

Abstract
Fusarinine C (FSC), a siderophore‐based chelator coupled with the model peptide c(RGDfK) (FSC(succ‐RGD)3), revealed excellent targeting properties in vivo using positron emission tomography (PET). Here, we report the details of radiolabeling conditions and specific activity as well as selectivity for 68Ga.
68Ga labeling of FSC(succ‐RGD)3 was optimized regarding peptide concentration, pH, temperature, reaction time, and buffer system. Specific activity (SA) of [68Ga]FSC(succ‐RGD)3 was compared with 68Ga‐1,4,7‐triazacyclononane, 1‐glutaric acid‐4,7 acetic acid RGD ([68Ga]NODAGA‐RGD). Stability was evaluated in 1000‐fold ethylenediaminetetraacetic acid (EDTA) solution (pH 7) and phosphate‐buffered saline (PBS). Metal competition tests (Fe, Cu, Zn, Al, and Ni) were carried out using [68Ga]‐triacetylfusarinine C.
High radiochemical yield was achieved within 5 min at room temperature, in particular allowing labeling with 68Ga up to pH 8 with excellent stability in 1000‐fold EDTA solution and PBS. The 10‐fold to 20‐fold lower concentrations of FSC(succ‐RGD)3 led to the same radiochemical yield compared with [68Ga]NODAGA‐RGD with SA up to 1.8 TBq/µmol. Metal competition tests showed high selective binding of 68Ga to FSC.
FSC is a multivalent siderophore‐based bifunctional chelator allowing fast and highly selective labeling with 68Ga in a wide pH range and results in stable complexes with high SA. Thus it is exceptionally well suited for the development of new 68Ga‐tracers for in vivo molecular imaging with PET.
Keywords: fusarinine C, RGD, 68Ga, radiolabeling, positron emission tomography (PET)
Introduction
Positron emission tomography (PET) is one of the most prominent molecular imaging modalities, mainly based on the clinical utility of [18F]fluorodeoxyglucose. The potential to radiolabel molecules with a high specificity towards a biological target offers tremendous opportunities both for clinical as well as preclinical applications. However, the use of 18F and other radionuclides such as 11C relies on the availability of cyclotrons in the vicinity of the nuclear medicine department and even as the capacities of production and distribution are increasing; the technology is hampered by the bottleneck of cyclotron‐based radionuclide production. Therefore, the last ten years have seen a tremendous interest in other radionuclides, in particular with improved availability based on generator elution. The most promising candidate is 68Ga, having a very suitable half‐life of 68 min, a high positron yield (89%) and therefore can be seen as a metallic alternative to 18F.1, 2
For labeling of molecular probes with 68Ga, a coordinating system is required. Therefore, the vector molecule should be conjugated with a bifunctional chelator. In contrast to 99mTc, which is widely used in clinical settings for single photon emission computed tomography and where a great number of structurally different chelating agents are available, less variety can be found for 68Ga, even though research in this field is very active increasing the number of chelating systems constantly.2 Until now, the most widely applied chelating system for radiolabeling of biomolecules with 68Ga is 1,7,7,10‐tetraazacyclododecane‐1,4,7,10‐tetraacetic acid (DOTA). However, DOTA is not ideally suited for Ga3+ having an effective ionic radius of 62 pm, comparable to Fe3+ (64.5 pm) and Zr4+ (72 pm) but considerably smaller than other radiometals used in the context of DOTA labeling such as In3+, Y3+, or lanthanides (La3+) with >80 pm.3 An alternative chelating system for 68Ga is 1,4,7‐triazacyclononane‐1,4,7‐triacetic acid (NOTA), which has a smaller cavity and allows binding of the Ga3+ cation with higher kinetic stability and labeling yield than its corresponding DOTA counterpart. Some triazacyclononane derivatives (e.g., NODAGA,4 1,4,7‐triazacyclononane phosphinic acid (TRAP),5 and 1,4,7‐triazacyclononane‐1,4‐bis[methylene(hydroxymethyl)phosphinic acid]‐7‐[methylene(2‐carboxyethyl)phosphinic acid] (NOPO)6, 7) are increasingly utilized in the development of 68Ga tracers.
