

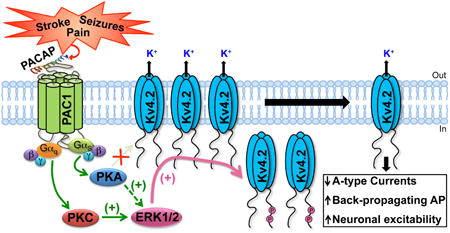
Abstract
The endogenous neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) is secreted by both neuronal and non-neuronal cells in the brain and spinal cord, in response to pathological conditions such as stroke, seizures, chronic inflammatory and neuropathic pain. PACAP has been shown to exert various neuromodulatory and neuroprotective effects. However, direct influence of PACAP on the function of intrinsically excitable ion channels that are critical to both hyperexcitation as well as cell death, remain largely unexplored. The major dendritic K+ channel Kv4.2 is a critical regulator of neuronal excitability, back-propagating action potentials in the dendrites, and modulation of synaptic inputs. We identified, cloned and characterized the downstream signaling originating from the activation of three PACAP receptor (PAC1) isoforms that are expressed in rodent hippocampal neurons that also exhibit abundant expression of Kv4.2 protein. Activation of PAC1 by PACAP leads to phosphorylation of Kv4.2 and downregulation of channel currents, which can be attenuated by inhibition of either PKA or ERK1/2 activity. Mechanistically, this dynamic downregulation of Kv4.2 function is a consequence of reduction in the density of surface channels, without any influence on the voltage-dependence of channel activation. Interestingly, PKA-induced effects on Kv4.2 were mediated by ERK1/2 phosphorylation of the channel at two critical residues, but not by direct channel phosphorylation by PKA, suggesting a convergent phosphomodulatory signaling cascade. Altogether, our findings suggest a novel GPCR-channel signaling crosstalk between PACAP/PAC1 and Kv4.2 channel in a manner that could lead to neuronal hyperexcitability.
Keywords: Pituitary adenylate cyclase-activating peptide (PACAP), G protein-coupled receptor (GPCR), Extracellular-signal-regulated kinase (ERK), Protein kinase A (PKA), Potassium channel, Kv4.2, Ion channel modulation
Graphical abstract
1. Introduction
Pituitary adenylate cyclase-activating polypeptide (PACAP), first isolated as a hypothalamic hormone (Miyata et al., 1989), is known to exert neurotrophic and neuromodulatory effects. It exists in two biologically active forms: as a 38-amino acid peptide PACAP38 or its post-translationally truncated N-terminal form, PACAP27 (Miyata et al., 1990). PACAP is expressed in brain regions including the thalamus, hypothalamus, amygdala, cortex, cerebellum and hippocampus, where PACAP38 is the more prevalent of the two forms (Koves et al., 1991, Arimura et al., 1994, Arimura, 1998). In the brain, PACAP exerts its biological effects by binding to its specific receptor PAC1 (gene name – Adcyap1r1) and activating diverse intracellular signaling (Spengler et al., 1993, Blechman and Levkowitz, 2013). PACAP38-mediated PKA activation is reported to modulate synaptic plasticity by phosphorylation of the NMDA-type ionotropic glutamate receptors (GluNs) that enhances channel activity in sympathetic preganglionic, cortical and hippocampal neurons (Wu and Dun, 1997, Yaka et al., 2003). Furthermore, PACAP38 has been shown to alter dendritic spine morphology by inducing shrinkage of spine heads in hippocampal neurons (Gardoni et al., 2012). Neuronal excitability in hippocampal CA1 neurons was found to be elevated upon PACAP38 application (Di Mauro et al., 2003), although the underlying mechanism is not understood. The mRNA and protein levels of PACAP are reported to be elevated following hyperexcitable insults such as cerebral ischemia and kainate-induced seizures in rats (Nomura et al., 2000, Chen et al., 2006b, Stumm et al., 2007). Such insults lead to an increase in the intrinsic excitability of neurons (Meier et al., 1992, Schiene et al., 1996, Fan et al., 2008); however, the precise molecular mechanisms governing such alterations in neuronal excitability are poorly understood.
Voltage-gated K+ (Kv) channels are critical regulators of the amplitude, frequency and duration of action potentials (APs), and thereby control neuronal excitability (Birnbaum et al., 2004, Johnston et al., 2010). Different Kv channels with specific biophysical properties are localized in different sub-cellular compartments of mammalian brain neurons, in order to perform these functions (Lai and Jan, 2006, Vacher et al., 2008). The Kv4.2 channel constitutes the major dendritic Kv channel with characteristic rapidly activating and inactivating A-type currents (IA) in mammalian brain neurons (Hoffman et al., 1997). These channels are critical in controlling the threshold and magnitude of neuronal membrane excitability as they limit back-propagating APs in the dendrites (Hoffman et al., 1997, Johnston et al., 2000, Yuan et al., 2002, Chen et al., 2006a). Accordingly, up-regulation of Kv4.2-mediated IA provides a neuroprotective mechanism for ischemia-induced excitotoxicity and neuronal damage (Deng et al., 2011), and downregulation of Kv4.2 currents lead to increased excitability of neurons in hyperexcitable and chronic pain conditions (Hu et al., 2006, Barnwell et al., 2009). Kv4.2 expression and localization is altered upon seizure induction in rats and elimination of Kv4.2 channels lowers the seizure threshold in knockout (Kcnd2-/-) mice (Tsaur et al., 1992, Lugo et al., 2008, Barnwell et al., 2009). In addition, Kv4.2 is enriched in dendritic spines where they function as integrators of synaptic information by altering NMDA receptor activation and subunit composition (Kim et al., 2007, Jung et al., 2008). Most of the modifications in Kv4.2 channel localization and function have been attributable to phosphorylation of the channel protein by several protein kinases (Hoffman and Johnston, 1998, Schrader et al., 2002, Schrader et al., 2006, Hammond et al., 2008, Schrader et al., 2009, Lin et al., 2011).
Since hyperexcitable conditions such as ischemic stroke, seizures and chronic pain lead to increases in local PACAP levels in the region of assault, as well as altered neuronal excitability, we hypothesized that PACAP could presumably influence Kv4.2 channel function. We found that multiple PAC1 isoforms are expressed in rodent brain neurons, and their activation by PACAP leads to elevation of intracellular cAMP levels and downstream activation of PKA and ERK1/2. Furthermore, distinct PKA, PKC, Ras and MEK1/2 signaling, as well as arrestin-2 signaling downstream of PAC1-Null or Hop1 or Hop2 isoform activation by PACAP converge on ERK1/2 activation. Subsequently, phosphorylation of Kv4.2 by ERK1/2 resulted in a decrease in the number of channels on the cell surface, leading to dynamic down-regulation of channel function. These results suggest a direct signaling crosstalk between PACAP and the major dendritic Kv channel in neurons, which could profoundly influence neuronal excitability.
2. Materials and Methods
2.1. Animals and ethics
All experiments involving the use of rats and mice were approved by the University of Iowa Institutional Animal Care and Use Committee, as well as by the Washington University Institutional Animal Care and Use Committee, and in strict accordance with the Guide for the Care and Use of Laboratory Animals described by the National Institutes of Health. Every effort was made to minimize the number of rats and mice used and their suffering.
2.2. Materials
Recombinant PACAP27 and PACAP38 were purchased from Bachem; PKA inhibitor KT5720 ((9R,10S,12S)-2,3,9,10,11,12-Hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, hexyl ester),PKC inhibitor BIM-I (2-[1-(3-Dimethylaminopropyl)indol-3-yl]-3-(indol-3-yl) maleimide), MEK1/2 inhibitor U0126 (1,4-Diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene), and PKA activator forskolin ([3R-(3α,4aβ,5β,6β,6aα,10α,10aβ,10bα)]-5-(Acetyloxy)-3-ethenyldodecahydro-6,10,10b-trihydroxy-3,4a,7,7,10a-pentamethyl-1H-naphtho[2,1-b]pyran-1-one) were purchased from R&D Systems – Tocris Bioscience; Hank's Balanced Salt Solution (HBSS), B27, Dulbecco's Modified Eagles Medium (DMEM), Trypsin-EDTA (0.05%), ProLong Gold, TRIzol, Alexa fluor-conjugated secondary antibodies, GlutaMAX, penicillin/streptomycin, Lipofectamine2000, Neurobasal media, and TOPO-TA cloning kit were purchased from Life Technologies. Site-directed mutagenesis kit was purchased from Agilent Technologies. All other chemicals, antibodies and reagents used in this study were purchased from Sigma, Bio-Rad, Roche Applied Science, PerkinElmer, Merck-Millipore, Cell Signaling Technologies, BD Biosciences, LI-COR Biosciences, VWR and Thermo-Fisher Scientific. All NeuroMab antibodies were purchased from the University of California at Davis and National Institutes of Health NeuroMab Facility through Antibodies Inc. 32P-labeled orthophosphate was obtained from Perkin-Elmer.
2.3. Primary culture of rat hippocampal neurons
Hippocampi from Sprague-Dawley rat embryos (E18 stage) were isolated and cultured according to the previously described protocols (Mohapatra et al., 2008, Shepherd et al., 2012). Cells were cultured with NeuroBasal medium containing B27 supplement, on poly-L-lysine coated petri dishes at a density of 100,000 cells per 35 mm dish for biochemical and cAMP ELISA experiments, and on poly-L-lysine coated 12 mm2 round glass coverslips at a density of 60,000 cells per 35 mm dish carrying 5 coverslips for electrophysiological and immunocytochemical experiments. For on-cell fluorescence immunocytochemical experiments neurons were plated at a density of 100,000 cells per well in 24-well plates that were coated with poly-L-lysine. Neurons were cultured at 37°C containing 5% CO2, with replacement of half of the media on a weekly basis. Immunocytochemical, biochemical and cAMP ELISA experiments were performed on neurons cultured for 14-16 days in vitro (DIV), and electrophysiological experiments were performed on neurons at 9-12 DIV.
2.4. Immunostaining of rodent brain sections and cultured rat hippocampal neurons
Whole brain sections (40 μm-thick, sagittal) from adult rats and mice were prepared as previously described (Shepherd et al., 2012). Fixed brain sections were blocked and permeabilized using 10% goat serum and 0.3% Triton X-100 in 0.1 M phosphate buffer (PB). Sections were then incubated with anti-Kv4.2 mouse monoclonal (1:500; clone K57/1; NeuroMab) and anti-PAC1 rabbit polyclonal (1:500; Thermo Fisher Scientific) antibodies at 4°C overnight. These antibodies have previously been characterized and validated for their specificity (Schulz et al., 2004, Menegola and Trimmer, 2006). After washing, sections were incubated with AlexaFluor 488-conjugated goat anti-mouse IgG and AlexaFluor 555-conjugated goat anti-rabbit IgG secondary antibodies (1:1000, Life Technologies), along with the nuclear dye DAPI (1:5000) for 3 h at 4°C with gentle agitation. Sections were then mounted onto glass slides under coverslips using ProLong Gold anti-fade mounting media (Life Technologies) and imaged using the LAS-X laser-scanning confocal imaging system (Leica) mounted on a TCS SPE DMI 4000B microscope equipped with 10× [numerical aperture (NA) 0.3] and 63× (NA 1.3) Apochromat objectives (Leica). Separate sets of brain sections were stained with the same combination of AlexaFluor conjugated secondary antibodies and DAPI, without any prior incubation with anti-Kv4.2 and anti-PAC1 antibodies, to determine the background immunofluorescence levels of brain sections. Multiple sections from multiple mouse/rat brains were immunostained and analyzed for Kv4.2 and PAC1 expression.
Immunostaining of cultured rat hippocampal neurons (CHNs; 14-16 DIV) grown on glass coverslips were performed as described previously (Mohapatra and Trimmer, 2006, Shepherd et al., 2012). Neurons were stained with anti-Kv4.2 mouse monoclonal (1:1000; clone K57/1; subtype-IgG1; NeuroMab), anti-PAC1 rabbit polyclonal (1:500; Thermo Fisher Scientific), and anti-glial fibrillary acidic protein (GFAP) mouse monoclonal (1:1000; clone 1B4; subtype-IgG2b; BD Biosciences) antibodies for 1 h at room temperature with gentle agitation. Secondary antibodies used for these staining were, AlexaFluor 555-conjugated goat anti-mouse IgG1, AlexaFluor 633-conjugated goat anti-mouse IgG2b, and AlexaFluor 488-conjugated goat anti-rabbit IgG secondary antibodies (1:2000, Life Technologies), along with the nuclear dye DAPI (1:5000) for 1 h at room temperature. After final washing, coverslips were mounted onto glass slides using ProLong Gold anti-fade mounting media (Life Technologies). Images were taken using an MRc-5 digital camera connected to a Zeiss AxioImager epifluorescence microscope, using the AxioVision software (Carl Zeiss), with a 63× Plan-Apochromat objective (NA 1.4; Carl Zeiss). Images were then transferred as.TIFF files to prepare overlapping images for each antibody staining in Photoshop software (Adobe Systems).
2.5. Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from hippocampus, cortex and cerebellum of adult mice and rats, as well as from cultured rat hippocampal neurons (CHNs) using TRIzol reagent (Life Technologies). Obtained RNA was reverse transcribed into cDNA using SuperScript III RT-PCR kit (Life Technologies) as per manufacturer's instructions. Primer set [5′-ATGAATGACAGCACAGCTC-3′ (forward) and 5′-TCAGGTGGCCAAGTTGTCG-3′ (reverse)] spanning the PAC1 variable region in the third intracellular loop, based on known mouse and rat PAC1 sequences (NCBI reference sequences: NM_007407.4 and NM_133511.2, respectively), was used. Identification of various PAC1 isoforms expressed in mouse and rat brain regions was done based on differences in the isoform-specific molecular weights. PCR products of PAC1-Null, PAC1-Hop1 and PAC1-Hop2 isoforms were confirmed by sequencing. Subsequently, the primer set 5′-ATGGCCAGAACCCTGC-3″ (forward) and 5′-TCAGGTGGCCAAGTTGTCG-3′ (reverse), based on known mouse PAC1 sequence (NCBI reference sequences: NM_007407.4) was utilized for the amplification of full-length PAC1 receptor isoforms from mouse hippocampus cDNA. Individual full-length PAC1 isoforms were cloned in TOPO-TA cloning vector, and subsequently sub-cloned in pYFP-N1 plasmid (Clontech) for expressing mPAC1-YFP protein.
2.6. On-cell fluorescence immunocytochemical assay on cultured hippocampal neurons
CHNs were subjected to on-cell Western assay for quantitatively determining the changes in the surface expression of neuronal Kv4.2 channels, as described previously (Lin et al., 2011). After specific drug treatments for different durations, neurons on 24-well plates were fixed with 4% PFA, blocked with Odyssey IR dye blocking buffer (LI-COR Biosciences), and then incubated with anti-Kv4.2 mouse monoclonal antibody (1:200; clone K57/1; NeuroMab) overnight at 4°C. After washing 3 times with the blocking solution cells were incubated with anti-mouse IgG conjugated with IR Dye-800 (1:5000; LI-COR Biosciences) for 1 h at room temperature. Cells were then washed and permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature, followed by addition of anti-MAP2 rabbit polyclonal antibody (1:1000; Sigma) in blocking buffer and further incubation for 1 h at room temperature. After washing, cells were incubated with anti-rabbit IgG conjugated with IR Dye-680 (1:5000; LI-COR Biosciences) for 1 h at room temperature. After final washing with PBS for 3 times, the relative infrared signal intensities at 700 and 800 nm were captured for each well using an Odyssey infrared imaging system (LI-COR Biosciences), and quantified using NIH ImageJ software. Surface Kv4.2 signal intensities were normalized to the respective MAP2 signal intensities for each well, and the relative levels of cell surface Kv4.2 under all drug treatment conditions were compared to untreated control conditions.
