

Abstract
Hypokalemia (low serum potassium level) is a common electrolyte imbalance that can cause a defect in urinary concentrating ability, i.e., nephrogenic diabetes insipidus (NDI), but the molecular mechanism is unknown. We employed proteomic analysis of inner medullary collecting ducts (IMCD) from rats fed with a potassium-free diet for 1 day. IMCD protein quantification was performed by mass spectrometry using a label-free methodology. A total of 131 proteins, including the water channel AQP2, exhibited significant changes in abundance, most of which were decreased. Bioinformatic analysis revealed that many of the down-regulated proteins were associated with the biological processes of generation of precursor metabolites and energy, actin cytoskeleton organization, and cell-cell adhesion. Targeted LC-MS/MS and immunoblotting studies further confirmed the down regulation of 18 selected proteins. Electron microscopy showed autophagosomes/autophagolysosomes in the IMCD cells of rats deprived of potassium for only 1 day. An increased number of autophagosomes was also confirmed by immunofluorescence, demonstrating co-localization of LC3 and Lamp1 with AQP2 and several other down-regulated proteins in IMCD cells. AQP2 was also detected in autophagosomes in IMCD cells of potassium-deprived rats by immunogold electron microscopy. Thus, enhanced autophagic degradation of proteins, most notably including AQP2, is an early event in hypokalemia-induced NDI.
Hypokalemia (low serum potassium level) is a very common electrolyte imbalance encountered in clinical medicine, occurring as a result of poor nutritional status, gastrointestinal diseases, and side effects from numerous medications. This potentially life-threatening condition adversely affects multiple organ systems, producing cardiac arrhythmias, muscle weakness and various abnormalities in renal function. One of the renal impairments caused by hypokalemia is a reduction in urinary concentrating ability and a lack of response to the antidiuretic hormone arginine vasopressin (AVP), resulting in nephrogenic diabetes insipidus (NDI; characterized by excessive thirst and excretion of large amounts of very dilute urine).
The ability to maintain water balance in humans is based on the function of kidneys to concentrate urine, a process which is regulated through the vasopressin/V2-receptor/aquaporin-2 axis. Approximately 180 L/day of blood entering the kidneys is filtered through glomeruli, however, only less than 1% of the filtered water is finally excreted as urine. In the kidneys, 90% of the filtered fluid is reabsorbed in the proximal tubules and thin ascending limb of Henle’s loop. The remaining 10% of the filtered fluid is reabsorbed in the connecting tubules and collecting ducts under the control of vasopressin, the water channel protein aquaporin-2 (AQP2), and other signal molecules1,2. The collecting duct system plays a critical role in fluid homeostasis since it is the site for the final stage of the urine concentrating process.
The ability of the collecting duct to concentrate urine depends predominantly on the functions of key proteins involved in two interdependent processes. First, the NKCC2 protein (bumetanide-sensitive Na+-K+-2Cl− cotransporter) functions to generate medullary interstitial hyperosmolality. Second, AQP2 and urea transporter proteins (UT-A1, UT-A3) regulate osmotic water and urea permeability of the renal collecting duct epithelium. AQP2 is expressed in principal cells of the cortical, outer, and inner medullary collecting ducts and is abundant in both the apical plasma membrane and subapical vesicles3. AQP2 is the primary target for vasopressin regulation of collecting duct water permeability4. The short-term regulation of AQP2 by vasopressin occurs in a period of minutes, accompanied by phosphorylation at serine 256 of AQP2, resulting in trafficking of AQP2-containing vesicles to the apical membrane and increasing water permeability of the principal cell5,6,7. The long-term vasopressin regulation, occurring over a period of hours to days, increases whole-cell abundance of AQP28,9,10.
Potassium deprivation has been reported to induce urinary concentrating defects through alterations in abundances of various proteins, including NKCC2 in thick ascending limb cells as well as AQP2 and/or urea transporter proteins in inner medullary collecting duct cells11,12,13,14. However, even though the induction of NDI by hypokalemia has been known for decades15, the mechanism remains a long-standing mystery.
While most studies of hypokalemia-induced NDI have focused on long-term effects (days to weeks) in the collecting duct, the onset of a urinary concentrating defect in potassium-deprived rats has been reported to be as early as 12–24 hours13. In order to capture the initial alterations that might lead to the pathogenesis of hypokalemia-induced NDI, we chose to investigate the proteome of the collecting duct from potassium-deprived rats at a time before a significant urinary concentrating defect and the consequent confounding alterations have already occurred. Since the inner medullary collecting duct (IMCD) is an important part of the collecting duct system and can be easily isolated in large amounts with high purity, it is more suitable for proteomic experiments than other parts of the collecting duct. Thus, mass spectrometry-based proteomics was employed to identify and quantify changes in abundance of IMCD proteins in this study. The proteomic findings revealed a reduction in abundance of AQP2, as well as proteins involved in mitochondrial energy metabolism, actin cytoskeleton organization, and cell-cell adhesion. The decreased abundance of these proteins was further confirmed by targeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) and immunoblotting.
Next, in an extension of our proteomic analysis, we employed both electron microscopy (EM) and immunofluorescence labeling of IMCD cells to identify the possible mechanism for the observed protein down-regulation. Both of these analyses revealed that the decreased abundance of identified proteins was related to autophagic protein degradation. Numerous studies have demonstrated induction of autophagy in kidney diseases16 and kidney tubular injury resulting from various conditions, e.g., nephrotoxins17 and ischemic injury18. This study is the first to report that autophagy is an early event involved in a common electrolyte imbalance-induced NDI.
Results
Animal model of hypokalemia-induced NDI
Rats were fed a potassium-free diet (NK) or normal chow diet for 1–3 days with monitoring of key physiological parameters. The earliest detectable change was in the urinary fractional excretion of potassium, which decreased significantly (59%) (P < 0.01) within 24 hours of initiating the potassium-free diet (Fig. 1A). For the other monitored parameters, significant changes of serum potassium (P < 0.05), urine volume (P < 0.05), and urine osmolality (P < 0.01) were detected by day 2, representing the earliest appearance of hypokalemia and a urinary concentrating defect (Fig. 1B–D). Decreased urinary excretion of potassium preceding the onset of hypokalemia is explained by the feedforward homeostatic control mechanism involving the gut dietary potassium sensor as described previously19,20,21 (see Discussion for further details).
Figure 1. Physiological parameters of a rat model of early hypokalemia-induced NDI.

Urinary fractional excretion of potassium (A), serum potassium (B), urine volume (C), and urine osmolality (D) were measured in rats receiving either a normal chow diet (■, n = 7) or a potassium-free diet (NK; □, n = 7) for 1, 2, and 3 days. The units for urinary fractional excretion of potassium (shown as %) are a ratio of urinary:plasma potassium concentration normalized to that of urinary:plasma creatinine concentration. (E) Immunoblotting of inner medullary proteins from control rats (+, ■, n = 7) and potassium-deprived rats (–, □, n = 7) after 1 day of potassium deprivation. The two bands of AQP2 (29 kDa non-glycosylated and 37 kDa glycosylated isoforms) are indicated with arrows.