An alternative to these cyclic aminocarboxylates and aminophosphinates is siderophores. These are low molecular mass chelators with a very high affinity for ferric ions and are utilized by bacteria, fungi, and plants for iron acquisition and storage.8 Because of the comparable ionic radius, charge, and electronic configuration, 68Ga3+ shows almost identical coordination chemistry to Fe3+ and can be used to radiolabel the corresponding siderpohore. One of the first siderophores used for labeling with 67/68Ga3+ was desferoxamine B (DFO) with high radiochemical yield (RCY).9 [67/68Ga]DFO‐octreotide showed high and specific tumor uptake in mice but disappointing data in patient because of strong protein binding in human plasma.10
Recently, we have reported that triacetylfusarinine (TAFC) (Figure 1), which forms hexacoordinate ferric complexes in a tris‐bidentate form involving the three hydroxamate functions of the molecule, had very good complexing properties for 68Ga resulting in high specific activities (SAs) and excellent metabolic stabilities.11, 12 In the light of these promising properties, we were interested whether fusarinine C (FSC), the deacetylated form of TAFC, could be used as a basis for preparing specifically targeted 68Ga‐labeled bioconjugates. To evaluate this hypothesis, a RGD peptide targeting the integrin α v β 3, which is overexpressed on activated endothelial cells during tumor neovascularization,13 was chosen as model targeting molecule. FSC(succ‐RGD)3 was synthesized and evaluated in vitro and in vivo.14 Our initial data showed that FSC can be used as bifunctional chelator for synthesis of trimeric bioconjugates allowing 68Ga labeling with excellent radiolabeling properties. Moreover, high affinity for integrin α v β 3 in vitro and high tumor uptake in vivo of [68Ga]FSC(succ‐RGD)3 proved highly promising properties of the FSC chelating scaffold. Here, we report on the optimization of radiolabeling conditions including metal competition tests and achievable SA of [68Ga]FSC(succ‐RGD)3 and compared them with the established monomeric [68Ga]NODAGA‐RGD.15
Figure 1.
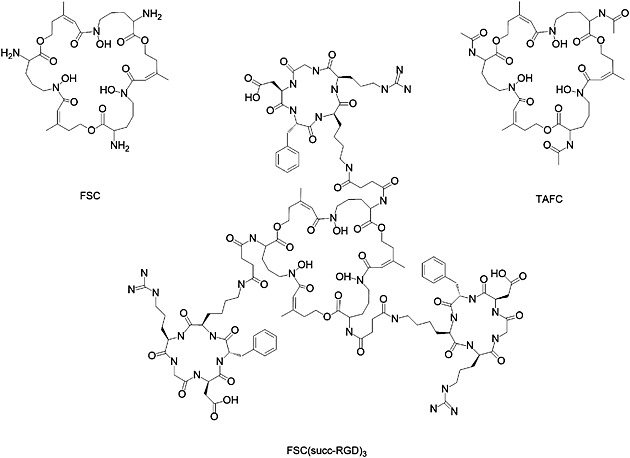
Structures of FSC, TAFC, and FSC(succ‐RGD)3.
Materials and methods
All described substances and solvents were of reagent grade and were used without further purification. NODAGA‐RGD was synthesized in our laboratory as described earlier.15 [Fe]Fusarinine C ([Fe]FSC) and TAFC were gifts from Prof. Hass and were prepared as described previously.13, 14 MilliQ water was used for preparing all reagent solutions. The 68Ga‐generator (IGG100) was purchased from Eckert & Ziegler Strahlen‐ und Medizintechnik AG (Berlin, Germany) with a nominal activity of 1100 MBq and was eluted with 0.1M HCl solution (Rotem Industries Ltd., Beer‐Sheva, Israel).