2.7. Site-directed mutagenesis, and culture and transfection of HEK293T cells
Recombinant rat Kv4.2 in pRBG4 vector (generously provided by Dr. James Trimmer) was used as a template to generate mutants using the QuickChange site-directed mutagenesis kit (Agilent Technologies) as per manufacturer's instructions. Alanine-substitution mutations were generated at PKA phosphorylation site (Kv4.2-S552A) and at ERK1/2 phosphorylation sites (Kv4.2-T602A, Kv4.2-T607A, Kv4.2-S616A, and Kv4.2-T602/607A/S616A triple mutant), as per previous identification and characterization (Adams et al., 2000, Anderson et al., 2000, Schrader et al., 2006, Hammond et al., 2008, Schrader et al., 2009, Seikel and Trimmer, 2009). Human embryonic kidney cells stably expressing T-antigen (HEK293T; ATCC) were cultured in DMEM containing GlutaMax, 10% fetal bovine serum (FBS, HyClone) and penicillin/streptomycin, in a humidified incubator at 37°C with 5% CO2. Cells were transiently co-transfected with plasmids containing wild-type (WT) or mutant Kv4.2 cDNAs, KChIP2 cDNA (generously provided by Dr. James Trimmer) and mPAC1-Null-YFP-N1 or mPAC1-Hop1-YFP-N1 or mPAC1-Hop2-YFP-N1 cDNAs (cloned from mouse hippocampus) in HEK293T cells using Lipofectamine2000 reagent, according to manufacturer's protocol. To determine the effect of Ras activation on PAC1-induced ERK1/2 phosphorylation, HEK293T cells were co-transfected with pcDNA3 plasmid containing the dominant negative form of Ras (N17-Ras or DN-Ras), along with plasmids containing each of the above-mentioned mPAC1 isoforms. In order to verify the role of arrestin signaling in PAC1-mediated ERK1/2 activation, HEK293T cells stably expressing Arrestin-2 (also referred in the literature as β-arrestin-1) siRNA or Arrestin-3 (also referred in the literature as β-arrestin-2) siRNA were used. Generation and characterization of these stable cell lines are described previously (Zhang et al., 2012). Efficacy of specific siRNA-mediated knockdown of Arrestin-2 and Arrestin-3 in these stable cells lines was verified using previously designed specific primer sets and quantitative RT-PCR protocols (Zhang et al., 2012). These stable cells were transiently transfected with plasmids containing either mPAC1-Null-YFP-N1 or mPAC1-Hop1-YFP-N1 or mPAC1-Hop2-YFP-N1 cDNAs. All biochemical and electrophysiological experiments were performed 42-48 h post-transfection.
2.8. Assay for intracellular cAMP production
Intracellular cAMP production in CHNs and transfected HEK293T cells was quantified using a cAMP ELISA kit (Cell Signaling). Following treatment (20 min at 37°C) with forskolin (10 μM), PACAP27 or PACAP38 (10 or 100 nM each) or untreated/control cells were lysed and cAMP concentrations were determined according to the manufacturer's protocol. Total protein concentrations were obtained using BCA assay (Pierce). Intracellular cAMP levels were expressed as femtomoles of cAMP per μg of total protein. Experiments were repeated in 4 or more cultures of hippocampal neurons and HEK293T cells.
2.9. Immunoblotting
Lysates were prepared from CHNs and transfected HEK293T cells (with or without drug treatments) and size-fractionated on 10% SDS-PAGE gels as described previously (Mohapatra and Trimmer, 2006, Shepherd et al., 2012). Separated proteins were then transferred to a nitrocellulose membrane and blocked using 4% non-fat milk powder in Tris-buffered saline (TBS) or 5% BSA for phosphorylated proteins. Membranes were probed using anti-mouse phosphorylated ERK1/2 at T202/Y204 (p-ERK1/2, 1:1000; Cell Signaling) and anti-rabbit total ERK1/2 (t-ERK1/2, 1:1000; Cell Signaling) primary antibodies, and subsequently incubated with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit IgG secondary antibodies (1:10,000; Antibodies Inc.). After washing three times with TBS, immunoreactive bands were developed with enhanced chemiluminescence reagent (PerkinElmer), onto X-ray films (Kodak Biomax). Experiments were repeated in 3 or more batches of CHNs and transfected HEK293T cells.
2.10. Metabolic 32P-labeling
Metabolic labeling with 32P was performed as previously described (Merrill et al., 2013). DIV 12-13 CHNs on culture dishes were washed and incubated in medium containing phosphate-free RPMI (MP Biomedical), 1% dialyzed FBS and 0.5 mCi·mL−1 32P-labeled orthophosphate (PerkinElmer) for 4 h at 37°C. During the last 20 m in of this incubation, neurons were treated with vehicle, 100 nM PACAP38 or 10 μM forskolin. Cells were then washed, lysed and immunoprecipitated with either anti-Kv4.2 antibody (5 μg per reaction; clone K57/1; NeuroMab) or anti-protein phosphatase 2A (PP2A) antibody (5 μg per reaction; Millipore) in the presence of phosphatase inhibitors. Lysates were analyzed by phosphorimaging and immunoblotting for total Kv4.2 (1:1000; clone K57/1; NeuroMab), and the signal intensities quantified using the NIH Image J software. Experiments were repeated in CHNs from 4 different culture batches.
2.11. Electrophysiology and data analyses
Outward K+ currents were recorded from CHNs and transfected HEK293T cells using the whole-cell voltage-clamp mode of the patch-clamp technique at room temperature, as described previously(Mohapatra et al., 2009, Shepherd et al., 2012). Patch pipettes were pulled from borosilicate glass tubings (TW150F-4; World Precision Instruments) using a PC-10 puller (Narishige). Pipettes were fire polished using a microforge (World Precision Instruments) to obtain a pipette resistance of 2-5 MΩ when filled with internal solution containing (in mM): 140 KCI, 5 NaCI, 1 CaCI2, 1 MgCI2, 10 HEPES, 10 EGTA, and 0.4 Na-ATP, pH 7.3. The extracellular buffer contained (in mM): 140 NaCI, 5 KCI, 2 CaCI2, 1 MgCI2, 10 HEPES, and 10 glucose, pH 7.4. For CHN recordings, 0.5 μM tetrodotoxin was added to the external solution to block fast-activating Na+ currents, and 10 μM MK801 was added to block NMDAR-mediated currents. We observed NMDAR-like EPSCs in cultured rat hippocampal neurons while recording neuronal A-type K+ (IA) currents during our preliminary experiments, presumably due to significant spontaneous glutamate release and neuronal activity in 9-12 DIV cultures. Since the isolation of IA currents were done digitally from total neuronal outward K+ currents (IK), these NMDAR-mediated currents interfered with accurate isolation of IA currents, due to which MK801 was applied in the extracellular buffers in all experiments on cultured hippocampal neurons. Cells were treated with 100 nM PACAP38 in external solution for 20 mins at 37°C or pre/co-treated for 10 min with 1 μM KT5720 or 1 μM U0126 before addition of PACAP38 for 15 min. Currents were recorded using a Digidata 1440A data acquisition system (Molecular Devices) and Axopatch 200B amplifier, with a sampling rate of 5 kHz and filtering at 1 kHz using a digital Bessel filter. Currents were compensated from whole-cell capacitance and series resistance (60-70%), and leak-subtractions were performed off-line. pCLAMP 10 software (Molecular Devices) was used for data acquisition and Clampfit 10 (Molecular devices), Origin 7 (Microcal Software), Excel (Microsoft) and Prism6 (GraphPad Software) were used for data analysis and preparing traces/figures.
For isolation of neuronal IA currents, total IK currents were first obtained with step-depolarization from -100 mV to +80 mV for 500 ms in +10 mV increments, from a holding potential of -100 mV. Subsequently, delayed rectifier K+ (IDR) currents were obtained by applying a -30 mV pre-pulse for 500 ms before step-depolarization from -100 mV to +80 mV for 500 ms in +10 mV increments, in order to inactivate IA. Isolation of IA was then done by digitally subtracting IDR from IK, as described previously (Norris and Nerbonne, 2010). For voltage-dependent activation of Kv4.2 currents in HEK293T cells, cells were held at -100 mV followed by step-depolarization from -100 to +80 mV for 500 ms with +10 mV increments. Current densities were calculated by dividing peak IA (CHNs) or Kv4.2 (in HEK293T cells) currents at each voltage step with the original cell capacitance. Conductance (G) was determined using the formula G = I/(V-EK) where I is the peak current, V is the membrane potential and EK is the reversal potential of K+ (-85 mV). Conductance-voltage curves were obtained by fitting normalized conductance (G/Gmax) with the Boltzmann equation, and half-maximal conductances (G1/2) were determined.
2.12. Statistical analysis
One-way ANOVA with multiple comparison post-hoc tests, and unpaired Student's t-test (Microsoft Excel and GraphPad Prism6) were used for testing the significance of data obtained in control vs various drug treatment groups. p<0.05 was considered statistically significant.
3. Results
3.1. PAC1 isoform expression in rodent brain and activation of downstream signaling
Expression of the PACAP receptor, PAC1, in rodent brain has previously been reported utilizing in-situ hybridization and immunohistochemical staining (Spengler et al., 1993, Joo et al., 2004). The PAC1 mRNA in vertebrates has been shown to undergo extensive alternative splicing that can affect ligand binding, G-protein coupled signaling and receptor heteromerization (Spengler et al., 1993, Chatterjee et al., 1996, Pantaloni et al., 1996, Ushiyama et al., 2007). We chose to perform an unbiased analysis of the expression of PAC1 in rat and mouse brain. Our RT-PCR analysis with specific primers targeting the variable region of PAC1 identified splice variants corresponding to two different molecular weights (Fig. 1A). These two bands were observed in the cortex, hippocampus and cerebellum of both rat and mouse, as well as in cultured rat hippocampal neurons (CHNs). Upon sequencing of these PCR products/bands we confirmed that the lower band was PAC-1-Null and the upper band contained both PAC1-Hop1 and PAC1-Hop2 isoforms. PAC1 has been previously described to be expressed in CA1, CA3 and dentate gyrus of the hippocampal formation (Joo et al., 2004). Consistent with this report, we observed diffuse immunoreactivity of PAC1 in these areas of both mouse (Fig. 1B) and rat brain (Supplementary Fig. 1), with relatively greater staining intensity in the outer molecular layer of the dentate gyrus, stratum oriens and stratum radiatum regions. These regions in the hippocampal formation are enriched with distal dendrites and dendritic spines, where Kv4.2, a major neuronal dendritic ion channel, is highly expressed (Hoffman et al., 1997, Johnston et al., 2000, Rhodes et al., 2004), as seen with Kv4.2 immunolabeling (Fig. 1B). The PAC1 and Kv4.2 immunostaining overlaps to a large extent, indicating that they are presumably co-expressed within the same population of neurons and might therefore be amenable to signaling crosstalk. In order to further confirm this assertion we performed immunocytochemical staining of cultured rat hippocampal neurons with Kv4.2 and PAC1 antibodies, and found that both these proteins are expressed in the somata and dendrites of these neurons (Supplementary Fig. 2). In addition to neurons PAC1 is also expressed in astrocytes/glial cells, as verified by co-labeling with the astro-glial marker GFAP (Supplementary Fig. 2). Although our RT-PCR results show relatively low levels of all PAC1 isoforms in mouse cortex, no visible differences were observed in PAC1 immunostaining patterns and intensities in both rat and mouse cortex (data not shown).
Figure 1.
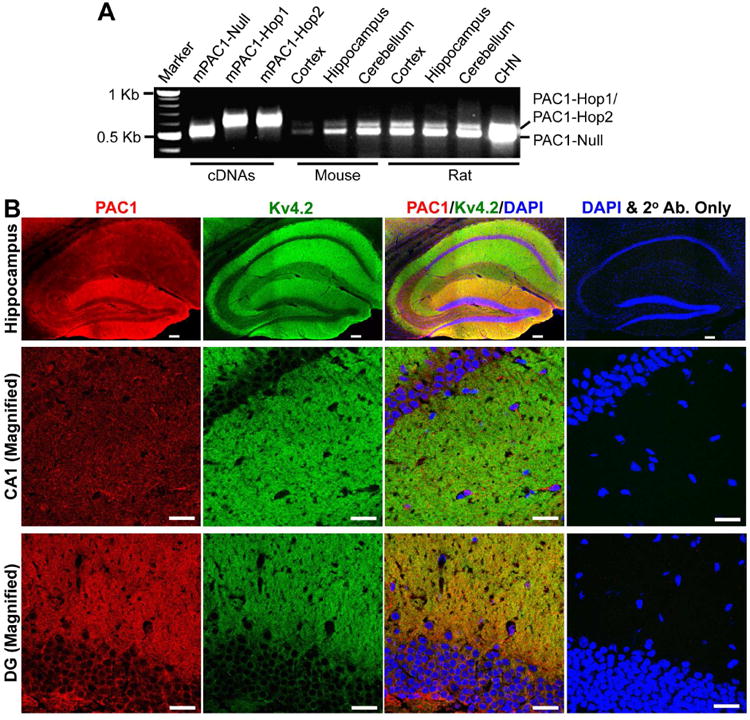
PAC1 is expressed in mouse and rat brain. A, Representative image showing the PAC1 mRNA expression in various mouse and rat brain regions, and in cultured rat hippocampal neurons (CHN) from RT-PCR analysis using specific primers spanning the variable region of PAC1. The Null, Hop1 and Hop2 isoforms were identified based on transcript size, followed by sequencing of the specific PCR products. cDNAs of mouse PAC1-Null, PAC1-Hop1 and PAC1-Hop2 were used as positive controls in these RT-PCR experiments. Numbers on the left denote the relative mobility of molecular weight markers. B, Representative confocal photomicrographs of the hippocampal formation in sections from mouse brain (40 μm; sagittal) immunostained with antibodies against Kv4.2 (green) and PAC1 (red), and DAPI (blue). Kv4.2 is expressed predominantly in the outer molecular layer of the dentate gyrus, as well as in stratum oriens and stratum radiatum layers, which contain the distal dendrites of neurons in CA1 and CA3 regions (magnified images shown in middle and bottom rows. PAC1 immunoreactivity is more intense in the outer molecular layer of the dentate gyrus, followed by CA1 and CA3 pyramidal cell body and dendritic layers. Images on the right column of each row are taken from mouse brain sections that were incubated only with the same secondary antibodies (2° Ab.) and DAPI, without the primary antibodies against Kv4.2 and PAC1. Scale bar – 100 μm for images in the top row (magnification: 10×), and 25 μm for middle- and bottom-row images (magnification: 63×).