Immunoblotting of proteins from the entire inner medulla demonstrated a 23% decrease in total AQP2 protein (P < 0.05) after 1 day of potassium deprivation (Fig. 1E), in agreement with previous studies12,13. As an additional novel finding in the present study, even larger decreases were detected for phosphorylation of AQP2 at both vasopressin-sensitive sites22, pS256 (59.6%) and pS261 (43.4%). The decreases in AQP2 protein abundance and phosphorylation at day 1 should have been accompanied by an early urinary concentration defect (i.e., reflected by a decrease in urine osmolality), which was not actually detected until day 2 (Fig. 1D). However, since the urine osmolality was an average value from the 24-h collection, it did not represent the actual urine osmolality at the final hours of each collecting period. When urine osmolality was measured in urine recovered from bladders of some of the potassium-deprived rats at 24 hours (i.e., from those animals that had full bladders during kidney tissue harvesting), lower urine osmolality was detected compared to the controls (Supplementary Fig. 1), suggesting that a urinary concentrating defect was present near the end of day 1 in those animals. Cyclic adenosine monophosphate (cAMP) levels in IMCD cells were not significantly different between the potassium-deprived and control rats after 1 day (NK: 8.6 ± 1.0 vs. Control: 10.7 ± 0.52 fmol/μg, P > 0.05). These results confirmed that, even before the appearance of frank hypokalemia, early molecular changes occurred in IMCD cells after only 1 day of potassium deprivation.
Identification of global molecular changes by proteomics and bioinformatics
For proteomic analysis, we selected a time point of 1 day after initiation of potassium deprivation, the time at which the initial molecular changes had started to occur without any change in serum potassium level. A total of 3,396 unique peptides, 2,303 from SEQUEST and 2,635 from InsPecT (Fig. 2A), were identified in IMCD cells with a false discovery rate (FDR) of <1% using a target-decoy analysis (Supplementary data 1). A total of 2,368 unambiguous peptides (i.e., those matching a single protein) were assigned to 802 proteins by the ProMatch program23. The quantification of unambiguous peptides was determined from the extracted-ion chromatograms using QUOIL software24. A histogram of the base-2 logarithm of abundance ratios for all quantified peptides between the potassium-deprived and control groups (Fig. 2B) reveals that the abundances of the majority of peptides were not changed substantially, as confirmed by the total protein intensities in a representative coomassie-stained gel (Fig. 2C). However, a slightly left-skewed distribution (skewness = −0.12, P < 0.05) of the histogram is also observed, reflecting a slight tendency toward decreased abundance among the total group of quantified peptides. Relative abundance at the protein level was then calculated for each protein based on the median abundance ratio of the corresponding peptides in control and potassium-depleted states (Supplementary data 2). A volcano plot in Fig. 2D illustrates the average log2 protein abundance ratios (of those proteins observed in all three biological replicates, total of 475 proteins) versus significance, expressed in –log10 (P-value). Proteins exhibiting significant changes in abundance were identified using a dual statistical criterion: (i) P < 0.05 [−log10 (P-value) > 1.3] by two-tailed t-test and (ii) average |log2 (NK/Control)| ≥ 0.58 (based on a reported 95% confidence interval [95% CI] of the label-free LC-MS/MS technique25). Based on this stringent analysis, we identified 128 proteins that significantly decreased in abundance, while only 3 proteins were significantly increased in IMCD cells of potassium-deprived rats (Supplementary tables 1 and 2). Note that AQP2 was significantly decreased by 28% in this proteomic experiment, in agreement with the previous immunoblotting result shown in Fig. 1E, and confirming the early response to potassium deprivation. A complete list of protein identification, protein quantification, and associated Gene Ontology (GO) terms from this study is provided in a publicly accessible online database at https://hpc.nih.gov/ESBL/Database/HypoK/.
Figure 2. Proteomic analysis of IMCD proteins from the potassium-deprived and control rats.

(A) A total of 3,396 unique peptides were identified. (B) Distribution of log2peptide abundance ratios between two groups for all quantified peptides. (C) A Coomassie-stained gel demonstrates an equal loading amount of total IMCD proteins from the two groups for the proteomic analysis. (D) A volcano plot of 475 proteins observed in all three biological replicates. A horizontal solid line indicates P-value of 0.05 [−log(P-value) of 1.3]. Vertical dashed lines indicate 95% CI of protein abundance ratios [|log2(NK/Control)| ≥ 0.58]25).
The proteins exhibiting significant changes in abundance before the onset of hypokalemia-induced NDI were further investigated using the DAVID bioinformatics resource26. The highly-represented annotations were grouped into clusters (using a list of all IMCD-expressed transcripts as a background). The most significantly enriched clusters included proteins with the functional GO terms “generation of precursor metabolites and energy”, “guanyl nucleotide binding”, “actin cytoskeleton organization”, and “fascia adherens” (Fig. 3 and Supplementary table 3).
Figure 3. Highly-represented functional annotation clusters of the significantly changed proteins.
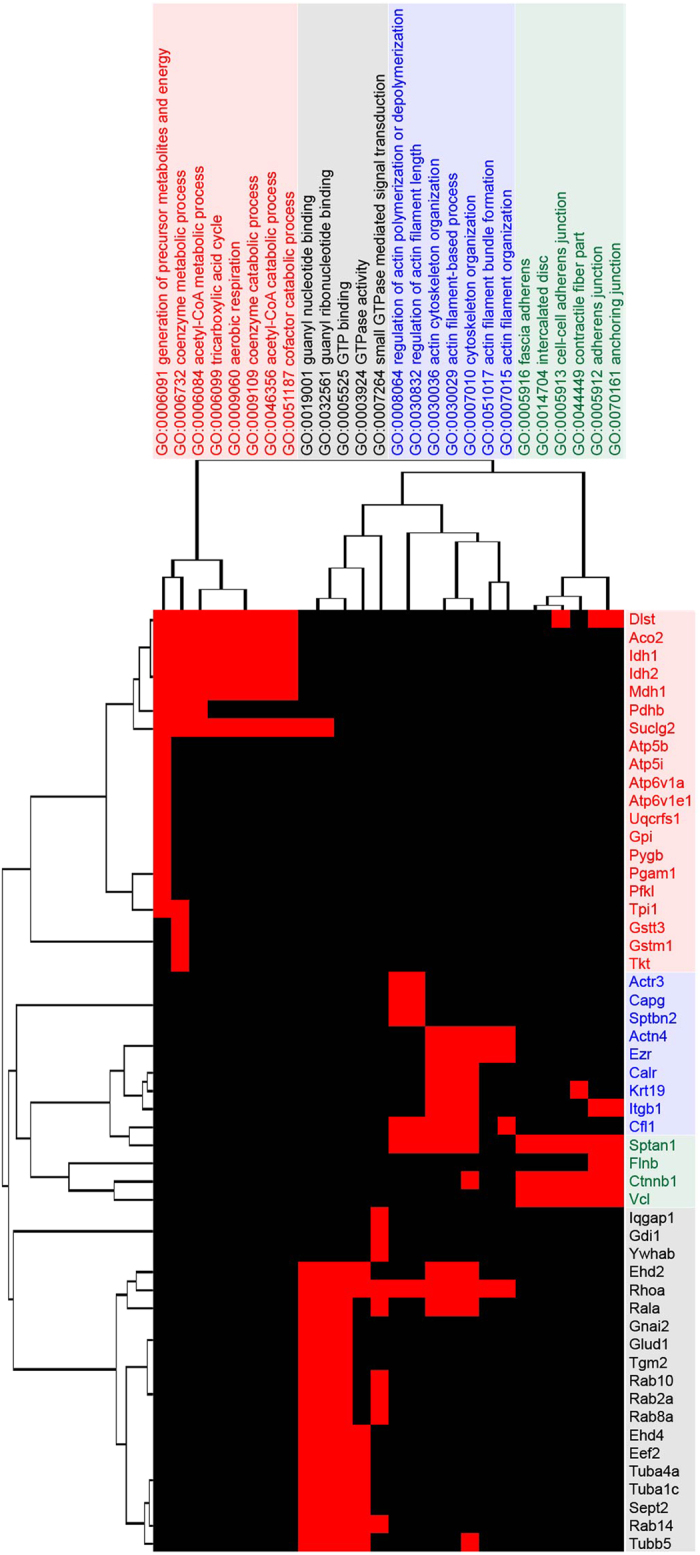
Vertical dendrogram demonstrates four over-represented clusters (P < 0.05) of GO terms. Horizontal dendrogram shows the list of proteins for each cluster.