Analytical methods
Analytical reversed‐phase high‐performance liquid chromatography (RP‐HPLC) analysis was performed with an UltiMate 3000 RS HPLC pump, an UltiMate 3000 RS column compartment (column oven temperature was set at 25°C), an UltiMate 3000 ultraviolet (UV)–vis variable wavelength detector (Dionex, Gemering, Germany), and a Raytest radiometric detector (Raytest GmbH, Straubenhardt, Germany). A Vydac 218 TP5215, 150 × 3.0 mm column (SRD, Vienna, Austria), flow rate 1.0 mL/min, and UV–vis detection at 220/410 nm were employed with the following acetonitrile (CH3CN)/H2O/0.1% trifluoroacetic acid (TFA) gradient: 0–0.5 min 0% CH3CN, 0.5–7.0 min, 0–55% CH3CN.
Radiolabeling efficiency and radiochemical purity were additionally analyzed by instant thin‐layer chromatography (ITLC) on ITLC silica gel strips with two different mobile phases: 0.1 M aqueous sodium citrate (pH 5) (TLC1) and with a 1:1 (v/v) mixture of 1 M aqueous ammonium acetate and methanol as a mobile phase (pH 7) (TLC2). The strips were scanned using a mini‐scan radio TLC scanner with a flow‐count detector (LabLogic, Sheffield, UK).
Synthesis of FSC(succ‐RGD)3
Detailed chemical synthesis of FSC(succ‐RGD)3 is described in reference.14 Briefly, the side‐chain protected c(‐Arg(Pbf)‐Gly‐Asp(OtBu)‐DPhe‐Lys(Z)‐) (protecting group: Pbf: 2,2,4,5,7‐pentamethyl‐3‐hydrobenzofuran‐6‐sulfonyl, OtBu: O‐tert‐butyl, Z: benzyloxycarbonyl) was synthesized using a Fmoc protection strategy, followed by cyclization and lysine (Lys) sidechain deprotection. The amino group of Lys was derivatized by succinic anhydride (succ). An excess of c(‐Arg(Pbf)‐Gly‐Asp(OtBu)‐DPhe‐Lys(succ)‐) was conjugated with [Fe]FSC without further purification. The side‐chain deprotection using TFA resulted in [Fe]FSC(succ‐RGD)3. The removal of iron from [Fe]FSC(succ‐RGD)3 was accomplished by dissolving the conjugate in disodium ethylenediaminetetraacetic acid (EDTA) solution (25 mM, pH 4). The crude product was purified via preparative RP‐HPLC and lyophilized. Matrix‐assisted laser desorption ionization time‐of‐flight mass spectrometry [M + H]+ = 2785.0 [C126H183N33O39; exact mass: 2782.3 (calculated).
Radiolabeling of FSC(succ‐RGD)3 with Ga‐68
For the optimization of the labeling conditions, the following parameters were investigated: peptide concentration, pH, temperature, buffer systems, and time dependencies of radioactivity incorporation. To a fraction of 1 mL of the generator eluate containing the highest activity (approximately 400–500 MBq in 0.1 M HCl), a solution of sodium acetate (NaOAc, 1.9 M) or 4‐(2‐hydroxyethyl) piperazine‐1‐ethanesulfonic acid (HEPES) buffer (1 M) was added and mixed by brief shaking, resulting in a pH ranging from 1.1 to 6. Subsequently, 100 μL of the corresponding solution was mixed with 20 μL of FSC(succ‐RGD)3 with concentrations ranging from 0.04 to 45 μM. For pH 7 and pH 8, the radiolabeling protocol was changed slightly: 20 μL of stock solutions of FSC(succ‐RGD)3 were mixed with 100 μL 68Ga eluate, then HEPES sodium salt solution (1 M) was added to adjust to appropriate pH. The reaction mixtures were incubated at room temperature (RT) or heated to 70°C and subsequently cooled down in a water bath. At different time points (2, 3, 5, 15, and 20 min), 5 μL of the labeling solution was removed to detect the reaction rate. RCYs were determined via analytical RP‐HPLC (tR = 6.6 min) and Radio‐TLC (TLC1: [68Ga]FSC(succ‐RGD)3 stayed at the origin, and free 68Ga moved with the front; TLC2: 68Ga‐colloid stayed at the origin and [68Ga]FSC(succ‐RGD)3 moved with the front).