We next verified if PAC1 expressed in hippocampal neurons are functional. Stimulation of PAC1 by its ligand PACAP has been shown to activate GαS-coupled signaling for the downstream activation of adenylate cyclase (AC), resulting in production of cAMP (Spengler et al., 1993, Lu et al., 1998). We therefore used an ELISA-based approach to quantify cAMP levels in CHNs after treatment with PACAP38 (10 and 100 nM, 20 min) or its N-terminal truncated version PACAP27 (10 and 100 nM, 20 min). Both PACAP27 and PACAP38 led to a significant increase in cAMP levels (181.52 ± 16.6 and 189.44 ± 4.7 fmol/μg protein for 10 nM PACAP27 and 10 nM PACAP38, respectively), as compared to control/untreated conditions (0.39 ± 0.12 fmol/μg protein; Fig. 2A). No significant dose-dependence of PACAP-induced increases in intracellular cAMP levels was observed in CHNs (Fig. 2A). Surprisingly, elevation in cAMP was higher upon PACAP treatment than with forskolin (10 μM, 20 min), a direct activator of AC (103.12 ± 17.25 fmol/μg; Fig. 2A). Another hallmark of PACAP/PAC1 signaling that has been widely reported in multiple cell types (Villalba et al., 1997, Blechman and Levkowitz, 2013, May et al., 2014) is the activation of ERK1/2, as determined by elevated levels of phospho-ERK1/2. We therefore performed immunoblot analysis to determine the effect of PACAP on ERK1/2 phosphorylation in CHNs. Increasing concentrations of both PACAP27 and PACAP38 dramatically enhanced ERK1/2 phosphorylation levels (Fig. 2B). Specifically, the majority of the phosphorylation occurred in the ERK2 isoform (p42 band) upon PACAP exposure. Similar to cAMP production, we did not observe a clear dose-dependence of the PACAP-induced increase in ERK1/2 phosphorylation levels in CHNs, although PACAP38 showed some trend (Fig. 2B-C). These results suggest that the PAC1 is functionally expressed in hippocampal neurons, and that its activation by PACAP leads to increased intracellular cAMP production (precursor for PKA activation) and activation of ERK1/2.
Figure 2.
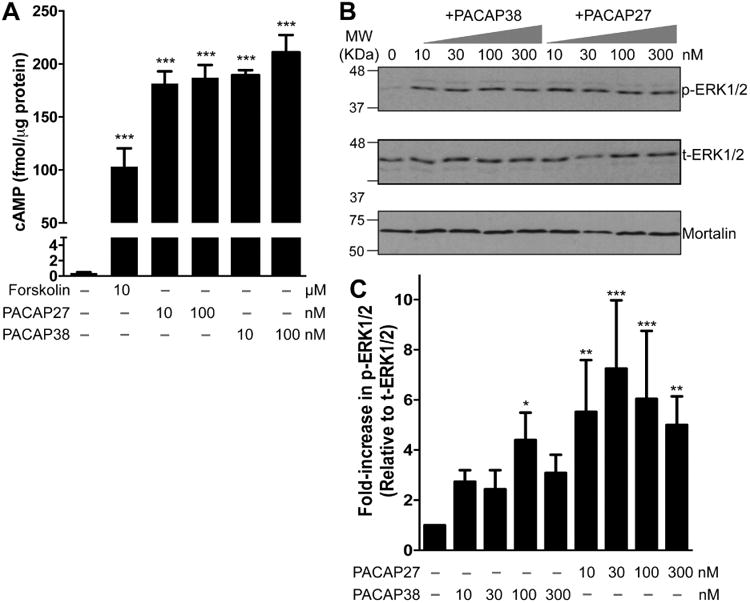
Functional expression of PAC1 in cultured rat hippocampal neurons. A, ELISA-based quantification of intracellular cAMP levels following treatment with PACAP27 (10 and 100 nM), PACAP38 (10 and 100 nM), and forskolin (10 μM) for 20 min. Increased intracellular cAMP levels upon PACAP treatment are indicative of functional Gαs-coupled PAC1 expression in these neurons. Data are presented as mean ± SEM (n = 4). *** denotes p<0.05, significantly different compared to control or untreated condition (One-way ANOVA with Dunnett's multiple comparisons test). B, Representative immunoblot analysis of ERK1/2 phosphorylation in neurons without or with the treatment of either PACAP27 (10, 30, 100 and 300 nM; 20 min) or PACAP38 alone (10, 30, 100 and 300 nM; 20 min). Numbers on the left denote the relative mobility of molecular weight markers. t-ERK1/2 and p-ERK1/2 denote total-ERK1/2 and phospho-ERK1/2, respectively. The bottom row shows sample normalization for same samples probed with anti-mortalin antibody. D, Quantification of p-ERK1/2 levels in neurons from experiments as shown in panel C. Data are presented as mean ± SEM fold-increase in p-ERK1/2 levels relative to t-ERK1/2 levels, normalized to control or untreated conditions, as detailed in methods (n = 4-5). * denotes p<0.05, ** denotes p<0.01 and *** denotes p<0.01, significantly different compared to control or untreated condition (One-way ANOVA with Dunnett's multiple comparisons test).
3.2. PACAP exposure leads to enhanced Kv4.2 phosphorylation and reduction in cell surface levels of the channel in hippocampal neurons
Given that PACAP treatment activates PKA and ERK1/2 in hippocampal neurons, we next sought to determine if Kv4.2 could serve as a downstream substrate for protein phosphorylation. Numerous reports have previously shown that Kv4.2 can undergo phosphorylation by PKA, PKC, ERK1/2 and other MAP kinases (Schrader et al., 2002, Yuan et al., 2002, Schrader et al., 2006, Hammond et al., 2008, Schrader et al., 2009, Seikel and Trimmer, 2009). In order to assess the global phosphorylation state of Kv4.2, we performed metabolic labeling of CHNs with 32P, followed by PACAP application, and subsequently immunoprecipitated the Kv4.2 protein. Separate sets of 32P labeled CHNs were used for immunoprecipitation of protein phosphatase 2A (PP2A) to serve as negative controls. From these samples the extent of Kv4.2 phosphorylation, relative to total channel protein levels were determined by phosphor-imaging and immunoblotting, respectively. Both forskolin- and PACAP38 treated-lysates showed more intense 32P signals as compared to controls (bottom row blot, Fig. 3A). Upon normalization to total immunoprecipitated Kv4.2 protein (top row blot, Fig. 3A), PACAP38 treatment showed a significant increase (1.67 ± 0.07 fold) in Kv4.2 phosphorylation levels compared to untreated or control conditions, while forskolin produced a 1.25 ± 0.12 fold increase (not significantly different from control (Fig. 3B). These results indicate that PACAP promotes phosphorylation of the Kv4.2 channel protein in CHNs.
Figure 3.
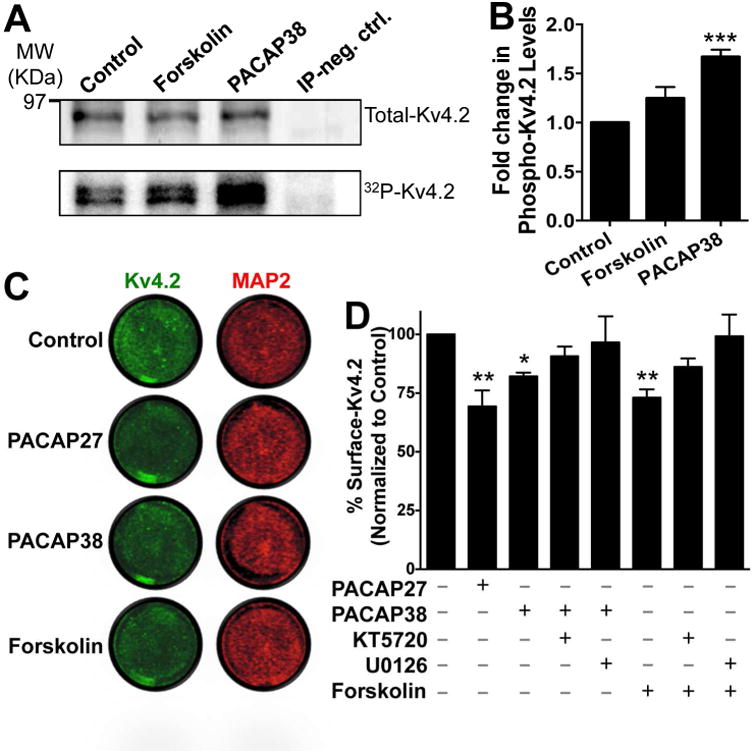
PACAP treatment of cultured rat hippocampal neurons leads to increased phosphorylation and a decrease in the surface expression level of Kv4.2 protein. A, Representative immunoblot (top row) and phosphor-imaging (bottom row) images of immunoprecipitated Kv4.2 protein from cultured hippocampal neurons pre-incubated with 32P-labeled orthophosphate. Number on the left denotes the relative mobility of protein molecular weight marker. Treatment of neurons with PACAP38 (100 nM) or forskolin (10 μM) for 20 min led to increased 32P signal intensities of Kv4.2 protein band, which were quantified (B) using the NIH Image J software. IP neg. Ctrl, denotes sample obtained with immunoprecipitation using anti-PP2A catalytic subunit antibody from cultured hippocampal neurons pre-incubated with 32P-labeled orthophosphate, in order to serve as a negative control. Data in panel B are expressed as mean ± SEM fold change in 32P signal intensities of Kv4.2 normalized to the total immunoprecipitated Kv4.2 protein in each individual treatment group (n = 4). *** denotes p<0.001, significantly different compared to control or untreated conditions (One-way ANOVA with Dunnett's multiple comparisons test). C, Representative images of cultured neurons from on-cell fluorescence immunocytochemical experiments, immunostained with anti-Kv4.2 antibody (green; before permeabilization) and anti-MAP2 antibody (red; after permeabilization), under control or untreated conditions, and upon treatment with PACAP (100 nM each) or forskolin (10 μM) for 20 min. Infrared signal intensities for each set of images were acquired using the same exposure time, and surface Kv4.2 signal intensities, relative to MAP2 signal intensities, were calculated using NIH ImageJ. Data are presented as mean ± SEM of % surface Kv4.2 levels normalized to control or untreated conditions in panel D (n = 5 for PACAP groups, and 4 for forskolin groups). Exposure of neurons to PACAP27 or PACAP38 or forskolin significantly decreased the % of surface Kv4.2 levels, an effect that was absent upon pre/co-treatment of neurons with the inhibitors of PKA (KT5720; 1 μM) and MEK1/2 (U0126; 1 μM). * denotes p<0.05 and ** denotes p<0.01, significantly different compared to control or untreated conditions (One-way ANOVA with Tukey's multiple comparisons test).
Phosphorylation of Kv4.2 by PKA has been shown to induce rapid internalization of the channel protein from the cell surface (Hoffman and Johnston, 1998, Kim et al., 2007, Lin et al., 2011). However, EKR1/2 phosphorylation of Kv4.2 has been suggested to have no influence on cell surface density of the channel protein, primarily based on comparison of constitutive levels of cell surface protein in WT and mutations at ERK1/2 phosphorylation sites (Schrader et al., 2006, Schrader et al., 2009), although direct evidence in support of this is lacking. We next verified whether PACAP/PAC1 activation, which could activate ERK1/2 and PKA in CHNs (Fig. 2A-C), interfered with the extent of cell surface Kv4.2 channel protein levels. Using a quantitative on-cell fluorescence immunocytochemical antibody-based infrared intensity assay (Lin et al., 2011), we determined that exposure of CHNs to PACAP27 and PACAP38 (100 nM, 20 min) led to a significant reduction in cell surface Kv4.2 protein levels in neurons (70.35 ± 6% and 80.54 ± 2.24% of untreated control levels for PACAP27 and PACAP38 treatment conditions, respectively; Fig. 3C-D). Interestingly, no significant reduction in cell surface Kv4.2 protein levels was observed when cells were pre/co-treated with PACAP38 and pharmacological inhibitors of either PKA (1 μM KT5720) or MEK1/2 (1 μM U0126). Exposure of neurons to forskolin (10 μM, 20 min), the activator of cAMP/PKA, also led to a similar reduction in cell surface Kv4.2 protein levels (64.28 ± 9.16% compared to untreated control conditions), which was again absent when cells were pre/co-treated with pharmacological inhibitors of either PKA (1 μM KT5720) or ERK1/2 (by inhibition of MEK1/2; 1 μM U0126; Fig. 3C-D). These results indicate that PACAP/PAC1 signaling in hippocampal neurons could lead to dynamic phosphorylation of Kv4.2 protein, and PKA/ERK-dependent internalization of the channel protein from the cell surface.
3.3. PACAP acutely reduces hippocampal A-type currents (IA)
Having established that PACAP can influence the phosphorylation state of Kv4.2, we next determined its effect on channel activation. Currents through Kv4.2 constitute the majority of neuronal IA currents (Hoffman et al., 1997, Johnston et al., 2000, Chen et al., 2006a). A number of studies have shown that exposure to forskolin or 8-Br-cAMP leads to altered localization of Kv4.2 and reduction in IA currents in hippocampal neurons, mainly by PKA-phosphorylation of the channel protein (Hoffman and Johnston, 1998, Hammond et al., 2008, Lin et al., 2011), and to some extent by ERK1/2-phosphorylation of the channel protein (Schrader et al., 2009). We used two-step voltage protocols in order to isolate IA from hippocampal neurons (Norris and Nerbonne, 2010). PACAP38 application led to a significant reduction in IA current density at more depolarizing potentials as compared to control neurons (Fig. 4A-B). The current density at +80 mV upon treatment with 100 nM PACAP38 for 20 min was 38.1 ± 7.6 pA/pF as compared to 74.1 ± 9.3 pA/pF under control conditions. Analysis of relative current densities at physiologically relevant voltages showed a significant reduction in IA densities upon PACAP38 treatment at all voltages from -30 mV to +40 mV (Fig. 4C). Since PACAP38-induced internalization of cell surface Kv4.2 protein was sensitive to PKA/ERK1/2 activity, we next performed whole-cell IA recordings from PACAP38-treated CHNs that were pre/co-treated with pharmacological inhibitors of either PKA (1 μM KT5720; Fig. 4, D) or ERK1/2 (1 μM U0126; Fig. 4E). Both these inhibitors completely attenuated the PACAP38-induced decrease in IA densities (Fig. 4D-E), indicating the involvement of PKA and ERK1/2 signaling in dynamic modulation of IA in hippocampal neurons.
Figure 4.