Data verification by targeted LC-MS/MS and immunoblotting
In order to confirm the observed findings from the proteomic and bioinformatic analyses, a targeted LC-MS/MS approach was utilized as an independent measurement. A new animal experiment was performed for this study using the same upstream experimental setup as for the non-targeted LC-MS/MS experiments. A list of protein names, peptide sequences, precursor m/z ratios, retention times, and integrated peak areas of the 16 proteins selected for analysis by targeted LC-MS/MS is provided in Supplementary data 3.
Consistent with the non-targeted LC-MS/MS technique, the targeted analysis confirmed a reduction in the abundances of all the selected proteins from the discovery proteomic studies (Supplementary Fig. 2A). For instance, we validated down-regulation of 9 proteins in the “generation of precursor metabolites and energy” cluster (Atp6v1e1, Atp5b, Atp6v1a, Atp5i, Ndufs3, Aco2, Idh1, Idh2, and Mdh1), all of which are known mitochondrial proteins. (Beyond the mentioned cluster, there were 22 more mitochondrial proteins that were identified as significantly decreased by the non-targeted LC-MS/MS studies.) Down-regulation was also confirmed for the major proteins in the “actin cytoskeleton organization” and “fascia adherens” clusters (Itgb1, Actn4, Capg, Ctnnb1, Ezr, Sptan1, and Vcl). In addition to the targeted LC-MS/MS results, immunoblotting confirmed the significant down-regulation of three proteins in the “fascia adherens” and “guanyl nucleotide binding” clusters [β-catenin (Ctnnb1) by 30.3%, cortactin (Cttn) by 88.7%, and RhoA by 25%, respectively) in IMCD cells of potassium-deprived rats (Supplementary Fig. 2B).
The early decline (after only 1 day of potassium deprivation) in the abundance of the proteins identified by proteomic analysis could be explained by either an increase in protein degradation or a reduction in protein production of short half-life proteins (i.e., t1/2 shorter than 24 hours). Based on cross-referencing with the t1/2 values at basal conditions for the mouse collecting duct proteins as reported by Sandoval et al.27, the majority of the down-regulated proteins was estimated to possess t1/2 longer than 24 hours (Fig. 4A and Supplementary table 4), thereby eliminating a decrease in protein production as the likely mechanism for the down-regulation of the majority of identified proteins. However, AQP2 and some of the other down-regulated proteins (e.g., Cdh16 and Itgb1) have reported half-lives shorter than 24 hours. Therefore, we quantified changes in mRNA levels of Aqp2 and five representative transcripts from the highly-represented GO clusters (Cdh16, Itgb1, Vcl, Mdh1, and Atp5b). Figure 4B demonstrates no significant differences between the potassium-deprived and control groups for these six transcripts, indicating that the observed decreases in abundance of these proteins are not regulated at the transcriptional level. Thus, other post-transcriptional processes, such as decreased rate of translation and/or increased protein degradation, are responsible for the observed reductions in protein abundance.
Figure 4. Estimated half-lives and transcript quantification of down-regulated proteins.
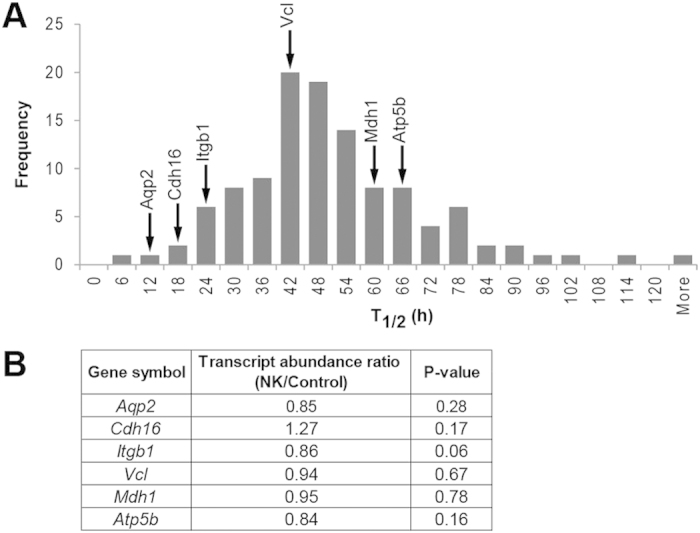
(A) A distribution of the estimated half-lives for the down-regulated proteins identified in this proteomic study (based on mouse collecting duct proteins27). The estimated half-lives of AQP2 and five representative proteins are indicated by arrows (A) and their transcript abundance ratios (NK/Control) as determined by qRT-PCR are shown (B).
Structural abnormalities of IMCD in early hypokalemia-induced NDI
To elucidate the possible mechanism for the protein down-regulation identified by proteomic analysis, we first performed structural and ultrastructural analyses of the IMCD after 1 day of potassium deprivation. Histopathology of the IMCD showed disruption between tubular cells, and occasional collapsed tubules in the potassium-deprived conditions (Fig. 5B vs. the control in 5A). The observed changes were widespread, occurring in all of the examined fields. Electron micrographs revealed distinct ultrastructural changes in the intercellular junctions of the IMCD from potassium-deprived rats (Fig. 5D vs. the control in 5C), including a thick electron dense deposition and disorganized filaments at tight junctions. These observed ultrastructural changes were also widespread, occurring in all cells examined. Moreover, less distinct zonula adherens with thick electron dense deposition was observed in some cells. Electron micrographs of the IMCD from potassium-deprived rats also revealed electron dense depositions within the mitochondrial matrix and swelling of cristae in the majority of mitochondria in all cells (Fig. 5F), as compared to well-defined, sharp mitochondrial cristae in control IMCD (Fig. 5E). Strikingly, single-membrane autophagolysosomes containing electron dense membrane structures (Fig. 5G) and double-membrane autophagosome vesicles or phagophores (early stage of autophagosome formation) were identified by electron microscopy in every observed IMCD cell of the potassium-deprived rats; there were 2–3 autophagosomes/autophagolysosomes per cell, compared to 1 of these structures per 10 cells in control rats (P < 0.05). Damaged mitochondria engulfed by autophagosomes (mitophagy) (Fig. 5H) were found occasionally (but was not quantified) in the potassium-deprived group, however mitophagy was not seen in the control group. The structures of the endoplasmic reticulum, Golgi apparatus, and other organelles were not detectably affected. All findings were based on the observation of at least 10 random cells (electron microscopy) or microscopic fields (light microscopy) from two potassium-deprived rats and two control rats. The swelling and other structural changes to mitochondria we observed could be associated with an activation of apoptosis, which could also be responsible for the down-regulation of proteins identified in this study. To investigate a potential activation of apoptosis in response to potassium deprivation, we performed immunoblotting of both intact and cleaved forms of caspase 3 (Supplementary Fig. 3). The result demonstrated no statistically significant increase in the cleaved form of caspase 3, indicating that apoptosis does not seem to play a major role in the protein degradation process found in this study.