Radiolabeling of NODAGA‐RGD with 68Ga
The optimal labeling condition of NODAGA‐RGD with 68Ga was adopted according to reference.15 Briefly, to a fraction of 1 mL of the generator eluate containing the highest activity (approximately 400–500 MBq in 0.1 M HCl), a solution of NaOAc (1.9 M, 180 μL) was added and mixed by brief shaking, resulting in a final solution with pH 5. Then 100 μL of this solution was mixed with 20 μL NODAGA‐RGD with concentrations ranging from 0.6 to 10 μM. The labeling mixtures were allowed to react for 15 min at RT. RCYs were determined via analytical RP‐HPLC (tR = 5.3 min).
Competition experiments
For the investigation of the influence of metals during 68Ga‐radiolabeling, the acetylated version of FSC, TAFC was used as model compound and radiolabeled with 68Ga adding different metals in concentrations ranging from 3 to 30 000 μM. The metals [Fe(NO3)3, FeSO4, Al2(SO4)3, ZnSO4, CuSO4, Ni(NO3)3 and SnCl2] were dissolved in 0.1 M HCl to yield stock solutions in a concentration of 84 mM. The stock solutions were diluted to 8.4 mM, 840, 84 and 8.4 μM, respectively. These solutions were mixed with a freshly eluted 68Ga fraction (v:v 1:1). Radiolabeling was performed as described in the preceding texts. The final volume of all labeling solutions was 140 μL with a final TAFC concentration of 3 μM and final metal concentrations ranged from 3 μM to 30 mM, the reactions were carried out at pH 4.5 for 15 min. As a control the influence of Fe3+ on 68Ga‐labeling of NODAGA‐RGD was tested following the same protocol. RCYs were determined by TLC 1.
Stability assay
Investigation of the stability of [68Ga]FSC(succ‐RGD)3 in 1000‐fold EDTA solution (pH 7) was carried out by incubating the radiotracer for 30, 60, and 120 min at 37°C. An aliquot of 100 μL of [68Ga]FSC(succ‐RGD)3 (50 μM) was mixed with 100 μL of EDTA solution (50 mM). The pH was adjusted to 7. At selected time points 10 μL aliquots were analyzed directly via analytical RP‐HPLC.
Investigation of the stability of [68Ga]FSC(succ‐RGD)3 at different concentrations were carried out by diluting the radiotracer (50 μM) with PBS to give FSC(succ‐RGD)3 concentrations ranging from 50 μM to 5 nM. The samples were analyzed at selected time points (10 and 60 min) via analytical RP‐HPLC.
Results and discussion
In this study, we focused on the optimization of radiolabeling conditions to reach the highest SA of [68Ga]FSC(succ‐RGD)3, to determine the stability of [68Ga]FSC(succ‐RGD)3 under different conditions, and to evaluate the influence of various metals on 68Ga labeling.
Buffer system
The buffer system can have great influence on 68Ga‐labeling yield and thus on SA, which has already been reported.16 In our study, sodium acetate solution and HEPES buffer were chosen to compare the influence of the buffer on FSC(succ‐RGD)3 labeling. As shown in Figure 2 higher RCYs of [68Ga]FSC(succ‐RGD)3 were achieved using HEPES buffer at low precursor concentrations (< 0.6 μM) at RT (15 min reaction time). Comparable RCYs for both buffer systems were found at 0.6 μM precursor concentration, but lower RCYs for HEPES at higher precursor concentrations (>0.6 μM). The concentration of precursor required for quantitative 68Ga incorporation was 1.2 μM when using sodium acetate and 3 μM in the case of HEPES buffer at RT. At elevated temperature (70°C), quantitative 68Ga incorporation was achieved at 0.6 μM precursor concentration using sodium acetate. This indicates an advantage of the sodium acetate systems for 68Ga labeling of FSC conjugates in particular at increased temperature.
Figure 2.