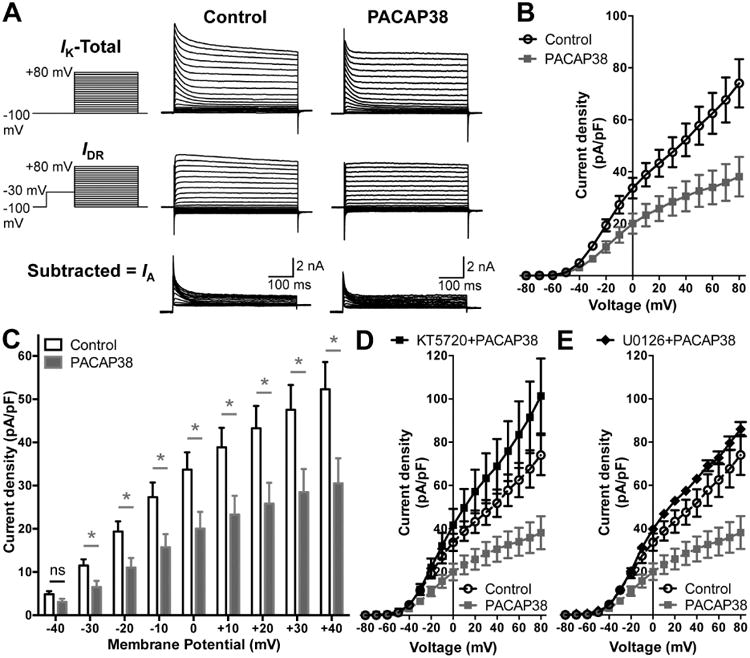
Reduction of A-type K+ (IA) currents in hippocampal neurons following PACAP38 exposure. A, Representative traces of whole-cell voltage-clamp recordings showing electrophysiological isolation of IA in cultured rat hippocampal neurons. Total outward K+ currents (IK) were obtained with step-depolarization from -100 mV to +80 mV for 500 ms in +10 mV increments, from a holding potential of -100 mV. Delayed rectifier currents (IDR) were obtained by applying a -30 mV pre-pulse for 500 ms before step-depolarization from -100 mV to +80 mV for 500 ms in +10 mV increments, in order to inactivate IA. Isolation of IA was achieved by subtracting IDR from IK. PACAP38 treatment (100 nM, 20 min) led to reduction in IA amplitude, as quantified in panel B showing the current density – voltage plots, and re-plotted showing PACAP effects at physiologically-relevant voltages (-40 mV to +40 mV) in panel C. Data in panels B and C are presented as mean ± SEM obtained from multiple batches of cultured neurons without (control) or with PACAP38 treatment (n = 12 for both groups), ns: not significantly different, and * denotes p<0.05, significantly different compared to control or untreated conditions (Unpaired Student's t-test). D-E, current density – voltage plots of IA currents recorded from neurons with PACAP38 treatment (100 nM, 20 min) along with the inhibitors of PKA (KT5720; 1 μM) or MEK1/2 (U0126; 1 μM), showing attenuation of the PACAP38-induced decrease in IA density. Current density data for control and PACAP38 treatment from panel B are re-plotted here as dashed lines for direct comparison. Data are presented as mean ± SEM obtained from multiple batches of cultured neurons (n = 10 for KT5720+PACAP38; and n = 8 for KT5720+PACAP38 treatment groups).
3.4. Cloned PAC1 isoforms from mouse hippocampus exhibit similar functional signaling characteristics under heterologous expression
Kv4.2 is the major, but not the only channel responsible for the generation of IA in hippocampal neurons. Dendritic Kv4.3 channels and axonal Kv3.3, Kv3.4 and Kv1.4 channels also contribute to IA currents in these neurons (Rudy and McBain, 2001, Vacher et al., 2008, Carrasquillo et al., 2012, Carrasquillo and Nerbonne, 2014). In order to understand the specific modulation of Kv4.2 by PACAP-mediated channel phosphorylation, heterologous expression of Kv4.2 channel and PAC1 receptor in mammalian cells is preferred. First, utilizing specific primer sets we cloned the full-length cDNAs of PAC1 isoforms from mouse hippocampus. Upon sequencing, three isoforms were identified, PAC1-Null, PAC1-Hop1 and PAC1-Hop2 (Supplementary Fig. 3, and Fig. 5A), which were consistent with our prior observations (Fig. 1A). These individual isoforms were then sub-cloned into peYFP-N1 expression vectors for further characterization. The nucleotide sequence of the PAC1-Null isoform was identical to the previously cloned PAC1-Null from mouse brain (NCBI reference sequence: NM_001025372.2; (Hashimoto et al., 1996). The PAC1-Hop1 isoform cloned from mouse hippocampus shows an insertion of 84 nucleotides, corresponding to 28 amino acid residues, in the 3rd intracellular loop (between the 5th and 6th transmembrane domains of the protein, Supplementary Fig. 3, and Fig. 5A), consistent with the mouse PAC1-Hop1 sequence from GeneBank (NCBI reference sequence: NM_007407.4). However, our cloned PAC1-Hop1 from mouse hippocampus showed four nucleotide substitutions, out of which two were silent, leading to no change in the amino acid translation. The other two substitutions, at nt413 and nt679, result in changes in amino acids Asp to Gly at residue 138 and Lys to Arg at residue 227 of PAC1, respectively (Supplementary Fig. 3). The cloned PAC1-Hop2 isoform from mouse hippocampus was identical to the PAC1-Null isoform, except for the insertion of 81 nucleotides, corresponding to 27 amino acid residues, in the same 3rd intracellular loop region (Supplementary Fig. 3, and Fig. 5A).
Figure 5.
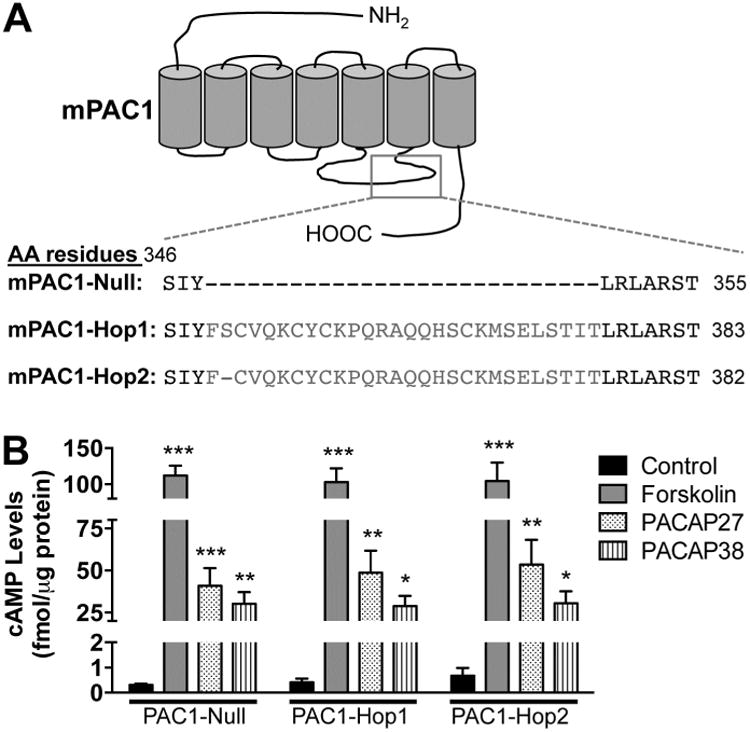
Increased intracellular cAMP production in HEK293T cells overexpressing cloned mouse brain PAC1 isoforms. A, Schematic showing the region in the 3rd intracellular loop where splicing occurs, alongside the amino acid residues for each of the three isoforms expressed in mouse hippocampus. B, ELISA-based quantification of cAMP levels following treatment with PACAP27 (100 nM), PACAP38 (100 nM) or forskolin (10 μM) for 20 min in HEK293T cells transfected with plasmids carrying YFP-tagged mouse PAC1-Null, PAC1-Hop1 or PAC1-Hop2 cDNAs. Controls for each PAC1 isoform transfection groups denote cells treated with vehicle (in 0.1% BSA solutions that is used to prepare PACAP stock solutions). Data are presented as mean ± SEM (n = 3 for forskolin groups, and 5 for untreated/control and PACAP treatment groups). * denotes p<0.05, ** denotes p<0.01, and *** denotes p<0.001, significantly different compared to control or untreated conditions for respective PAC1 isoforms (One-way ANOVA with Tukey's multiple comparisons test).
We next confirmed that the PACAP/PAC1-mediated signaling cascades activated in neurons were also recapitulated upon heterologous expression in HEK293T cells. For this purpose, we co-transfected Kv4.2 and its accessory subunit KChIP2 with either of the three PAC1 isoforms that we had cloned from mouse hippocampus. cAMP levels were measured following 20 min application of PACAP27 (100 nM), PACAP38 (100 nM) or forskolin (10 μM) in HEK293T cells expressing Kv4.2 + KCHIP2 and YFP-tagged PAC1-Null or PAC1-Hop1 or PAC1-Hop2 (Fig. 5B). PACAP27 and PACAP38 elevated cAMP levels to a similar extent, as did forskolin, the direct activator of AC, in cells transfected with all three cloned PAC1 isoforms (Fig. 5B). Control levels of cAMP were approximately 0.3 – 0.7 fmol/μg under each of the three transfected conditions. We next determined if PACAP stimulates ERK1/2 activation in this heterologous system. PACAP did not activate ERK1/2 in cells expressing YFP in addition to Kv4.2 and KChIP2, as indicated by no detectable phospho-ERK1/2 signals (Fig. 6A). Transfection of cells with YFP-tagged PAC1-Null or PAC1-Hop1 or PAC1-Hop2, along with Kv4.2 and KChIP2 led to activation of ERK1/2 upon PACAP27 or PACAP38 treatments, as determined by increased levels of p-ERK1/2 (Fig. 6B) that are significantly increased, compared the respective control/un-treated conditions (Fig. 6C).
Figure 6.
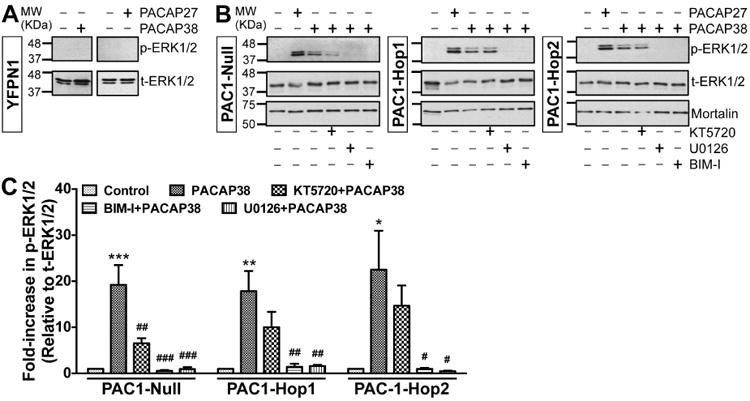
Increased ERK1/2 phosphorylation in HEK293T cells overexpressing cloned mouse brain PAC1R isoforms, and the underlying intracellular cascades. Representative immunoblots of lysates obtained from HEK293T cells transfected with plasmid carrying YFP-N1 (panel A) or YFP-tagged PAC1-Null, PAC1-Hop1 or PAC1-Hop2 (panel B) cDNAs. Cells were treated with either PACAP27 or PACAP38 alone (100 nM; 20 min) or pre/co-treated with specific inhibitors of PKA (KT5720; 1 μM) or MEK1/2 (U0126; 1 μM) or PKC (BIM-I; 500 nM) 10 min before PACAP38 treatment. Numbers on the left denote the relative mobility of molecular weight markers. t-ERK1/2 and p-ERK1/2 denote total-ERK1/2 and phospho-ERK1/2, respectively. The bottom immunoblot rows show sample normalization for the same samples probed with anti-mortalin antibody. C, Quantification of p-ERK1/2 levels in cells from experiments as shown in panel B. Data are presented as mean ± SEM fold-increase in p-ERK1/2 levels relative to t-ERK1/2 levels, normalized to control or untreated conditions, as detailed in methods (n = 3 for BIM-I groups, and 4-5 for all other groups). * denotes p<0.05, ** denotes p<0.01, and *** denotes p<0.001, significantly different compared to respective control or untreated conditions; and # denotes p<0.05, ## denotes p<0.01, and ### denotes p<0.001, significantly different compared to respective PACAP38 treatment conditions (One-way ANOVA with Tukey's multiple comparisons test).
Multiple intracellular signaling cascades underlying ERK1/2 activation have been shown for PAC1 isoforms in different cell types (Macdonald et al., 2005, Broca et al., 2009, Blechman and Levkowitz, 2013, May et al., 2014). Our results from mouse hippocampal PAC1 isoforms expressed in HEK293T cells show that PACAP38-mediated activation of ERK1/2 could be significantly attenuated by the PKA inhibitor KT5720 (1 μM) in Null, but not in Hop1 and Hop2 isoforms (Fig. 6B-C). However, the PKC inhibitor BIM-I (0.5 μM) and MEK1/2 inhibitor U0126 (1 μM) that inhibits ERK1/2 activation, completely attenuated ERK1/2 phosphorylation in all three isoforms (Fig. 6B-C). It is known that activation of PKA and PKC could lead to phosphorylation and activation of Ras, which in combination with Rap1 leads to activation of MEK1/2, and MEK1/2 phosphorylates ERK1/2 for its activation (Villalba et al., 1997, Blechman and Levkowitz, 2013, May et al., 2014). To verify the involvement of Ras, we co-transfected HEK293T cells with a dominant-negative Ras (N17-Ras or DN-Ras), which led to complete attenuation of PACAP38-induced ERK1/2 phosphorylation in all three isoforms (Fig. 7A). PACAP/PAC1-induced ERK1/2 activation has also been shown to be mediated in a G-protein-independent manner, via arrestin-2 (also referred to as (β-arrestin-1) signaling in INS-1E (β cells and pancreatic islets (Broca et al., 2009). Accordingly, we investigated the role of arrestin-2 in PACAP-induced ERK1/2 activation by transfecting HEK293T cells stably expressing either arrestin-2 or arrestin-3 siRNA (the later one as a negative control) with individual PAC1 isoforms. These stable siRNA arrestin knockdown HEK293T cells were previously characterized to have a stable reduction of either arrestin-2 or arrestin-3 expression (Zhang et al., 2012). Furthermore, we performed qRT-PCR and determined that there was a 60-70% reduction in arrestin-2 and arrestin-3 mRNA levels only in HEK293T-arrestin-2-siRNA and HEK293T-arrestin-3-siRNA stable cell lines, respectively, as compared to HEK293T cells, similar to the previously observed results (Zhang et al., 2012). PACAP38 treatment (100 nM, 20 min) led to strong ERK1/2 phosphorylation in HEK293T cells stably expressing arrestin-3 siRNA with each individual PAC1 isoform, without any difference between individual isoforms (Fig. 7B), indicating no involvement of arrestin-3 in PACAP/PAC1-mediated ERK1/2 activation. However, PACAP38 treatment of cells stably expressing arrestin-2 siRNA failed to induce any detectable ERK1/2 phosphorylation upon transient transfection of Hop1 and Hop2 isoforms, but not the Null isoform (Fig. 7B). In cells stably expressing arrestin-2 siRNA and transient transfection of PAC1-Null isoform, PACAP38 treatment led to similar extent of ERK1/2 phosphorylation, as observed in cells without any siRNA expression (Fig. 6B) or in cells expressing arrestin-3 siRNA (Fig. 7B). Collectively, these results indicate that differences exist amongst PAC1 isoforms for downstream intracellular signaling leading to ERK1/2 phosphorylation, in terms of the involvement of PKA and arrestin-2 signaling; however, these signaling cascades converge on PKC-Ras-MEK1/2 signaling downstream of activation of all three isoforms (Fig. 7C). Since PAC1-Null is the most widely and abundantly expressed isoform in the brain (Fig. 1A), and relatively multi-faceted downstream signaling results from its activation, we used this isoform for all subsequent experiments in HEK293T cells.