Figure 5. Structural abnormalities of IMCD in early potassium-deprived rats.

Compared to control IMCD (A), histopathology in potassium-deprived IMCD (B) shows disruption between IMCD cells (arrows) and a collapsed tubular lumen (*), Toluidine Blue, original magnification 400x. (C) An electron micrograph (EM) of the intercellular junction of control IMCD cells demonstrates a thin tight junction (arrow) and thin delicate adherens junction (arrow head). (D) A tight junction with disorganized filaments as well as thick electron dense deposition (arrow) and ill-defined adherens junction with thick electron dense deposition (arrow head) were observed in potassium-deprived IMCD cells. (E) Normal mitochondria in control IMCD cells. (F) Abnormal mitochondria in potassium-deprived IMCD show swollen cristae with the focal electron dense deposition (arrow head). Autophagolysosomes (G, arrow heads) and mitophagy (H, arrow heads) in IMCD cells of the potassium-deprived rats.
Co-localization of the down-regulated proteins with autophagosomes/autophagolysosomes
Based on the ultrastructural analysis revealing the extensive presence of autophagic structures in IMCD cells of potassium deprived rats, we further investigated the potential role of autophagy in down-regulation of the proteins detected by proteomic analysis. Co-localization studies were performed by immunofluorescence using antibodies against markers specific for autophagosomes (LC3) and lysosomes (Lamp1). First, numerous puncta containing AQP2 were seen in IMCD cells of the potassium-deprived rats, with significant co-localization with both LC3 and Lamp1 (Fig. 6A), indicating that AQP2 was located in autophagolysosomes. Representative scatter plots demonstrate the high degree of co-localization between AQP2 and LC3 or Lamp1 after potassium deprivation (Fig. 6B). A bar graph summarizes Pearson correlation coefficients of AQP2 and LC3 or Lamp1 from 30 co-localized puncta (Fig. 6C). Other down-regulated proteins identified, including Itgb1, Vcl, Ctnnb1, Ezrin-Moesin-Radixin (ERM), Cttn, and RhoA were also co-localized with AQP2 and Lamp1 in a punctate pattern, also indicating their localization in autophagolysosomes (Fig. 7 and Supplementary Fig. 4). Scatter plots demonstrate the degree of co-localization between Itgb1 and Lamp1 (Pearson correlation coefficient, R = 0.90), Vcl and Lamp1 (R = 0.74), Ctnnb1 and Lamp1 (R = 0.70) as well as ERM and Lamp1(R = 0.65) in potassium-free condition (Fig. 7). No co-localization of these proteins with LC3/Lamp1 was observed in control rats.
Figure 6. Co-localization of AQP2 with autophagy markers.

(A) The inner medulla sections of control rats (C) and rats fed with a potassium-free diet for 1 day (NK) were triple-labeled against AQP2 (red), Lamp1 (pink), and LC3 (green). Insets demonstrate significant co-localization as a group of puncta, which were not observed in control sections. Scale bar = 4 μm. (B) Representative scatter plots demonstrate the high degree of co-localization between AQP2 and LC3 (r2 = 0.83, P < 0.05) or Lamp1 (r2 = 0.73, P < 0.05) after potassium deprivation. (C) A bar graph summarizes Pearson correlation coefficients of AQP2 and LC3 or Lamp1. (D) By immunogold EM, AQP2 (small gold particles, arrows) and LC3β (large gold particles, arrow heads) localize in autophagosomes in IMCD cells of potassium-deprived rats. (E) AQP2 (small gold particles, arrows) and cathepsin-D (large gold particles, arrow heads) also localize in similar membrane-bound organelles.
Figure 7. Co-localization of actin cytoskeletal and cell adhesion proteins with autophagy markers.

The inner medulla sections of control rats (C) and rats fed with a potassium-free diet for 1 day (NK) were triple-labeled against AQP2 (red), Lamp1 (pink), and down-regulated proteins including Itgb1, Vcl, Ctnnb1, and ERM (green). Insets demonstrate the areas where significant co-localization was observed as a group of puncta, which was not observed in control sections. Scale bar = 4 μm. Scatter plots demonstrate the degree of co-localization between lysosomal marker (Lamp1) and Itgb1 (r2 = 0.81, P < 0.05), Vcl (r2 = 0.54, P < 0.05), Ctnnb1 (r2 = 0.49, P < 0.05), or ERM (r2 = 0.42, P < 0.05) after potassium deprivation.
The immunofluorescence results were confirmed by double-label immunogold electron microscopy with AQP2 and LC3 (Fig. 6D), Lamp1 (Supplementary Fig. 5), or another lysosomal marker, cathepsin D (Fig. 6E). AQP2 and LC3 or AQP2 and cathepsin D localize in similar membrane-bound organelles throughout the kidney IMCD cells of potassium deprived rats. Similar structures were not frequently observed in control rats. The presence of AQP2 in autophagosomes and autophagolysosomes in the IMCD of potassium-deprived rats supports the role of autophagic protein degradation as the mechanism responsible for the down-regulation of IMCD proteins in the early stage of hypokalemia-induced NDI.
Potassium re-feeding ameliorates autophagic degradation of AQP2 induced by potassium deprivation
Potassium replenishment is known to reverse hypokalemia-induced NDI in clinical patients28,29, and also in experimental animals fed a potassium-deficient diet12,30. In order to further investigate the role of autophagy induced by potassium deprivation in producing the NDI phenotype, we performed a potassium re-feeding experiment. Following 1 day of potassium deprivation, rats were switched to a standard diet (i.e., containing the normal potassium content) for an additional 3 days. As with the potassium deprivation experiments, the earliest detectable change after potassium re-feeding was in the urinary fractional excretion of potassium, which was rapidly normalized within 24–48 hours (compare Fig. 8A to Fig. 1A). As expected, potassium re-feeding also reversed both the development of hypokalemia and the NDI phenotype, normalizing serum potassium level (Figs 8B vs. 1B), urine volume (Figs 8C vs. 1C) and urine osmolality (Figs 8D vs. 1D). Concomitantly, potassium re-feeding restored the abundances of AQP2 and pS256 AQP2 in the inner medulla to normal levels (Fig. 8E). In addition, the autophagy markers, LC3 and LAMP1, were no longer detectable and/or no longer co-localized with AQP2 after the refeeding (Fig. 8F). Thus, the recovery of the normal phenotype coincided with the restoration of AQP2 levels and the halting of its degradation via autophagy.