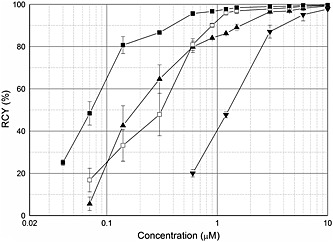
Influence of temperture and buffering system on the radiochemical yield of radioligand. [68Ga]FSC(succ‐RGD)3: 70°C, NaOAc, pH 4.5 (—■—), RT, NaOAc, pH 4.5 (—□—), RT, HEPES buffer, pH 4.5 (—▲—); [68Ga]NODAGA‐RGD: RT, NaOAc, pH 5 (—▼—).
Specific activity
We have reported the feasibility of labeling NODAGA‐RGD with 68Ga at ambient temperature.15 As shown in Figure 3, fairly high concentrations of NODAGA‐RGD are required to achieve quantitative labeling at RT. Comparing RCYs of [68Ga]FSC(succ‐RGD)3 with [68Ga]NODAGA‐RGD approximately 10‐fold to 20‐fold lower precursor concentrations for labeling our FSC conjugate were required, which provided much higher SA because of the lower excess of precursor required for labeling. Table 1 shows the RCY and corresponding calculated SA of [68Ga]FSC(succ‐RGD)3 and [68Ga]NODAGA‐RGD at different precursor concentrations. The SAs and RCYs were calculated by normalizing the starting activities of these set of experiments to approximately 50 MBq in 100‐μL eluate. Considering a reproducible RCY > 80%, a SA up to 1.8 TBq/µmol was observed for [68Ga]FSC(succ‐RGD)3, approximately 18‐fold higher than [68Ga]NODAGA‐RGD where only 0.1 TBq/µmol were achieved with a RCY of 87%. This allows preparation of 68Ga‐labeled tracers targeting low capacity sites for clinical but also preclinical applications, where variation of SA over a wider range may be beneficial to achieve optimal targeting. Additionally regulatory toxicity concerns can be more easily addressed. The maximal achievable SA in this study is close to the extraordinary high SA reported for phosphinic acid functionalized polyazacycloalkanes such as NOPO and TRAP.17
Figure 3.
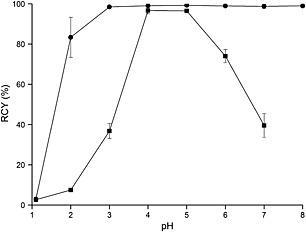
Influence of pH on the radiochemical yield of [68Ga]FSC(succ‐RGD)3: RT, 45 μM (—●—), RT,1.5 μM (—■—).
Table 1.
Radiochemical yields and calculated specific activities for [68Ga]FSC(succ‐RGD)3 and [68Ga]NODAGA‐RGD
| Precursor amount | Precursor concentration | Radiochemical yield | Specific activity |
|---|---|---|---|
| (nmol) | (μM) | (%) | (GBq/µmol) |
| [68Ga]FSC(succ‐RGD)3 | |||
| 0.0045 | 0.04 | 25.1 ± 1.3 | 2230 ± 118 |
| 0.009 | 0.07 | 48.4 ± 5.6 | 2150 ± 247 |
| 0.018 | 0.14 | 80.7 ± 4.0 | 1792 ± 89 |
| 0.036 | 0.3 | 86.6 ± 0.1 | 962 ± 1.6 |
| 0.072 | 0.6 | 95.6 ± 1.0 | 531 ± 5.5 |
| 0.11 | 0.9 | 96.7 ± 0.2 | 352 ± 0.7 |
| [68Ga]NODAGA‐RGD | |||
| 0.072 | 0.6 | 20.0 ± 1.8 | 111 ± 10 |
| 0.14 | 1.2 | 47.7 ± 1.4 | 136 ± 4 |
| 0.36 | 3 | 87.0 ± 3.1 | 97 ± 3.4 |
| 0.72 | 6 | 95.1 ± 2.9 | 53 ± 1.6 |
| 1.20 | 10 | 97.7 ± 0.4 | 33 ± 0.1 |
Values presented as mean ± SD (n ≥ 3 of separate labeling experiments) at typical time of injection (30 min after start of labeling), activity used 40 MBq.