Figure 7.
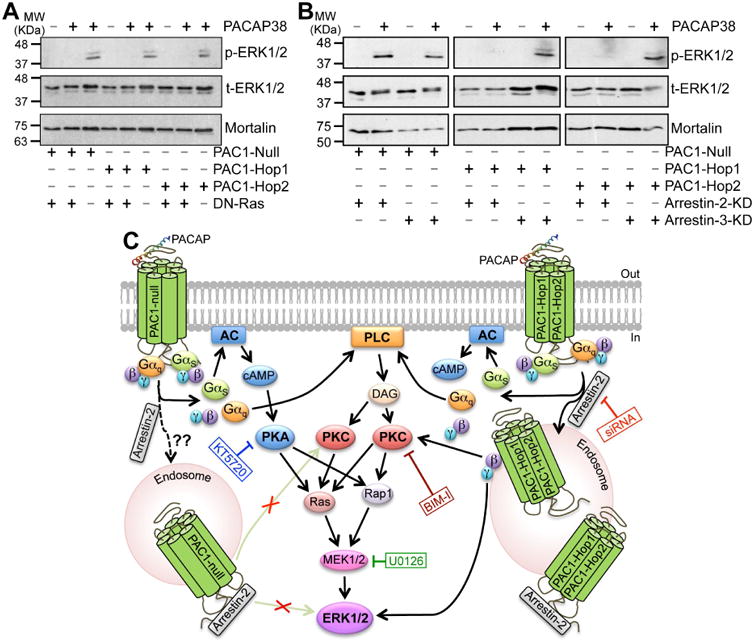
Ras and arrestin-2-dependence of PACAP38/PAC1-mediated ERK1/2 activation. A, representative immunoblots (from 3 independent experiments) of lysates obtained from HEK293T cells transfected with plasmid carrying YFP-tagged PAC1-Null, PAC1-Hop1 or PAC1-Hop2 cDNAs, with or without the plasmid carrying dominant-negative Ras, N17Ras (DN-Ras), as indicated below the blot panels. Lysates were collected from untreated cells, and from cells with PACAP38 treatment (100 nM; 20 min), as indicated on top of the blot panels. Numbers on the left denote the relative mobility of molecular weight markers. B, representative immunoblots (from 3 independent experiments) of lysates obtained from HEK293T cells stably expressing siRNA for arrestin-2 (arrestin-2-KD) or arrestin-3 (arrestin-2-KD), further transiently transfected with plasmid carrying YFP-tagged PAC1-Null, PAC1-Hop1 or PAC1-Hop2 cDNAs, as indicated below the blot panels. Lysates were collected from untreated cells, and from cells with PACAP38 treatment (100 nM; 20 min), as indicated on top of the blot panels. Numbers on the left denote the relative mobility of molecular weight markers. t-ERK1/2 and p-ERK1/2 denote total-ERK1/2 and phospho-ERK1/2, respectively, and the bottom row immunoblots show sample normalization for the same samples probed with anti-mortalin antibody for both panels a and b. PACAP38 treatment failed to induce any detectable ERK1/2 phosphorylation in DN-Ras transfected cells in all three PAC1 isoforms. Interestingly, PACAP38 treatment led to strong ERK1/2 phosphorylation in HEK293T cells stably expressing arrestin-3 siRNA with each individual PAC1 isoforms. However, PACAP38 treatment did not lead to any detectable ERK1/2 phosphorylation in cells stably expressing arrestin-2 siRNA with PAC1-Hop1 and Hop2, but not with the Null isoform. C, graphical scheme depicting the intracellular signaling cascades activated upon PACAP-stimulation of PAC1-Null or Hop1 or Hop2 receptors, as determined from results shown in Fig. 5B and 6A-C and 7A-B. Black arrows indicate stimulation/activation, and texts in blue, green, red and dark red boxes connected to blunt-ended lines indicate inhibition of specific signaling components with pharmacological modulators or dominant-negative overexpression or siRNA expression. Abbreviations for AC, cAMP, DAG, ERK1/2, MEK1/2, PLC, PKA and PKC are consistent with those described elsewhere in the manuscript.
3.5. PACAP acutely reduces Kv4.2 currents in transfected HEK293T cells
We next performed voltage-clamp electrophysiological analysis of Kv4.2 currents, with or without PACAP treatment in HEK293T cells. PACAP38 treatment (100 nM) of HEK293T cells expressing Kv4.2, KChIP2 and mPAC1-Null-YFP significantly decreased Kv4.2 current density as compared to control/untreated conditions (Fig. 8A-B). We determined the time-course of the PACAP-induced reduction in Kv4.2 currents, which showed a progressive decrease in the current density that was maximal at 20 min of PACAP38 treatment (Fig. 8A-B). Mean current density at +80 mV in control cells was 325.4 ± 33.7 pA/pF, whereas it was reduced to 265.0 ± 36.6 pA/pF, 207.5 ± 33.8 pA/pF, 121.6 ± 25.8 pA/pF, and 159.8 ± 40.3 pA/pF in cells treated with 100 nM PACAP38 for 5, 10, 20 and 30 min, respectively (Fig. 8B). We also determined the voltage-dependence of Kv4.2 activation from the conductance-voltage relationship. There was no significant difference in the half-maximal channel conductance upon PACAP38 treatment (G1/2 = -2.7 ± 0.8 mV, k = 18.2 ± 0.8 mV; and G1/2 = -3.1 ± 0.8 mV, k = 15.5 ± 1.2 mV for control and PACAP38-20 min treatment conditions, respectively; Fig. 8C). These G1/2 values for Kv4.2 are in line with the previously published G1/2 activation value for the channel currents in HEK293T cells co-expressing KChIP2 (Liu et al., 2014). Similarly, we determined the voltage-dependence of activation of IA, from recordings obtained with cultured hippocampal neurons (Fig. 4A-B), with or without PACAP38 treatment (100 nM, 20 min). No significant difference was observed in the activation G1/2 values for control vs PACAP38-treated neurons (Fig. 8D). We also determined the voltage-dependence of Kv4.2 channel inactivation, and found no significant difference in the half-maximal inactivation potentials for control vs PACAP38 treatment conditions (not shown). These results, in combination with our observations on PACAP38-mediated rapid internalization of cell surface Kv4.2 levels (Fig. 3C-D), suggest that downregulation of channel function primarily occurs due to a decrease in the number of surface channels, rather than any alteration in voltage-dependence of channel activation.
Figure 8.
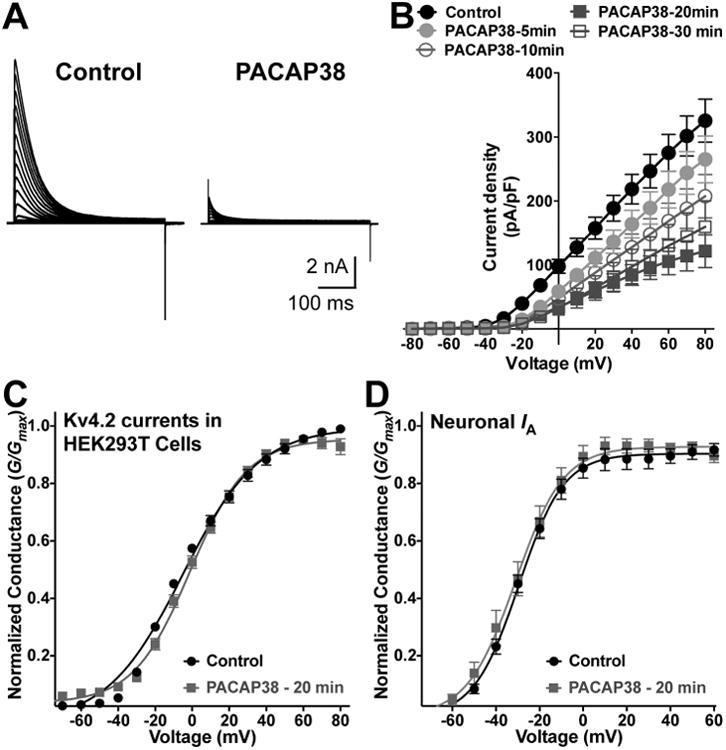
PACAP38-mediated reduction of Kv4.2 currents is resulted from a decrease in current density, without influencing the voltage-dependence of channel activation. A, Representative traces of whole-cell voltage-clamp recordings from HEK293T cells transfected with plasmids containing Kv4.2, KChlP2 and PAC1-Null cDNAs, without (control) or with PACAP38 (100 nM) for 20 min. Cells were step-depolarized from -100 mV to +80 mV for 500 ms with +10 mV increments from a holding potential of -100 mV. B, Current density – voltage plots of Kv4.2 currents from cells without (control) or with PACAP38 (100 nM) treatment for 5, 10, 20, and 30 min. Data are presented as mean ± SEM [n = 17 cells for control, and 10, 12, 18 and 8 cells for 5, 10, 20 and 30 min PACAP38 treatments, respectively]. C-D, Conductance – voltage curves for Kv4.2 currents recorded from HEK293T cells (C), and for IA currents recorded from cultured hippocampal neurons (D), without or with PACAP38 treatment (100 nM, 20 min). The half-maximal activation potential (G1/2) and slope factor (k) calculated from these traces are: G1/2 = -2.7 ± 0.8 mV, k = 18.2 ± 0.8; and G1/2 = -3.1 ± 0.8 mV, k = 15.5 ±1.2 for control and PACAP38 treatment conditions, respectively (panel C); and G1/2 = -29.8 ± 0.7 mV, k = 10.2 ± 0.6; and G1/2 = -31.0 ± 1.4 mV, k = 11.1 ± 1.2 for control and PACAP38 treatment conditions, respectively (panel D). Data are presented as mean ± SEM [n = 17 and 18 cells for control and PACAP38 treatment groups, respectively].
3.6. PACAP-induced reduction of Kv4.2 currents is dependent on both PKA and ERK1/2 activity
Our results showed that all three isoforms of PAC1 expressed in mouse brain neurons mediate cAMP and ERK signaling. Also, PACAP38 application led to significant reduction in IA and Kv4.2 current amplitudes. We next investigated if such functional modulation of Kv4.2 currents was dependent on these kinases. Pre/co-treatment of HEK293T cells co-expressing Kv4.2, KCHIP2 and PAC1-Null-YFP with the PKA inhibitor KT5720 (1 μM) for 20 min significantly attenuated the PACAP38-mediated decrease in Kv4.2 current density, compared to KT5720 (1 μM) application alone (Fig. 9A-B). Furthermore, pre/co-treatment of U0126 (1 μM) for 20 min, an inhibitor of ERK1/2 activation, completely abolished the PACAP38-mediated decrease in Kv4.2 currents, compared to application of U0126 (1 μM) alone (Fig. 9A, and C). Pre/co-treatment of cells with both KT5720 and U0126 together for 20 min also led to complete attenuation of PACAP38-mediated decrease in Kv4.2 current density (Fig. 9A, and D), similar to the U0126 + PACAP38 treatment condition. It must be noted here that application of KT5720 and U0126 together did not produce any additive effects on PACAP38-induced reduction in Kv4.2 currents, as compared to conditions when these inhibitors were applied individually.
Figure 9.
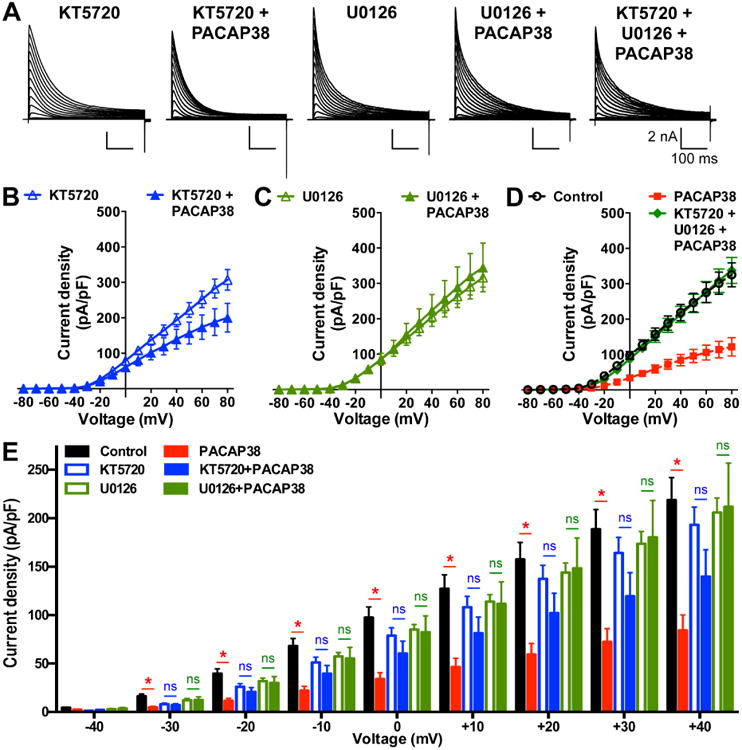
PACAP38-mediated reduction in Kv4.2 current density is dependent on the activity of both PKA and ERK1/2. A, Representative traces of whole-cell voltage-clamp recordings from HEK293T cells transfected with plasmids containing Kv4.2, KChlP2 and PAC1-Null cDNAs, with PKA inhibitor KT5720 alone (1 μM), PACAP38 (100 nM) + (KT5720, 1 μM), ERK1/2 inhibitor U0126 (1 μM) alone, PACAP38 (100 nM) + (U0126, 1 μM), and PACAP38 (100 nM) + KT5720 (1 μM) + U0126 (1 μM) for 20 min. Cells were step-depolarized from -100 mV to +80 mV for 500 ms with +10 mV increments from a holding potential of -100 mV. B-D, Current density – voltage plots of Kv4.2 currents from cells with various drug treatments, as shown in panel A. Data are presented as mean ± SEM [n = 10 and 13 cells for KT5720 and KT5720 + PACAP38, respectively (B); 10 and 12 cells for U0126 and U0126 + PACAP38, respectively (C); and 11 cells for KT5720 + U0126 + PACAP38 (D) groups]. Current density data for control and 20 min PACAP38 treatment conditions from Fig. 7B are re-plotted in panel D as dotted lines for comparison. E, Comparative plot of Kv4.2 current densities calculated from panels B-C and Fig. 7B, showing indicated peptide/drug effects at physiologically-relevant voltages (-40 mV to +40 mV). ns: not significantly different, and * denotes p<0.05, significantly different compared to respective control or KT5720 alone or U0126 alone treatment conditions (Unpaired Student's t-test).
Upon closer look at the Kv4.2 current densities at relatively less depolarizing and physiologically-relevant voltages (-40 mV to +40 mV) it became clear that co-application of KT5720 or U0126 with PACAP38 led to no significant decrease in Kv4.2 current densities at these voltages (Fig. 9E). These results, in combination with our observations from immunoblot experiments (Fig. 7B-C), indicate that these two kinases presumably converge on ERK1/2 to influence Kv4.2 channel function.
