkul K, Sevco TJ, Kwak DH, Liu X. ChemBioChem. 2014;15:665. doi: 10.1002/cbic.201300794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hillwig ML, Liu X. Nat Chem Biol. 2014;10:921. doi: 10.1038/nchembio.1625. [DOI] [PubMed] [Google Scholar]
- 13.Micallef ML, Sharma D, Bunn BM, Gerwick L, Viswanathan R, Moffitt MC. BMC Microbiol. 2014;14:213. doi: 10.1186/s12866-014-0213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park A, Moore RE, Patterson GML. Tetrahedron Lett. 1992;33:3257. [Google Scholar]
- 15.Stratmann K, Moore RE, Bonjouklian R, Deeter JB, Patterson GML, Shaffer S, Smith CD, Smitka TA. J Am Chem Soc. 1994;116:9935. [Google Scholar]
- 16.Richter JM, Ishihara Y, Masuda T, Whitefield BW, Llamas T, Pohjakallio A, Baran PS. J Am Chem Soc. 2008;130:17938. doi: 10.1021/ja806981k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maimone TJ, Ishihara Y, Baran PS. Tetrahedron. 2015;71:3652. doi: 10.1016/j.tet.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raveh A, Carmeli S. J Nat Prod. 2007;70:196. doi: 10.1021/np060495r. [DOI] [PubMed] [Google Scholar]
- 19.Smitka TA, Bonjouklian R, Doolin L, Jones ND, Deeter JB, Yoshida WY, Prinsep MR, Moore RE, Patterson GML. J Org Chem. 1992;57:857. [Google Scholar]
- 20.Bonitz T, Alva V, Saleh O, Lupas AN, Heide L. PLoS One. 2011;6:e27336. doi: 10.1371/journal.pone.0027336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steffan N, Grundmann A, Yin WB, Kremer A, Li SM. Curr Med Chem. 2009;16:218. doi: 10.2174/092986709787002772. [DOI] [PubMed] [Google Scholar]
- 22.Ding Y, Williams RM, Sherman DH. J Biol Chem. 2008;283:16068. doi: 10.1074/jbc.M801991200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ivleva NB, Golden SS. Methods Mol Biol. 2007;362:365. doi: 10.1007/978-1-59745-257-1_26. [DOI] [PubMed] [Google Scholar]
- 24.Tanner ME. Nat Prod Rep. 2015;32:88. doi: 10.1039/c4np00099d. [DOI] [PubMed] [Google Scholar]
- 25.Mahmoodi N, Tanner ME. Chem Bio Chem. 2013;14:2029. doi: 10.1002/cbic.201300385. [DOI] [PubMed] [Google Scholar]
- 26.Caballero E, Avendano C, Menendez JC. J Org Chem. 2003;68:6944. doi: 10.1021/jo034703l. [DOI] [PubMed] [Google Scholar]
- 27.Luk LY, Qian Q, Tanner ME. J Am Chem Soc. 2011;133:12342. doi: 10.1021/ja2034969. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.