Figure 8. Responses of potassium-deprived rats to potassium re-feeding.

Urinary fractional excretion of potassium (A), serum potassium (B), urine volume (C), and urine osmolality (D) were measured in rats receiving a normal chow diet (■, n = 7) for 4 days or a potassium-free diet for 1 day followed by a normal chow diet on days 2, 3, and 4 (“re-fed” rats; □, n = 7, red arrows indicate the start of refeeding). The units for urinary fractional excretion of potassium (shown as %) are a ratio of urinary:plasma potassium concentration normalized to that of urinary:plasma creatinine concentration. (E) Normalized abundance ratios for AQP2 and pS256AQP2, quantified by immunoblotting of inner medullary proteins from potassium-deprived rats (■, n=7) and re-fed rats (□, n = 7) at day 4. The normalized ratios were obtained by dividing the band intensities from each experimental animal by the average intensity of the control group (control for either potassium-deprived rats or re-fed rats). (F) The inner medulla sections of control rats (C) and re-fed rats were probed using antibodies against LC3 (green), Lamp1 (pink), and AQP2 (red).
Discussion
Previous studies of hypokalemia-induced NDI in animal models have mostly investigated the long-term effects of potassium deprivation (several days to weeks), focusing on a limited number of target molecules in the kidney such as AQP2, urea transporters, sodium channels, the transcriptional activator TonEBP, and cAMP11,12,13,14,31,32,33. However, a study by Amlal et al. demonstrated that urinary concentrating defects induced by potassium deprivation can actually be detected much earlier (i.e., within a day)13. Thus, in the present study, we devised a strategy to better understand the pathogenesis of this condition by (i) investigating changes that occur in the initial stage of hypokalemia-induced NDI and (ii) applying an unbiased discovery approach (i.e., mass spectrometry-based proteomics). While serum potassium, urine volume, and cAMP levels remained unchanged at day 1, the early occurrence of a urinary concentrating defect (at day 1) was apparent in our study (Supplementary Fig. 1); in addition, early reduction in AQP2 protein abundance and phosphorylation and urinary potassium excretion (at day 1) were also detectable.
The reason that these early changes could occur in the absence of a decreased serum potassium level is the function of a feedforward regulatory system, originating with the gut dietary potassium sensor19,20,21. Although some major elements of the entire system are not yet completely understood, potassium sensing cells in the gut splanchnic bed (especially ones that supply stomach19 and hepato-portal vein34) transmit potassium intake signals to the brain, probably the hypothalamus, which subsequently relays signals to the pituitary. Hypophysectomy ameliorates the feedforward regulation of the kidney in response to changes in dietary potassium level, but the pituitary peptide(s) responsible for this regulation has not been identified21,35. As a result of this neuroendocrine feedforward system, a rapid response to decreased dietary potassium intake occurs21, producing an early decrease in urinary potassium excretion (significant on day 1, Fig 1A), thereby conserving body potassium20,30.
An early reduction in AQP2 protein was also previously demonstrated by Amlal et al.13, occurring along the entire collecting duct system as early as the first 24–48 hours after starting a potassium-deficient diet, however, the mechanism for AQP2 down-regulation remained unknown. The present study is the first to demonstrate that AQP2 in IMCD cells is sequestered in autophagosomes/autolysosomes as an early event following potassium deprivation. Since down-regulation of AQP2 is sufficient to induce a urinary concentrating defect36,37, it is at least plausible, if not highly probable, that autophagic degradation is the mechanism for down-regulation of AQP2 in hypokalmemia-induced NDI. Furthermore, this finding of early hypokalemia-induced autophagy in the IMCD is supported by a previous study showing that lysosomal enzyme activities in the renal inner medulla increased after 1 day of potassium depletion38.
Autophagy is a highly conserved and regulated catabolic process that involves the sequestration and transport of macromolecules and organelles to the lysosome for degradation39. In response to cellular stress, the basal activity of this normal process is increased to maintain cellular homeostasis. Autophagy is not simply a bulk degradation process but it can also be highly selective40,41,42,43, and in fact, we observed no change in abundance for a majority (>70%) of the proteins identified in the proteomic analysis. In addition to AQP2, 127 other proteins, including at least three groups of functionally-related proteins, appeared to be selectively down-regulated at this very early time point, prior to the appearance of NDI features. For example, proteomic analysis revealed down-regulation of 41 mitochondrial proteins, including a group of 19 proteins involved in energy production, which was also supported by the evidence of mitochondrial damage and mitophagy from our EM analysis. These findings are consistent with the study by Aithal et al. which demonstrated defective mitochondrial energy production as well as depletion of the mitochondrial content of potassium, magnesium and calcium in the renal medulla of rats fed with a potassium-deficient diet for 5 weeks44.
The other groups of down-regulated proteins identified by proteomic analysis were involved in actin cytoskeleton organization and cell-cell adhesion (Table 1). The co-localization with autophagy markers for six proteins representing these two groups indicated that autophagy also appears to play a role in selective down-regulation of these groups of proteins. These findings were also supported by histopathology and EM analysis showing disruption of cell-cell junctions between IMCD cells and disorganized actin filaments at tight junctions. Similarly, a previous in vitro study demonstrated that potassium depletion inhibited formation of tight junctions, desmosomes and bundled stress fibers in Madin Darby canine kidney (MDCK) epithelial cells45.
Table 1. A list of cell-cell adhesion and cytoskeleton related proteins which correlate with proteomic analysis and kidney histopathology.
| Functions | Proteins |
|---|---|
| Cadherin-based adherens | β-catenin (Cttnb1)*, vinculin (Vcl)*, cadherin-16 (Cdh16), α-actinin-4 (Actn4) |
| Focal adhesion protein | integrin β-1 (Itgb1)*, vinculin (Vcl)*, filamin-B (Flnb), α-actinin-4 (Actn4) |
| Actin cytoskeleton organization | ezrin (Ezr)*, transforming protein RhoA precursor (RhoA)*, integrin β-1 (Itgb1)*, calreticulin precursor (Calr), EH domain-containing protein 2 (Ehd2), keratin, type I cytoskeletal 19 (Krt19), spectrin alpha chain, non-erythrocytic 1 (Sptan1), ras-related protein Ral-A (Rala) |
*Proteins co-localized with AQP2 and Lamp1 in IMCD of potassium-deprived rats as demonstrated by immunofluorescence in this study.