3.7. PACAP-induced reduction of Kv4.2 currents is dependent on phosphorylation of the channel protein by ERK1/2, but not PKA
Previous studies have shown that Kv4.2 can be phosphorylated by PKA at S552, and by ERK1/2 at residues T602, T607 and S616 in the cytoplasmic C-terminus of the channel protein (Adams et al., 2000, Anderson et al., 2000). The kinase inhibitors used in our study could have off-target effects, and alternatively, PACAP/PAC1/ERK1/2-induced reduction in Kv4.2 currents could also be mediated by phosphorylation of other proteins, but not of Kv4.2 directly. To examine whether the PACAP effect involves direct phosphorylation of Kv4.2 channel, we introduced phospho-disruptive mutations at the PKA (S552A) and ERK phosphorylation sites (T602, T607, and S616A). Upon expression in HEK293T cells, along with KChIP2 and PAC1-Null-YFP, these mutants did not exhibit any significant difference in their current densities, as compared to the WT channel (Fig. 10A-D, and 11A-D). Interestingly, the PKA phosphorylation site mutant, Kv4.2-S552A, exhibited a significant reduction in current density upon PACAP38 application (100 nM, 20 min; Fig. 10A-B), to an extent similar to that observed for Kv4.2-WT channels (Fig. 10A-D). These results are not in direct agreement with observations from experiments on Kv4.2-WT channel with KT5720 application (Fig. 9B and E). However, based on our immunoblot results showing attenuation of ERK1/2 phosphorylation with KT5720 pre/co-application in the PAC1-Null isoform, these observations in the Kv4.2-S552A mutant clearly indicate that PKA activation converges on ERK1/2 activation to exert functional effects on Kv4.2 channel.
Figure 10.
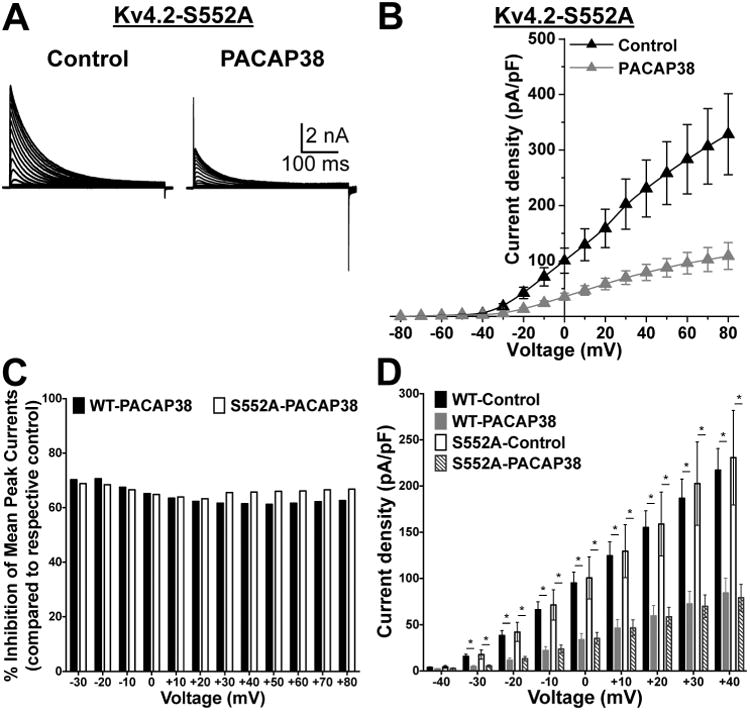
The PACAP38-mediated reduction in Kv4.2 currents is conserved in channel with a phospho-disruptive mutation at the PKA-phosphorylation site. A, Representative traces of whole-cell voltage-clamp recordings, using similar voltage protocol as used in Fig. 8, from HEK293T cells co-transfected with plasmids containing the cDNA of phospho-disruptive mutant Kv4.2 at PKA site, Kv4.2-S552A along with plasmids containing the cDNAs of KChIP2 and PAC1-Null. B, Current density – voltage plots of Kv4.2-S552A channel, with or without PACAP38 treatment (100 nM, 20 min). Data are presented as mean ± SEM (n = 7-8 cells). C, The proportion of PACAP38-induced current reduction for Kv4.2-WT and Kv4.2-S552A mutant channels are presented as percent inhibition of mean peak currents, normalized to respective mean peak currents recorded from untreated control cells, at physiologically-relevant voltages, as calculated from the data presented in Fig. 8B and 9B. D, comparative plot of Kv4.2-WT and Kv4.2-S552A current densities at physiologically-relevant voltages, calculated from the panel 8B and Fig. 7F, showing significant reduction in current densities at -40 mV to +40 mV upon PACAP38 application in both WT and S552A mutant channel. * denotes p<0.05, significantly different compared to respective WT and S552A control cells (Unpaired Student's t-test).
Figure 11.
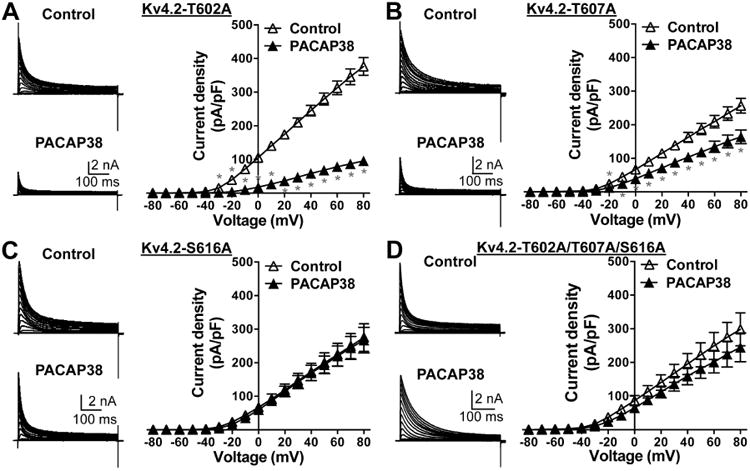
The PACAP38-mediated reduction in Kv4.2 currents is attenuated in channels with phospho-disruptive mutations at ERK1/2-phosphorylation sites. Representative traces of whole-cell voltage-clamp recordings (using similar voltage protocol as in Fig. 8) from HEK293T cells transfected with plasmids containing Kv4.2-T602A (A) or Kv4.2-T607A (B) or Kv4.2-S616A (C) or Kv4.2-T602A/T607A/S616A triple mutant (D) cDNAs, along with the plasmids containing KChlP2 and PAC1-Null cDNAs. Current density – voltage plots for these mutant channels, without or with PACAP38 treatment (100 nM, 20 min) are given next to the current traces under each panel. Data are presented as mean ± SEM (n = 5-7 cells for A, 7 cells for B, 10-11 cells for C, and 7-9 cells for D). * denotes p<0.05, significantly different for PACAP38 treatment conditions compared to current densities at respective voltage pulses under control conditions (Unpaired Student's t-test).
We next investigated the role of ERK1/2 phosphorylation of Kv4.2 in the PACAP-induced reduction in channel function/density. Of the 3 amino acid residues in Kv4.2 that can be phosphorylated by ERK1/2, we found that the Kv4.2-T602A mutant exhibited a significant PACAP38-mediated decrease in channel current density, to an extent similar to that observed in WT channels (Fig. 11A). Although the Kv4.2-T607A mutant exhibited a significant PACAP38-mediated decrease in channel current density, the extent of this decrease was lesser compared to WT channels (Fig. 11B). Interestingly, the Kv4.2-S616A mutant did not show any PACAP38-induced reduction in channel current density (Fig. 11C), and a similar observation was made for the Kv4.2-T602/T607/S616A triple mutant channel (Fig. 11D).
Altogether, these results suggest that PACAP38-mediated activation of PKA and PKC converges on ERK1/2 activation, which subsequently leads to phosphorylation of Kv4.2. Phosphorylation of Kv4.2 at S616, and to some extent at T607, result in internalization of channel proteins from the cell surface, thereby leading to significant downregulation of channel function.
4. Discussion
Our study identified the expression of PAC1 isoforms in different rodent brain regions, where they are mostly co-expressed in Kv4.2-immunoreactive neurons. The PAC1 ligands, PACAP27 and PACAP38, activate multiple G-protein signaling mechanisms that result in elevated levels of cAMP, indicating increased PKA activity, as well as PKA/PKC/Ras-dependent activation of ERK1/2. Furthermore, PACAP exposure enhances Kv4.2 phosphorylation levels, which is accompanied by a rapid reduction in cell surface levels of the channel protein, leading to downregulation of Kv4.2-based IA currents in hippocampal neurons and under heterologous expression. Interestingly, this reduction in Kv4.2 current was independent of the voltage-dependence of channel activation; rather it was dependent on ERK1/2-, but not PKA-mediated phosphorylation of the channel protein. These findings suggest a novel convergent phosphorylation-dependent mechanism by which PACAP could function as a neuromodulator to increase neuronal excitability.
Isoform-specific differences in PAC1 signaling have been reported by several groups, but the results are often conflicting (Spengler et al., 1993, Nicot and DiCicco-Bloom, 2001, Blechman and Levkowitz, 2013, Yan et al., 2013). A number of studies have reported that the PAC1-Null isoform signals preferentially through GαS-coupled mechanisms resulting in AC activation, whereas the Hop1 isoform signals through the Gαq pathway resulting in inositol-triphosphate production mediated by phospholipase-Cβ (PLCβ) activation (Spengler et al., 1993, Nicot and DiCicco-Bloom, 2001). In contrast, other studies have reported that all PAC1 variants can activate AC (Spengler et al., 1993, Blechman and Levkowitz, 2013). These differences could be attributed to developmental changes in expression, as well as differences in the species and tissue types studied (Grimaldi and Cavallaro, 1999, Jamen et al., 2002, Yan et al., 2013). Our study shows functional expression of GαS-coupled signaling, resulting in elevated cAMP levels for all three PAC1 isoforms detected in rodent brain, a finding in line with the previous reports (Spengler et al., 1993, Blechman and Levkowitz, 2013). Interestingly, we found that PACAP38-induced ERK1/2 phosphorylation is largely dependent on PKA activity in the Null isoform, but not in Hop1 and Hop2 isoforms. However, our results suggest that PKC and Ras activity are the major downstream determinants of ERK1/2 activation in all three PAC1 isoforms. Previous studies have shown a similar PKC dependence of ERK1/2 activation for the human PAC1-Hop1 isoform (May et al., 2014), and Ras-dependent ERK1/2 activation upon PACAP38 exposure of rat cerebellar granule neurons (Villalba et al., 1997), where Kv4.2 is abundantly expressed (Vacher et al., 2008). Another interesting finding from our study is the involvement of a GPCR-independent, arrestin-2-dependent endocytic pathway that leads to ERK1/2 activation in PAC1-Hop1 and Hop2 isoforms, but not in the Null isoform. Arrestin-2-mediated potentiation of ERK1/2 by PACAP has previously been reported in pancreatic β-cells (Broca et al., 2009). Isoform-specific differences in arrestin dependence have not been elucidated before. The ability of the Null isoform to activate such diverse signaling pathways that converge on ERK1/2 phosphorylation could presumably provide a rationale for its abundant expression over the other two isoforms in mammalian brain, where it could influence the expression, localization and function of several ion channels and receptors.
The influence of PACAP-activated signaling mechanisms on modulation of voltage-gated channels that are critical to the intrinsic excitability of neurons had not been well explored previously. Since PAC1 is localized to neuronal dendrites, and previous studies have shown modulation of NMDA-type glutamate receptors by PACAP (Yaka et al., 2003, Macdonald et al., 2005), we chose to explore such signaling crosstalk between PACAP and Kv4.2 channel, which is a critical regulator of dendritic excitability. Downregulation of Kv4.2 expression and IA current density upon prolonged (24 h) exposure to PACAP in mouse olfactory neuroepithelial cells and neurons have been shown. These effects on Kv4.2 expression are sensitive to pharmacological inhibition of PLC and PKC (Han and Lucero, 2005, 2006). In contrast, PACAP38 has been shown to exert no modulating effects on IA currents in rat cerebellar granule neurons (Mei et al., 2004), in which Kv4.2 is abundantly expressed (Vacher et al., 2008). Our findings suggest that Kv4.2 currents could be rapidly downregulated by PACAP/PAC1 signaling that is dependent on convergent PKA/ERK1/2-mediated phosphoregulation of the channel. The precise mechanisms underlying internalization of Kv4.2 are not known. A dileucine motif in the C-terminus of the channel has previously been shown to be critical for forskolin-mediated internalization of surface Kv4.2 channels (Hammond et al., 2008). AMPA-mediated internalization of Kv4.2 channels from dendritic spines was also shown to be mediated by clathrin-dependent endocytosis (Kim et al., 2007). Additionally, it has been suggested that PKA phosphorylation-induced internalization of Kv4.2 is dependent on its interaction with the A-kinase anchoring protein 5 (AKAP5, also known as AKAP79/150) scaffold, which carries the regulatory subunits of PKA (Lin et al., 2011). However, ERK1/2 phosphorylation/activation are independent of any direct influence from AKAP5 scaffolding or signaling. Therefore, the precise molecular and cellular mechanisms underlying ERK1/2-mediated internalization of cell surface Kv4.2 protein remains to be elucidated.
Previous studies have shown that Kv4.2 can undergo rapid PKA-dependent phosphorylation at residue S552, following an increase in neuronal activity or treatment with forskolin, which led to a decrease in the cell surface density of Kv4.2 protein and reduction in the total channel currents (Hammond et al., 2008, Lin et al., 2011). Our results show that PACAP activation of PAC1 increased cAMP production to a similar or greater extent than forskolin treatment, and pharmacological blockade of PKA activity led to no significant reduction of Kv4.2 current density by PACAP38 at physiologically-relevant membrane potentials. However, the phospho-disruptive mutant Kv4.2-S552A channel exhibits PACAP38-induced reduction of current density similar to that observed in WT channels, suggesting no direct phosphorylation of Kv4.2 by PKA downstream of PACAP/PAC1 activation. Furthermore, our biochemical analysis in HEK293T cells showed attenuation of PACAP-mediated ERK1/2 phosphorylation/activation in the major/abundant PAC1 isoform Null by pharmacological blockade of PKA, which suggests that PKA signaling converges on ERK1/2 activation, in order to downregulate Kv4.2 current density. Phosphorylation of Kv4.2 by ERK1/2 at residues T602, T607 and S616 has previously been shown in hippocampal neurons (Schrader et al., 2006). Other studies have also shown that activation of ERK1/2 [both by direct pharmacological modulators, as well as downstream of metabotropic glutamate receptor 5 (mGluR5) activation] leads to phosphorylation of Kv4.2, in particular at the residue S616, resulting in reduced IA current density in spinal cord dorsal horn neurons (Hu et al., 2006, Hu et al., 2007, Hu and Gereau, 2011). Our results are consistent with these observations, and suggest that the PACAP/PAC1-mediated reduction in Kv4.2 current is mediated by direct phosphorylation of the channel protein by ERK1/2. Furthermore, our observations also suggest S616 as the critical ERK1/2 phosphorylation site, as has been shown for mGluR5-mediated phosphorylation of Kv4.2 (Hu et al., 2007, Hu and Gereau, 2011). Since, PACAP did not significantly alter the voltage-dependence of channel activation/inactivation, and PACAP treatment led to a PKA/ERK1/2-dependent decrease in the cells surface channel numbers, the reduction of channel density can be attributed mostly to the later.