Influence of pH
Gallium adopts oxidation state + III in aqueous solution and acts as a hard Lewis acid with high charge density and small ionic radius, which can form stable complexes with hard Lewis bases, such as hydroxide, alkoxide, halogen, and ammonia moieties. If Ga3+ concentration exceeds the nanomolar range in aqueous solution, it can hydrolyze to different insoluble colloidal gallium hydroxide species which occurs at a pH of approximately 5, while at a pH of 8 (or higher) highly soluble [Ga(OH)4]‐ ions are formed. Radiolabeling procedures with Ga3+ are usually carried out at pH 2–4 (e.g., DOTA‐TOC pH 3.5–4.018), whereas labeling at higher pH is also possible (e.g., NODAGA‐RGD at pH 5) depending on the chelating system applied.19 Šimeček's group6 recently reported that NOPO, a phosphinic acid functionalized polyazacycloalkane, can be quantitatively labeled at pH 7 using 10 μM labeling precursor at 95°C but reported a dramatic decrease of RCY at pH 8. In our study, the influence of pH was investigated at RT (Figure 3), where FSC(succ‐RGD)3 in 1.5 μM concentration could be labeled quantitatively at pH 4 and 5. A particularly interesting feature of FSC(succ‐RGD)3 is that it can be labeled readily at a substantially higher pH values using a higher concentration of precursor. In our study, quantitative labeling was achieved using 45 μM precursor concentration at pH 7 and 8, respectively. The advantage of radiolabeling at pH 7 and 8 is the potential to radiolabel precursors with 68Ga that only dissolve in neutral or basic solutions or are unstable at acidic pH levels.
Influence of reaction time
68Ga, with a short half‐life of 68 min, also requires minimizing radiolabeling time. Figure 4 shows the influence of the reaction time on the RCY of [68Ga]FSC(succ‐RGD)3. A RCY of 95.7% was achieved within 5 min at pH 4.5 at RT and did not increase with prolonged reaction time.
Figure 4.
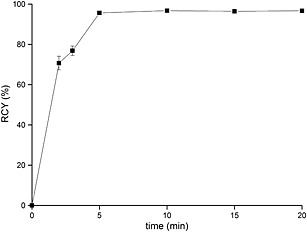
Influence of reaction time on the radiochemical yield of [68Ga]FSC(succ‐RGD)3 (1.5 μM, RT, pH 4.5).
Metal ions
The influence of metal ions on 68Ga labeling of chelators like NOTA, DOTA, TRAP‐Pr, or NOPO has been studied by Šimeček et al.20 TAFC was used for metal competition experiments in our study as a model FSC‐derivative. The influence of different concentrations of metal ions (Fe3+, Fe2+, Al3+, Zn2+, Cu2+, Ni3+, and Sn2+) on 68Ga labeling of TAFC was studied, as comparison for the influence of Fe3+ NODAGA‐RGD was used (Figure 5). Labeling was performed at 3‐μM precursor concentration to ensure quantitative reaction conditions. Particular focus was put on iron as a very common metal impurity in generator eluates but also as FSC and TAFC are siderophores with high affinity for iron. We found no influence on labeling of TAFC even if the concentration of Fe3+ was equal to the concentration of TAFC and the concentration of 68Ga3+ much lower in the labeling solution. This indicates a higher affinity of FSC for Ga3+ possibly attributed to the harder Lewis acid property of Ga3+. At higher concentrations, RCY dropped significantly; for Fe2+, this effect on 68Ga labeling was less pronounced; quantitative labeling could still be achieved with 30 μM of Fe2+ concentration, 10 times higher than that of Fe3+. In contrast to TAFC, NODAGA‐RGD labeling with 68Ga was influenced by the presence of Fe3+, which was reflected by a greater variation in RCYs at the concentration of 3 μM Fe3+ and a dramatic RCY decrease at higher concentration. Al3+, with a trivalent charge and a proximity to Ga in the periodic table, had no influence on 68Ga labeling of TAFC up to a concentration of 300 μM. Zn2+ as the decay product of 68Ga accumulating in the generator did not decrease RCYs even at the concentration of 30 mM, which is highly unlikely to occur in a generator. Cu2+ and Ni3+ were also studied but had no influence on 68Ga labeling. Sn2+ was found to have a minor effect on 68Ga labeling at 3 μM, RCYs decrease to approximately 64% at 30 μM and remained stable up to 3 mM. Overall FSC‐derivatives have an excellent selectivity for 68Ga superior to NOTA‐derivatives and comparable to triazacyclononane–phophinates as reported previously.6, 7
Figure 5.