One question that arises from the results of this study is how potassium deprivation is linked to mitochondrial dysfunction and the other observed changes in cellular structures. It is well established that the inner mitochondrial membrane contains at least four different potassium channels and at least one K+/H+ antiporter46,47. These channels function in a “K+ cycle” to help regulate changes in mitochondrial matrix volume, membrane potential and rate of ATP synthesis, as well as to modulate the generation of reactive oxygen species (ROS) by mitochondria. In a previous study, intracellular potassium ([K+]i) in inner medullary cells was significantly decreased between 24 and 72 hours after initiating a low-potassium diet30. Thus, while we did not measure [K+]i in IMCD cells of our rats fed with a potassium-free diet (vs. a low-potassium diet in the aforementioned study), it is likely that [K+]i was decreased at the 24 hour time-point used in the present study. Based on these factors, we postulate that decreased [K+]i should alter mitochondrial potassium channel activities and potassium balance across the inner mitochondrial membrane. Altered mitochondrial potassium balance would then lead to perturbations in mitochondrial volume, membrane potential, and rates of ATP and ROS synthesis by mitochondria. Indeed, mitochondrial volume changes (i.e., swelling) were observed in our study (Fig. 5F). Thus, it is plausible that mitochondrial damage (from ROS and any of the other mitochondrial perturbations mentioned above) was induced at the early stage of potassium deprivation. This damage would initiate mitophagy and subsequent cellular ATP depletion followed by the downstream activation of autophagic flux, resulting in the structural defects and other changes that we observed. Consistent with this hypothesis, disruption of adherens junctions has been observed in various ATP-depletion models in vitro (cultured MDCK cells)48. In addition, autophagy has been reported in another ATP-depletion model, i.e., an ischemia model in the human renal proximal tubular cell line, HK-249.
Our data indicate that potassium deprivation lead to autophagic degradation of a specific subset of cellular proteins, including AQP2 and the other proteins mentioned above; from this study, it is not possible to determine whether the down-regulation of the latter proteins is linked in a direct or causal way to AQP2 degradation or the NDI phenotype. Although our results clearly showed that activation of autophagy (and mitophagy) in IMCD cells was an early event in response to potassium depletion, we have not definitively demonstrated that autophagic degradation is responsible for the NDI phenotype. In order to formally demonstrate that autophagy produces NDI, we would need to show that inhibition of autophagy prevents the urinary concentrating defect and other manifestations of NDI. Unfortunately, the inhibitors that are commonly used to block autophagy (chloroquine, 3-MA, etc.) are non-specific, only act at late stage in the autophagy process, and/or cannot be used for in vivo studies (i.e., animal studies)50,51,52,53,54, and results using these inhibitors are often quite variable both in vivo and in vitro55,56. Furthermore, no in vitro model exists that recapitulates the hypokalemia-induced NDI phenotype. The only other way to inhibit autophagy is to knockout various autophagy-related genes, which is not feasible using a rat model. One practical, although somewhat less direct, approach performed in this study was a re-feeding experiment. The findings demonstrated that the recovery of the normal phenotype after potassium replenishment coincided with the normalization of AQP2 levels and its co-localization with autophagy markers was abolished, indicating that autophagic degradation, at least for AQP2, was arrested. Nevertheless, definitive demonstration that autophagy is the mechanism responsible for hypokalemia-induced NDI will require a strategy which specifically blocks the early stage of autophagy and is suitable for in vivo use.
Methods
Brief descriptions of key experimental procedures are provided below. For complete details see supplementary material online.
Experimental Design and Statistical Rationale
Experimental animals
All experiments were approved by the Animal Care and Use Committee of the Faculty of Medicine, Thammasat University and were carried out in “accordance” with the approved guidelines. Experiments were conducted on male Sprague-Dawley rats weighing 180–200 g. Rats were randomly assigned into 2 groups: rats fed with a potassium-free diet (TD.88239, Harlan, WI, USA) or control rats fed with standard rat chow.
Sample preparation for proteomic analysis
IMCD tissue was prepared from renal inner medullas according to Stokes et al.57 with modifications58. Three independent pairs of samples were prepared using four potassium-deprived rats versus four control rats for each pair (IMCD tissue in each group was pooled prior to homogenization). Each pair of samples (100 μg protein per sample) was subjected to 1D SDS-PAGE and in-gel trypsinization was performed as previously described59.
LC-MS/MS analysis
Samples were analyzed using an LTQ Orbitrap XL mass spectrometer (Thermo Scientific, San Jose, CA). Mass spectra were searched using two different search algorithms, SEQUEST (Bioworks version 3.31 SP1)60 and InsPecT (version 20070905)61, with the rat RefSeq database (National Center for Biotechnology Information, March, 2010, 30734 entries) which also included a list of common contaminating proteins from other species (e.g., porcine trypsin). Precursor ion tolerance was 10 ppm, while fragment ion tolerance was 0.8 Da. Three missed trypsin cleavage sites were allowed. Static modifications included carbamidomethylation of cysteine (+57.021 Da). Variable modifications included oxidation of methionine (+15.995 Da). All datasets were filtered to achieve a false discovery rate of <1%, estimated based on target-decoy analysis62 based on an absolute Xcorr threshold of 0.4. The InsPecT search results were filtered to a random match probability <1%.
Label-free quantification
Peptide quantification by a label-free technique was performed using QUOIL software24. Only proteins with peptides quantified in all three biological replicates were used for statistical significance testing. The log2 of NK/Control protein abundance ratio was used as the basis for calculation of the average abundance change and statistical significance by unpaired t-test for each protein.
Bioinformatics
The DAVID bioinformatics resource (Database for Annotation, Visualization and Integrated Discovery, NIAID, http://david.abcc.ncifcrf.gov/)26 was used to extract the list of significantly enriched functional annotation clusters associated with the proteins exhibiting significantly altered abundance as a result of potassium deprivation.
Targeted LC-MS/MS
For targeted LC-MS/MS experiments, new IMCD samples were prepared as described above, except that one pair of samples (four potassium-deprived rats versus four control rats for each pair) was analyzed. Peptides from selected proteins (identified as down-regulated in the initial proteomic analysis) were chosen based on the following criteria: uniqueness to the targeted proteins; fully tryptic; and absence of potentially modified amino acids. Skyline63 was used to create an inclusion list of peptide m/z ratios for targeted proteomics experiments. Targeted LC-MS/MS analysis was performed using a Q-Exactive Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA) (see Full Methods in Supplementary Information). Peak areas for each peptide were extracted using Skyline software63.
Quantitative Real-Time PCR
The experiment was performed as previously described64. The rat primers are provided in Supplemental methods. All values were normalized to 18s rRNA.
Immunoblotting
Immunoblotting was carried out as previously described65. A list of antibodies used can be found in Supplemental methods. To normalize samples for gel loading, samples were first analyzed on a separate gel, stained with Coomassie blue, scanned, and quantified for final justification of protein concentration in each sample prior to immunoblotting experiments66.
Electron microscopy
Fresh renal inner medullas were prepared as described in Supplemental methods. The ultrathin sections were examined under a transmission electron microscope (FEI Tecnai G2 TWIN 200 kV).
Immunofluorescence
Immunofluorescence labeling of kidney sections was done as previously described67. A list of antibodies used can be found in Supplemental methods. Fluorescence images were acquired using an Axio Observer Z1 microscope in conjunction with ApoTome2 (Carl Zeiss, Jena, Germany).
Immunogold electron microscopy
The procedures have been described previously68. A list of antibodies used can be found in Supplemental methods. The sections were examined in a FEI Morgagni electron microscope.
Re-feeding experiment
An experiment was conducted on male Sprague-Dawley rats weighing 180–200 g. Fourteen rats were randomly assigned into 2 groups: 1) seven rats fed with a potassium-free diet for 1 day followed by standard rat chow for 3 days; and 2) seven rats fed with standard rat chow for 4 days. Urine and serum parameters were measured at study onset and daily. Kidneys were harvested and inner medullas were isolated, processed, and analyzed as described above.