Reduction in Kv4.2 currents leads to an increase in AP back-propagation, which ultimately results in enhanced excitability of mammalian brain neurons (Yuan et al., 2002, Chen et al., 2006a). Application of PACAP38 to CA1 hippocampal neurons has been shown to induce increased AP firing frequency (Di Mauro et al., 2003), although the underlying mechanism is not known. Findings from our study indicate that these effects could possibly result from PACAP38-mediated reduction in Kv4.2 currents. Interestingly, it was also shown that PACAP reduces the slow afterhyperpolarizing currents (sIAHP) in CA1 hippocampal pyramidal neurons, which resulted in increased AP firing frequency (Taylor et al., 2014). Therefore, in-depth future investigations are required to find out the relative contribution of PACAP-modulation of Kv4.2 vs sIAHP currents in the regulation of dendritic and overall neuronal excitability. Several studies have shown that PACAP expression is increased in and around the infarct area following cerebral ischemic stroke (Chen et al., 2006b). Furthermore, mice lacking a functional PACAP gene (Adcyap-/-) exhibit increased infarct volume upon induction of stroke, and intracerebroventricular administration of PACAP in both WT and Adcyap-/- mice can significantly reduce infarct volumes and the delayed inflammatory response following cerebral ischemia (Chen et al., 2006b, Ohtaki et al., 2006, Brifault et al., 2014). Rapid reperfusion following ischemic stroke leads to increased excitability of neurons, although the underlying intrinsic mechanisms are poorly understood (Witte and Stoll, 1997, Varelas and Mirski, 2001). Upregulation of Kv4.2-based IA currents in striatal neurons has been suggested to provide a neuroprotective mechanism following cerebral ischemia (Deng et al., 2011). Future studies are thus needed to explore the role of neuronal PACAP/PAC1-Kv4.2 signaling in alterations of intrinsic excitability associated with cerebral stroke-reperfusion injury.
The Kv4.2 channel has also been shown to be critical in spinal cord dorsal horn neurons for central sensitization of chronic pain conditions (Hu et al., 2006, Hu and Gereau, 2011). Increased ERK1/2 activation is observed in these neurons upon induction of chronic inflammatory and neuropathic pain (Ji et al., 2002, Zhuang et al., 2005, Zhang et al., 2014). Also, PACAP expression has been shown to be increased in spinal cord dorsal horn regions following induction of chronic inflammatory and neuropathic pain conditions (Mabuchi et al., 2004). Based on our findings, there is a high likelihood that PACAP-ERK1/2-Kv4.2 signaling axis could subserve a mechanism for the central sensitization of chronic inflammatory and neuropathic pain, which needs to be verified experimentally. In summary, our study suggests that PACAP, a critical neuropeptide that is endogenously released at elevated levels in multiple neuro-pathological conditions, could serve as a modulator of neuronal excitability, by dynamic modulation of Kv4.2 channel phosphorylation and function.
Supplementary Material
Supplementary Figure 1: Kv4.2 and PAC1 are co-expressed in rat brain. Representative confocal microscopic images of the hippocampal formation in sections from rat brain (40 μm; sagittal) immunostained with antibodies against Kv4.2 (green) and PAC1 (red), and DAPI (blue). Kv4.2 expression pattern in rat brain is similar to that of mouse with intense immunoreactivity in stratum oriens and stratum radiatum layers of CA1 and CA3 regions (magnified images shown in middle and bottom rows), as well as in the molecular layer of the dentate gyrus. Although no distinct localization was seen for PAC1, relatively less immunoreactivity is observed in the pyramidal cell body layer of CA1 and CA3 as compared to stratum oriens and stratum radiatum layers. Scale bar – 500 μm for top row, and 50 μm for middle- and bottom-row images.
Supplementary Figure 2: Kv4.2 and PAC1 are localized in neuronal soma and dendrites. Representative epifluorescence microscopic images of cultured rat hippocampal neuron immunostained with antibodies against PAC1 (green), Kv4.2 (red) and GFAP (blue). Images in the bottom row represent magnified views of the rectangular area marked on the respective top row images. PAC1 staining is observed in the neuronal soma and dendrites, as well as in the cell body and processes of astro/glial cells.
Supplementary Figure 3: cDNA cloning of PAC1-Null, PAC1-Hop1 and PAC1-Hop2 isoforms expressed in mouse hippocampus. The full-length nucleotide sequences of the PAC1 isoforms cloned from RNA obtained from mouse hippocampus are aligned as shown. Blue texts below the underlined sequence texts denote the transmembrane (TM) domains 1 to 7. Mutations resulting in a change in amino acid residues upon translation are denoted as white text with red highlight along with the corresponding alteration in the amino acid residue below that row. Silent mutations that do not affect the overall amino acid sequence are denoted in white text with green highlight. The variable intracellular region between 5th and 6th TM domains are shown as black text with grey highlight for PAC1-Hop1 and PAC1-Hop2 isoforms.
Highlights.
PACAP activation of hippocampal PAC1 isoforms leads to PKA and ERK1/2 activation.
The major dendritic K+ channel Kv4.2 is dynamically downregulated by PAC1/PKA/ERK1/2 signaling.
Convergence of PKA and ERK1/2 and phosphorylation of Kv4.2 by the later reduces channel density.
PACAP modulation of Kv4.2 is not influenced by voltage-dependence of channel activity.
Acknowledgments
This work was supported by NIH grants NINDS-NS069898 (to DPM), and NINDS-NS056244 (to SS) and NIDA-DA034929 (to MRB), as well as by start-up funds from the Office of the Vice President of Research, University of Iowa, and Washington University School of Medicine, Department of Anesthesiology and Washington University Pain Center (to DPM). The authors declare no competing financial interests. The authors wish to thank Dr. Robert W. Gereau IVth (Washington University School of Medicine) and Drs. Christopher J. Benson, Yuriy M. Usachev and Amy Lee (University of Iowa Carver College of Medicine) for their critical suggestions on this work.
Footnotes
Conflict of Interest Statement: The authors declare that they have no conflicts of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD. The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. Journal of neurochemistry. 2000;75:2277–2287. doi: 10.1046/j.1471-4159.2000.0752277.x. [DOI] [PubMed] [Google Scholar]
- Anderson AE, Adams JP, Qian Y, Cook RG, Pfaffinger PJ, Sweatt JD. Kv4.2 phosphorylation by cyclic AMP-dependent protein kinase. The Journal of biological chemistry. 2000;275:5337–5346. doi: 10.1074/jbc.275.8.5337. [DOI] [PubMed] [Google Scholar]
- Arimura A. Perspectives on pituitary adenylate cyclase activating polypeptide (PACAP) in the neuroendocrine, endocrine, and nervous systems. The Japanese journal of physiology. 1998;48:301–331. doi: 10.2170/jjphysiol.48.301. [DOI] [PubMed] [Google Scholar]
- Arimura A, Somogyvari-Vigh A, Weill C, Fiore RC, Tatsuno I, Bay V, Brenneman DE. PACAP functions as a neurotrophic factor. Annals of the New York Academy of Sciences. 1994;739:228–243. doi: 10.1111/j.1749-6632.1994.tb19825.x. [DOI] [PubMed] [Google Scholar]
- Barnwell LF, Lugo JN, Lee WL, Willis SE, Gertz SJ, Hrachovy RA, Anderson AE. Kv4.2 knockout mice demonstrate increased susceptibility to convulsant stimulation. Epilepsia. 2009;50:1741–1751. doi: 10.1111/j.1528-1167.2009.02086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiological reviews. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Blechman J, Levkowitz G. Alternative Splicing of the Pituitary Adenylate Cyclase-Activating Polypeptide Receptor PAC1: Mechanisms of Fine Tuning of Brain Activity. Frontiers in endocrinology. 2013;4:55. doi: 10.3389/fendo.2013.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brifault C, Gras M, Liot D, May V, Vaudry D, Wurtz O. Delayed Pituitary Adenylate Cyclase-Activating Polypeptide Delivery After Brain Stroke Improves Functional Recovery by Inducing M2 Microglia/Macrophage Polarization. Stroke; a journal of cerebral circulation. 2014 doi: 10.1161/STROKEAHA.114.006864. [DOI] [PubMed] [Google Scholar]
- Broca C, Quoyer J, Costes S, Linck N, Varrault A, Deffayet PM, Bockaert J, Dalle S, Bertrand G. beta-Arrestin 1 is required for PAC1 receptor-mediated potentiation of long-lasting ERK1/2 activation by glucose in pancreatic beta-cells. The Journal of biological chemistry. 2009;284:4332–4342. doi: 10.1074/jbc.M807595200. [DOI] [PubMed] [Google Scholar]
- Carrasquillo Y, Burkhalter A, Nerbonne JM. A-type K+ channels encoded by Kv4.2, Kv4.3 and Kv1.4 differentially regulate intrinsic excitability of cortical pyramidal neurons. The Journal of physiology. 2012;590:3877–3890. doi: 10.1113/jphysiol.2012.229013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo Y, Nerbonne JM. IA channels: diverse regulatory mechanisms. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry. 2014;20:104–111. doi: 10.1177/1073858413504003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee TK, Sharma RV, Fisher RA. Molecular cloning of a novel variant of the pituitary adenylate cyclase-activating polypeptide (PACAP) receptor that stimulates calcium influx by activation of L-type calcium channels. The Journal of biological chemistry. 1996;271:32226–32232. doi: 10.1074/jbc.271.50.32226. [DOI] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006a;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Samal B, Hamelink CR, Xiang CC, Chen Y, Chen M, Vaudry D, Brownstein MJ, Hallenbeck JM, Eiden LE. Neuroprotection by endogenous and exogenous PACAP following stroke. Regulatory peptides. 2006b;137:4–19. doi: 10.1016/j.regpep.2006.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng P, Pang ZP, Lei Z, Shikano S, Xiong Q, Harvey BK, London B, Wang Y, Li M, Xu ZC. Up-regulation of A-type potassium currents protects neurons against cerebral ischemia. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:1823–1835. doi: 10.1038/jcbfm.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Mauro M, Cavallaro S, Ciranna L. Pituitary adenylate cyclase-activating polypeptide modifies the electrical activity of CA1 hippocampal neurons in the rat. Neuroscience letters. 2003;337:97–100. doi: 10.1016/s0304-3940(02)01316-2. [DOI] [PubMed] [Google Scholar]
- Fan Y, Deng P, Wang YC, Lu HC, Xu ZC, Schulz PE. Transient cerebral ischemia increases CA1 pyramidal neuron excitability. Experimental neurology. 2008;212:415–421. doi: 10.1016/j.expneurol.2008.04.032. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Saraceno C, Malinverno M, Marcello E, Verpelli C, Sala C, Di Luca M. The neuropeptide PACAP38 induces dendritic spine remodeling through ADAM10-N-cadherin signaling pathway. Journal of cell science. 2012;125:1401–1406. doi: 10.1242/jcs.097576. [DOI] [PubMed] [Google Scholar]
- Grimaldi M, Cavallaro S. Functional and molecular diversity of PACAP/VIP receptors in cortical neurons and type I astrocytes. The European journal of neuroscience. 1999;11:2767–2772. doi: 10.1046/j.1460-9568.1999.00693.x. [DOI] [PubMed] [Google Scholar]
- Hammond RS, Lin L, Sidorov MS, Wikenheiser AM, Hoffman DA. Protein kinase a mediates activity-dependent Kv4.2 channel trafficking. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:7513–7519. doi: 10.1523/JNEUROSCI.1951-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Lucero MT. Pituitary adenylate cyclase activating polypeptide reduces A-type K+ currents and caspase activity in cultured adult mouse olfactory neurons. Neuroscience. 2005;134:745–756. doi: 10.1016/j.neuroscience.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Han P, Lucero MT. Pituitary adenylate cyclase activating polypeptide reduces expression of Kv1.4 and Kv4.2 subunits underlying A-type K(+) current in adult mouse olfactory neuroepithelia. Neuroscience. 2006;138:411–419. doi: 10.1016/j.neuroscience.2005.11.028. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Yamamoto K, Hagigara N, Ogawa N, Nishino A, Aino H, Nogi H, Imanishi K, Matsuda T, Baba A. cDNA cloning of a mouse pituitary adenylate cyclase-activating polypeptide receptor. Biochimica et biophysica acta. 1996;1281:129–133. doi: 10.1016/0005-2736(96)00056-9. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Johnston D. Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1998;18:3521–3528. doi: 10.1523/JNEUROSCI.18-10-03521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Alter BJ, Carrasquillo Y, Qiu CS, Gereau RWt. Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:13181–13191. doi: 10.1523/JNEUROSCI.0269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RWt. The kv4.2 potassium channel subunit is required for pain plasticity. Neuron. 2006;50:89–100. doi: 10.1016/j.neuron.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Gereau RWt. Metabotropic glutamate receptor 5 regulates excitability and Kv4.2-containing K(+) channels primarily in excitatory neurons of the spinal dorsal horn. Journal of neurophysiology. 2011;105:3010–3021. doi: 10.1152/jn.01050.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamen F, Puech R, Bockaert J, Brabet P, Bertrand G. Pituitary adenylate cyclase-activating polypeptide receptors mediating insulin secretion in rodent pancreatic islets are coupled to adenylate cyclase but not to PLC. Endocrinology. 2002;143:1253–1259. doi: 10.1210/endo.143.4.8739. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Hoffman DA, Magee JC, Poolos NP, Watanabe S, Colbert CM, Migliore M. Dendritic potassium channels in hippocampal pyramidal neurons. The Journal of physiology. 2000;525(Pt 1):75–81. doi: 10.1111/j.1469-7793.2000.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J, Forsythe ID, Kopp-Scheinpflug C. Going native: voltage-gated potassium channels controlling neuronal excitability. The Journal of physiology. 2010;588:3187–3200. doi: 10.1113/jphysiol.2010.191973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo KM, Chung YH, Kim MK, Nam RH, Lee BL, Lee KH, Cha CI. Distribution of vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide receptors (VPAC1, VPAC2, and PAC1 receptor) in the rat brain. The Journal of comparative neurology. 2004;476:388–413. doi: 10.1002/cne.20231. [DOI] [PubMed] [Google Scholar]
- Jung SC, Kim J, Hoffman DA. Rapid, bidirectional remodeling of synaptic NMDA receptor subunit composition by A-type K+ channel activity in hippocampal CA1 pyramidal neurons. Neuron. 2008;60:657–671. doi: 10.1016/j.neuron.2008.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves K, Arimura A, Gorcs TG, Somogyvari-Vigh A. Comparative distribution of immunoreactive pituitary adenylate cyclase activating polypeptide and vasoactive intestinal polypeptide in rat forebrain. Neuroendocrinology. 1991;54:159–169. doi: 10.1159/000125864. [DOI] [PubMed] [Google Scholar]
- Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nature reviews Neuroscience. 2006;7:548–562. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- Lin L, Sun W, Kung F, Dell'Acqua ML, Hoffman DA. AKAP79/150 impacts intrinsic excitability of hippocampal neurons through phospho-regulation of A-type K+ channel trafficking. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:1323–1332. doi: 10.1523/JNEUROSCI.5383-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YQ, Huang WX, Sanchez RM, Min JW, Hu JJ, He XH, Peng BW. Regulation of Kv4.2 A-Type Potassium Channels in HEK-293 Cells by Hypoxia. Frontiers in cellular neuroscience. 2014;8:329. doi: 10.3389/fncel.2014.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu N, Zhou R, DiCicco-Bloom E. Opposing mitogenic regulation by PACAP in sympathetic and cerebral cortical precursors correlates with differential expression of PACAP receptor (PAC1-R) isoforms. Journal of neuroscience research. 1998;53:651–662. doi: 10.1002/(SICI)1097-4547(19980915)53:6<651::AID-JNR3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Lugo JN, Barnwell LF, Ren Y, Lee WL, Johnston LD, Kim R, Hrachovy RA, Sweatt JD, Anderson AE. Altered phosphorylation and localization of the A-type channel, Kv4.2 in status epilepticus. Journal of neurochemistry. 2008;106:1929–1940. doi: 10.1111/j.1471-4159.2008.05508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi T, Shintani N, Matsumura S, Okuda-Ashitaka E, Hashimoto H, Muratani T, Minami T, Baba A, Ito S. Pituitary adenylate cyclase-activating polypeptide is required for the development of spinal sensitization and induction of neuropathic pain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:7283–7291. doi: 10.1523/JNEUROSCI.0983-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald DS, Weerapura M, Beazely MA, Martin L, Czerwinski W, Roder JC, Orser BA, MacDonald JF. Modulation of NMDA receptors by pituitary adenylate cyclase activating peptide in CA1 neurons requires G alpha q, protein kinase C, and activation of Src. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:11374–11384. doi: 10.1523/JNEUROSCI.3871-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May V, Buttolph TR, Girard BM, Clason TA, Parsons RL. PACAP-induced ERK activation in HEK cells expressing PAC1 receptors involves both receptor internalization and PKC signaling. American journal of physiology Cell physiology. 2014;306:C1068–1079. doi: 10.1152/ajpcell.00001.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei YA, Vaudry D, Basille M, Castel H, Fournier A, Vaudry H, Gonzalez BJ. PACAP inhibits delayed rectifier potassium current via a cAMP/PKA transduction pathway: evidence for the involvement of I k in the anti-apoptotic action of PACAP. The European journal of neuroscience. 2004;19:1446–1458. doi: 10.1111/j.1460-9568.2004.03227.x. [DOI] [PubMed] [Google Scholar]
- Meier CL, Obenaus A, Dudek FE. Persistent hyperexcitability in isolated hippocampal CA1 of kainate-lesioned rats. Journal of neurophysiology. 1992;68:2120–2127. doi: 10.1152/jn.1992.68.6.2120. [DOI] [PubMed] [Google Scholar]
- Menegola M, Trimmer JS. Unanticipated region- and cell-specific downregulation of individual KChIP auxiliary subunit isotypes in Kv4.2 knock-out mouse brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:12137–12142. doi: 10.1523/JNEUROSCI.2783-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill RA, Slupe AM, Strack S. N-terminal phosphorylation of protein phosphatase 2A/Bbeta2 regulates translocation to mitochondria, dynamin-related protein 1 dephosphorylation, and neuronal survival. The FEBS journal. 2013;280:662–673. doi: 10.1111/j.1742-4658.2012.08631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata A, Arimura A, Dahl RR, Minamino N, Uehara A, Jiang L, Culler MD, Coy DH. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochemical and biophysical research communications. 1989;164:567–574. doi: 10.1016/0006-291x(89)91757-9. [DOI] [PubMed] [Google Scholar]
- Miyata A, Jiang L, Dahl RD, Kitada C, Kubo K, Fujino M, Minamino N, Arimura A. Isolation of a neuropeptide corresponding to the N-terminal 27 residues of the pituitary adenylate cyclase activating polypeptide with 38 residues (PACAP38) Biochemical and biophysical research communications. 1990;170:643–648. doi: 10.1016/0006-291x(90)92140-u. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Misonou H, Pan SJ, Held JE, Surmeier DJ, Trimmer JS. Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels. 2009;3:46–56. doi: 10.4161/chan.3.1.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Siino DF, Trimmer JS. Interdomain cytoplasmic interactions govern the intracellular trafficking, gating, and modulation of the Kv2.1 channel. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:4982–4994. doi: 10.1523/JNEUROSCI.0186-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Trimmer JS. The Kv2.1 C terminus can autonomously transfer Kv2.1-like phosphorylation-dependent localization, voltage-dependent gating, and muscarinic modulation to diverse Kv channels. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:685–695. doi: 10.1523/JNEUROSCI.4620-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot A, DiCicco-Bloom E. Regulation of neuroblast mitosis is determined by PACAP receptor isoform expression. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4758–4763. doi: 10.1073/pnas.071465398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Ueta Y, Hannibal J, Serino R, Yamamoto Y, Shibuya I, Matsumoto T, Yamashita H. Induction of pituitary adenylate cyclase-activating polypeptide mRNA in the medial parvocellular part of the paraventricular nucleus of rats following kainic-acid-induced seizure. Neuroendocrinology. 2000;71:318–326. doi: 10.1159/000054552. [DOI] [PubMed] [Google Scholar]
- Norris AJ, Nerbonne JM. Molecular dissection of I(A) in cortical pyramidal neurons reveals three distinct components encoded by Kv4.2, Kv4.3, and Kv1.4 alpha-subunits. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:5092–5101. doi: 10.1523/JNEUROSCI.5890-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtaki H, Nakamachi T, Dohi K, Aizawa Y, Takaki A, Hodoyama K, Yofu S, Hashimoto H, Shintani N, Baba A, Kopf M, Iwakura Y, Matsuda K, Arimura A, Shioda S. Pituitary adenylate cyclase-activating polypeptide (PACAP) decreases ischemic neuronal cell death in association with IL-6. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7488–7493. doi: 10.1073/pnas.0600375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni C, Brabet P, Bilanges B, Dumuis A, Houssami S, Spengler D, Bockaert J, Journot L. Alternative splicing in the N-terminal extracellular domain of the pituitary adenylate cyclase-activating polypeptide (PACAP) receptor modulates receptor selectivity and relative potencies of PACAP-27 and PACAP-38 in phospholipase C activation. The Journal of biological chemistry. 1996;271:22146–22151. doi: 10.1074/jbc.271.36.22146. [DOI] [PubMed] [Google Scholar]
- Rhodes KJ, Carroll KI, Sung MA, Doliveira LC, Monaghan MM, Burke SL, Strassle BW, Buchwalder L, Menegola M, Cao J, An WF, Trimmer JS. KChIPs and Kv4 alpha subunits as integral components of A-type potassium channels in mammalian brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:7903–7915. doi: 10.1523/JNEUROSCI.0776-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends in neurosciences. 2001;24:517–526. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Schiene K, Bruehl C, Zilles K, Qu M, Hagemann G, Kraemer M, Witte OW. Neuronal hyperexcitability and reduction of GABAA-receptor expression in the surround of cerebral photothrombosis. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 1996;16:906–914. doi: 10.1097/00004647-199609000-00014. [DOI] [PubMed] [Google Scholar]
- Schrader LA, Anderson AE, Mayne A, Pfaffinger PJ, Sweatt JD. PKA modulation of Kv4.2-encoded A-type potassium channels requires formation of a supramolecular complex. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:10123–10133. doi: 10.1523/JNEUROSCI.22-23-10123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader LA, Birnbaum SG, Nadin BM, Ren Y, Bui D, Anderson AE, Sweatt JD. ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. American journal of physiology Cell physiology. 2006;290:C852–861. doi: 10.1152/ajpcell.00358.2005. [DOI] [PubMed] [Google Scholar]
- Schrader LA, Ren Y, Cheng F, Bui D, Sweatt JD, Anderson AE. Kv4.2 is a locus for PKC and ERK/MAPK cross-talk. The Biochemical journal. 2009;417:705–715. doi: 10.1042/BJ20081213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Rocken C, Mawrin C, Weise W, Hollt V, Schulz S. Immunocytochemical identification of VPAC1, VPAC2, and PAC1 receptors in normal and neoplastic human tissues with subtype-specific antibodies. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:8235–8242. doi: 10.1158/1078-0432.CCR-04-0939. [DOI] [PubMed] [Google Scholar]
- Seikel E, Trimmer JS. Convergent modulation of Kv4.2 channel alpha subunits by structurally distinct DPPX and KChIP auxiliary subunits. Biochemistry. 2009;48:5721–5730. doi: 10.1021/bi802316m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Loo L, Gupte RP, Mickle AD, Mohapatra DP. Distinct modifications in Kv2.1 channel via chemokine receptor CXCR4 regulate neuronal survival-death dynamics. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:17725–17739. doi: 10.1523/JNEUROSCI.3029-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg PH, Journot L. Differential signal transduction by five splice variants of the PACAP receptor. Nature. 1993;365:170–175. doi: 10.1038/365170a0. [DOI] [PubMed] [Google Scholar]
- Stumm R, Kolodziej A, Prinz V, Endres M, Wu DF, Hollt V. Pituitary adenylate cyclase-activating polypeptide is up-regulated in cortical pyramidal cells after focal ischemia and protects neurons from mild hypoxic/ischemic damage. Journal of neurochemistry. 2007;103:1666–1681. doi: 10.1111/j.1471-4159.2007.04895.x. [DOI] [PubMed] [Google Scholar]
- Taylor RD, Madsen MG, Krause M, Sampedro-Castaneda M, Stocker M, Pedarzani P. Pituitary adenylate cyclase-activating polypeptide (PACAP) inhibits the slow afterhyperpolarizing current sIAHP in CA1 pyramidal neurons by activating multiple signaling pathways. Hippocampus. 2014;24:32–43. doi: 10.1002/hipo.22201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaur ML, Sheng M, Lowenstein DH, Jan YN, Jan LY. Differential expression of K+ channel mRNAs in the rat brain and down-regulation in the hippocampus following seizures. Neuron. 1992;8:1055–1067. doi: 10.1016/0896-6273(92)90127-y. [DOI] [PubMed] [Google Scholar]
- Ushiyama M, Ikeda R, Sugawara H, Yoshida M, Mori K, Kangawa K, Inoue K, Yamada K, Miyata A. Differential intracellular signaling through PAC1 isoforms as a result of alternative splicing in the first extracellular domain and the third intracellular loop. Molecular pharmacology. 2007;72:103–111. doi: 10.1124/mol.107.035477. [DOI] [PubMed] [Google Scholar]
- Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiological reviews. 2008;88:1407–1447. doi: 10.1152/physrev.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas PN, Mirski MA. Seizures in the adult intensive care unit. Journal of neurosurgical anesthesiology. 2001;13:163–175. doi: 10.1097/00008506-200104000-00016. [DOI] [PubMed] [Google Scholar]
- Villalba M, Bockaert J, Journot L. Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17:83–90. doi: 10.1523/JNEUROSCI.17-01-00083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte OW, Stoll G. Delayed and remote effects of focal cortical infarctions: secondary damage and reactive plasticity. Advances in neurology. 1997;73:207–227. [PubMed] [Google Scholar]
- Wu SY, Dun NJ. Potentiation of NMDA currents by pituitary adenylate cyclase activating polypeptide in neonatal rat sympathetic preganglionic neurons. Journal of neurophysiology. 1997;78:1175–1179. doi: 10.1152/jn.1997.78.2.1175. [DOI] [PubMed] [Google Scholar]
- Yaka R, He DY, Phamluong K, Ron D. Pituitary adenylate cyclase-activating polypeptide (PACAP(1-38)) enhances N-methyl-D-aspartate receptor function and brain-derived neurotrophic factor expression via RACK1. The Journal of biological chemistry. 2003;278:9630–9638. doi: 10.1074/jbc.M209141200. [DOI] [PubMed] [Google Scholar]
- Yan Y, Zhou X, Pan Z, Ma J, Waschek JA, DiCicco-Bloom E. Pro- and anti-mitogenic actions of pituitary adenylate cyclase-activating polypeptide in developing cerebral cortex: potential mediation by developmental switch of PAC1 receptor mRNA isoforms. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:3865–3878. doi: 10.1523/JNEUROSCI.1062-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:4860–4868. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang NR, Planer W, Siuda ER, Zhao HC, Stickler L, Chang SD, Baird MA, Cao YQ, Bruchas MR. Serine 363 is required for nociceptin/orphanin FQ opioid receptor (NOPR) desensitization, internalization, and arrestin signaling. The Journal of biological chemistry. 2012;287:42019–42030. doi: 10.1074/jbc.M112.405696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang H, Shao H, Xue Q, Yu B. ERK MAP kinase activation in spinal cord regulates phosphorylation of Cdk5 at serine 159 and contributes to peripheral inflammation induced pain/hypersensitivity. PloS one. 2014;9:e87788. doi: 10.1371/journal.pone.0087788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Kv4.2 and PAC1 are co-expressed in rat brain. Representative confocal microscopic images of the hippocampal formation in sections from rat brain (40 μm; sagittal) immunostained with antibodies against Kv4.2 (green) and PAC1 (red), and DAPI (blue). Kv4.2 expression pattern in rat brain is similar to that of mouse with intense immunoreactivity in stratum oriens and stratum radiatum layers of CA1 and CA3 regions (magnified images shown in middle and bottom rows), as well as in the molecular layer of the dentate gyrus. Although no distinct localization was seen for PAC1, relatively less immunoreactivity is observed in the pyramidal cell body layer of CA1 and CA3 as compared to stratum oriens and stratum radiatum layers. Scale bar – 500 μm for top row, and 50 μm for middle- and bottom-row images.
Supplementary Figure 2: Kv4.2 and PAC1 are localized in neuronal soma and dendrites. Representative epifluorescence microscopic images of cultured rat hippocampal neuron immunostained with antibodies against PAC1 (green), Kv4.2 (red) and GFAP (blue). Images in the bottom row represent magnified views of the rectangular area marked on the respective top row images. PAC1 staining is observed in the neuronal soma and dendrites, as well as in the cell body and processes of astro/glial cells.
Supplementary Figure 3: cDNA cloning of PAC1-Null, PAC1-Hop1 and PAC1-Hop2 isoforms expressed in mouse hippocampus. The full-length nucleotide sequences of the PAC1 isoforms cloned from RNA obtained from mouse hippocampus are aligned as shown. Blue texts below the underlined sequence texts denote the transmembrane (TM) domains 1 to 7. Mutations resulting in a change in amino acid residues upon translation are denoted as white text with red highlight along with the corresponding alteration in the amino acid residue below that row. Silent mutations that do not affect the overall amino acid sequence are denoted in white text with green highlight. The variable intracellular region between 5th and 6th TM domains are shown as black text with grey highlight for PAC1-Hop1 and PAC1-Hop2 isoforms.