Additional Information
How to cite this article: Khositseth, S. et al. Autophagic degradation of aquaporin-2 is an early event in hypokalemia-induced nephrogenic diabetes insipidus. Sci. Rep. 5, 18311; doi: 10.1038/srep18311 (2015).
Supplementary Material
Acknowledgments
This work was supported by the Research Scholar Grant (RMU 5380032), Thailand Research Fund, Thammasat University, and the Chulalongkorn Academic Advancement into Its 2nd Century Project. The immunogold electron microscopic study is supported by the Danish Medical Research Council and Lundbeck Foundation. We thank Drs. Mark A. Knepper and Chung-Lin Chou (NHLBI, NIH, USA) for advice. We thank Nusara Chomanee and Christina Schmidt for assistance on electron microscopy, and Chatikorn Boonkrai for assistance on immunofluorescence microscopy. The authors thank Dr. N. Tony Eissa, Baylor College of Medicine, for very helpful discussion.
Footnotes
Author Contributions S.K., P.U. and T.P. designed the experiments. S.K., N.T., K.C., S-H.C. and R.A.F. performed the experiments. S.K., P.S., R.A.F. and T.P. analyzed the data. S.K., J.D.H., S.F., D.M.P., R.A.F. and T.P. wrote the manuscript and prepared the figures. All authors reviewed the manuscript.
References
- Hoffert J. D. et al. Dynamics of the G protein-coupled vasopressin V2 receptor signaling network revealed by quantitative phosphoproteomics. Mol Cell Proteomics 11, M111 014613, 10.1074/mcp.M111.014613 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. L., Miranda C. A. & Knepper M. A. Vasopressin and the regulation of aquaporin-2. Clin Exp Nephrol, 10.1007/s10157-013-0789-5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushimi K. et al. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature 361, 549–552, 10.1038/361549a0 (1993). [DOI] [PubMed] [Google Scholar]
- Knepper M. A. Molecular physiology of urinary concentrating mechanism: regulation of aquaporin water channels by vasopressin. Am J Physiol 272, F3–12 (1997). [DOI] [PubMed] [Google Scholar]
- Nielsen S. et al. Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-CD water channels to plasma membrane. Proc Natl Acad Sci USA 92, 1013–1017 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushimi K., Sasaki S. & Marumo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem 272, 14800–14804 (1997). [DOI] [PubMed] [Google Scholar]
- Katsura T., Gustafson C. E., Ausiello D. A. & Brown D. Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. Am J Physiol 272, F817–822 (1997). [PubMed] [Google Scholar]
- Nielsen S., DiGiovanni S. R., Christensen E. I., Knepper M. A. & Harris H. W. Cellular and subcellular immunolocalization of vasopressin-regulated water channel in rat kidney. Proc Natl Acad Sci USA 90, 11663–11667 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M. et al. Expression and distribution of aquaporin of collecting duct are regulated by vasopressin V2 receptor in rat kidney. J Clin Invest 94, 1778–1783, 10.1172/JCI117525 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanni S. R., Nielsen S., Christensen E. I. & Knepper M. A. Regulation of collecting duct water channel expression by vasopressin in Brattleboro rat. Proc Natl Acad Sci USA 91, 8984–8988 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkjaer M. L. et al. Altered expression of renal NHE3, TSC, BSC-1, and ENaC subunits in potassium-depleted rats. Am J Physiol Renal Physiol 283, F1376–1388, 10.1152/ajprenal.00186.2002 (2002). [DOI] [PubMed] [Google Scholar]
- Marples D., Frokiaer J., Dorup J., Knepper M. A. & Nielsen S. Hypokalemia-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla and cortex. J Clin Invest 97, 1960–1968, 10.1172/JCI118628 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlal H., Krane C. M., Chen Q. & Soleimani M. Early polyuria and urinary concentrating defect in potassium deprivation. Am J Physiol Renal Physiol 279, F655–663 (2000). [DOI] [PubMed] [Google Scholar]
- Jung J. Y. et al. Expression of urea transporters in potassium-depleted mouse kidney. Am J Physiol Renal Physiol 285, F1210–1224, 10.1152/ajprenal.00111.2003 (2003). [DOI] [PubMed] [Google Scholar]
- Relman A. S. & Schwartz W. B. The nephropathy of potassium depletion; a clinical and pathological entity. N Engl J Med 255, 195–203, 10.1056/NEJM195608022550501 (1956). [DOI] [PubMed] [Google Scholar]
- Fougeray S. & Pallet N. Mechanisms and biological functions of autophagy in diseased and ageing kidneys. Nat Rev Nephrol 11, 34–45, 10.1038/nrneph.2014.201 (2015). [DOI] [PubMed] [Google Scholar]
- Pallet N. et al. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 4, 783–791 (2008). [DOI] [PubMed] [Google Scholar]
- Kimura T. et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 22, 902–913, 10.1681/ASN.2010070705 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee F. N., Oh G., McDonough A. A. & Youn J. H. Evidence for gut factor in K+ homeostasis. Am J Physiol Renal Physiol 293, F541–547, 10.1152/ajprenal.00427.2006 (2007). [DOI] [PubMed] [Google Scholar]
- Greenlee M., Wingo C. S., McDonough A. A., Youn J. H. & Kone B. C. Narrative review: evolving concepts in potassium homeostasis and hypokalemia. Ann Intern Med 150, 619–625 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn J. H. Gut sensing of potassium intake and its role in potassium homeostasis. Semin Nephrol 33, 248–256, 10.1016/j.semnephrol.2013.04.005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller H. B. & Fenton R. A. Cell biology of vasopressin-regulated aquaporin-2 trafficking. Pflugers Arch 464, 133–144, 10.1007/s00424-012-1129-4 (2012). [DOI] [PubMed] [Google Scholar]
- Tchapyjnikov D. et al. Proteomic profiling of nuclei from native renal inner medullary collecting duct cells using LC-MS/MS. Physiol Genomics 40, 167–183, 10.1152/physiolgenomics.00148.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Wu W. W., Zeng W., Chou C. L. & Shen R. F. Label-free protein quantification using LC-coupled ion trap or FT mass spectrometry: Reproducibility, linearity, and application with complex proteomes. J Proteome Res 5, 1214–1223, 10.1021/pr050406g (2006). [DOI] [PubMed] [Google Scholar]
- Gunaratne R. et al. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc Natl Acad Sci USA 107, 15653–15658, 10.1073/pnas.1007424107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G. Jr. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4, P3 (2003). [PubMed] [Google Scholar]
- Sandoval P. C. et al. Proteome-wide measurement of protein half-lives and translation rates in vasopressin-sensitive collecting duct cells. J Am Soc Nephrol 24, 1793–1805, 10.1681/ASN.2013030279 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn J. N., Kowey P. R., Whelton P. K. & Prisant L. M. New guidelines for potassium replacement in clinical practice: a contemporary review by the National Council on Potassium in Clinical Practice. Arch Intern Med 160, 2429–2436 (2000). [DOI] [PubMed] [Google Scholar]
- Rastegar A. & Soleimani M. Hypokalaemia and hyperkalaemia. Postgrad Med J 77, 759–764 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linas S. L. et al. Mechanism of renal potassium conservation in the rat. Kidney Int 15, 601–611 (1979). [DOI] [PubMed] [Google Scholar]
- Beck N. & Webster S. K. Impaired urinary concentrating ability and cyclic AMP in K+-depleted rat kidney. Am J Physiol 231, 1204–1208 (1976). [DOI] [PubMed] [Google Scholar]
- Carney S., Rayson B. & Morgan T. A study in vitro of the concentrating defect associated with hypokalaemia and hypercalcaemia. Pflugers Arch 366, 11–17 (1976). [PubMed] [Google Scholar]
- Jeon U. S. et al. Downregulation of renal TonEBP in hypokalemic rats. Am J Physiol Renal Physiol 293, F408–415, 10.1152/ajprenal.00502.2006 (2007). [DOI] [PubMed] [Google Scholar]
- Morita H., Fujiki N., Miyahara T., Lee K. & Tanaka K. Hepatoportal bumetanide-sensitive K(+)-sensor mechanism controls urinary K(+) excretion. Am J Physiol Regul Integr Comp Physiol 278, R1134–1139 (2000). [DOI] [PubMed] [Google Scholar]
- Oh Y. T., Kim J. & Youn J. H. Role of pituitary in K+ homeostasis: impaired renal responses to altered K+ intake in hypophysectomized rats. Am J Physiol Regul Integr Comp Physiol 304, R1166–1174, 10.1152/ajpregu.00495.2012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek A., Fuchtbauer E. M., Kwon T. H., Frokiaer J. & Nielsen S. Severe urinary concentrating defect in renal collecting duct-selective AQP2 conditional-knockout mice. Proc Natl Acad Sci USA 103, 6037–6042, 10.1073/pnas.0511324103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortenoeven M. L., Pedersen N. B., Miller R. L., Rojek A. & Fenton R. A. Genetic ablation of aquaporin-2 in the mouse connecting tubules results in defective renal water handling. J Physiol 591, 2205–2219, 10.1113/jphysiol.2012.250852 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aithal H. N., Toback F. G., Dube S., Getz G. S. & Spargo B. H. Formation of renal medullary lysosomes during potassium depletion nephropathy. Lab Invest 36, 107–113 (1977). [PubMed] [Google Scholar]
- Levine B. & Kroemer G. Autophagy in the pathogenesis of disease. Cell 132, 27–42, 10.1016/j.cell.2007.12.018 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen T. & Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T. & Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012, 736905, 10.1155/2012/736905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. & Klionsky D. J. Mitochondria removal by autophagy. Autophagy 7, 297–300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Scott S. V., Oda M. N. & Klionsky D. J. Transport of a large oligomeric protein by the cytoplasm to vacuole protein targeting pathway. J Cell Biol 137, 609–618 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aithal H. N. & Toback F. G. Defective mitochondrial energy production during potassium depletion nephropathy. Lab Invest 39, 186–192 (1978). [PubMed] [Google Scholar]
- Rajasekaran S. A. et al. Na,K-ATPase activity is required for formation of tight junctions, desmosomes, and induction of polarity in epithelial cells. Mol Biol Cell 12, 3717–3732 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk A., Jarmuszkiewicz W. & Kunz W. S. Mitochondrial potassium channels. IUBMB Life 61, 134–143, 10.1002/iub.155 (2009). [DOI] [PubMed] [Google Scholar]
- Szabo I., Leanza L., Gulbins E. & Zoratti M. Physiology of potassium channels in the inner membrane of mitochondria. Pflugers Arch 463, 231–246, 10.1007/s00424-011-1058-7 (2012). [DOI] [PubMed] [Google Scholar]
- Bacallao R., Garfinkel A., Monke S., Zampighi G. & Mandel L. J. ATP depletion: a novel method to study junctional properties in epithelial tissues. I. Rearrangement of the actin cytoskeleton. J Cell Sci 107 (Pt 12), 3301–3313 (1994). [DOI] [PubMed] [Google Scholar]
- Suzuki C. et al. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun 368, 100–106, 10.1016/j.bbrc.2008.01.059 (2008). [DOI] [PubMed] [Google Scholar]
- Klionsky D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristol M. L. et al. Autophagy inhibition for chemosensitization and radiosensitization in cancer: do the preclinical data support this therapeutic strategy? J Pharmacol Exp Ther 344, 544–552, 10.1124/jpet.112.199802 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhasz G. Interpretation of bafilomycin, pH neutralizing or protease inhibitor treatments in autophagic flux experiments: novel considerations. Autophagy 8, 1875–1876 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein D. C., Gestwicki J. E., Murphy L. O. & Klionsky D. J. Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6, 304–312, 10.1038/nrd2272 (2007). [DOI] [PubMed] [Google Scholar]
- Yang Y. P. et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin 34, 625–635, 10.1038/aps.2013.5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 25, 717–729, 10.1101/gad.2016111 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva O. K. et al. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J Mol Med (Berl) 89, 877–889, 10.1007/s00109-011-0774-y (2011). [DOI] [PubMed] [Google Scholar]
- Stokes J. B., Grupp C. & Kinne R. K. Purification of rat papillary collecting duct cells: functional and metabolic assessment. Am J Physiol 253, F251–262 (1987). [DOI] [PubMed] [Google Scholar]
- Pisitkun T. et al. High-throughput identification of IMCD proteins using LC-MS/MS. Physiol Genomics 25, 263–276, 10.1152/physiolgenomics.00214.2005 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisitkun T., Shen R. F. & Knepper M. A. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci USA 101, 13368–13373, 10.1073/pnas.0403453101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J. R. 3rd, Eng J. K., McCormack A. L. & Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem 67, 1426–1436 (1995). [DOI] [PubMed] [Google Scholar]
- Tanner S. et al. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal Chem 77, 4626–4639, 10.1021/ac050102d (2005). [DOI] [PubMed] [Google Scholar]
- Elias J. E. & Gygi S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214, 10.1038/nmeth1019 (2007). [DOI] [PubMed] [Google Scholar]
- MacLean B. et al. . Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968, 10.1093/bioinformatics/btq054 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avihingsanon Y. et al. Measurement of urinary chemokine and growth factor messenger RNAs: a noninvasive monitoring in lupus nephritis. Kidney Int 69, 747–753, 10.1038/sj.ki.5000132 (2006). [DOI] [PubMed] [Google Scholar]
- Pisitkun T. et al. Akt and ERK1/2 pathways are components of the vasopressin signaling network in rat native IMCD. Am J Physiol Renal Physiol 295, F1030–1043, 10.1152/ajprenal.90339.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C. L., Ma T., Yang B., Knepper M. A. & Verkman A. S. Fourfold reduction of water permeability in inner medullary collecting duct of aquaporin-4 knockout mice. Am J Physiol 274, C549–554 (1998). [DOI] [PubMed] [Google Scholar]
- Yu M. J., Pisitkun T., Wang G., Shen R. F. & Knepper M. A. LC-MS/MS analysis of apical and basolateral plasma membranes of rat renal collecting duct cells. Mol Cell Proteomics 5, 2131–2145 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton R. A. et al. Acute regulation of aquaporin-2 phosphorylation at Ser-264 by vasopressin. Proc Natl Acad Sci USA 105, 3134–3139, 10.1073/pnas.0712338105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.