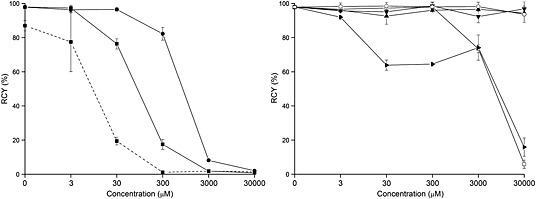
Influence of metals on TAFC and NODAGA‐RGD labeling with 68Ga. Left: [68Ga]FSC(succ‐RGD)3: Fe(NO3)3 (—■—), FeSO4 (—●—), [68Ga]NODAGA‐RGD: Fe(NO3)3 (‐‐‐■‐‐‐); Right: [68Ga]FSC(succ‐RGD)3: Ni(NO3)3 (—▼—), SnCl2 (—▸—), Al2(SO4)3 (—□—), ZnSO4 (—▲—), CuSO4 (—○—).
Stability
We already reported that [68Ga]FSC(succ‐RGD)3 was stable in PBS, FeCl3, DTPA solution and in fresh human serum at 37°C for 2 h.16 We additionally evaluated the stability of [68Ga]FSC(succ‐RGD)3 in EDTA solution because iron was removed quickly from [Fe]FSC(succ‐RGD)3 in 40‐fold EDTA solution at pH 4 during the synthesis of the conjugate. In our study, 1000‐fold EDTA was mixed with [68Ga]FeFSC(succ‐RGD)3 at pH 7 and analyzed at 30, 60, and 120 min with a HPLC system. There was almost no detectable decrease of RCY during the whole monitoring period, indicating a high stability at neutral pH.
Caraco et al. reported that dilution of DFO, an acyclic analogue of FSC, could cause the breakdown of the 67Ga3+–DFO complex. High RCYs can be achieved at concentrations >5 mM, while at nanomolar level (<50 nM) DFO does not act as a good chelating agent for gallium.21 In our study, 50 μM of [68Ga]FSC(succ‐RGD)3 was diluted in PBS to obtain FSC(succ‐RGD)3 in concentrations ranging from 50 μM to 5 nM and was analyzed at different time points (10 and 60 min after dilution) by RP‐HPLC. No dissociation of 68Ga from the FSC chelator was observed over 60 min, even at the lowest [68Ga]FSC(succ‐RGD)3 concentration, indicating superior stability to that of DFO particularly at low precursor concentrations.
Conclusion
The radiolabeling conditions and stabilities of the new siderophore‐based bifunctional chelator FSC coupled to the model peptide RGD were studied and showed that high RCY was achieved within 5 min at RT and within a wide pH range (3–8). In particular labeling at pH 7 and 8 was possible providing access to 68Ga‐labeled biomolecules that are unstable at acidic conditions. Specific activity achieved was 10–20 times higher compared with [68Ga]NODAGA‐RGD and was comparable to TRAP or NOPO conjugates. Excellent stability towards 1000‐fold EDTA challenge and PBS dilution at pH 7 was observed and metal competition tests showed high selectivity of FSC for complexing 68Ga. We conclude that FSC is exceptionally well suited for the development of 68Ga‐labeled tracer for in vivo molecular imaging with PET providing a high flexibility in labeling conditions and is an interesting alternative to currently used triazacyclononanes. The structural relationship to DFO also raises the possibility of using FSC for radiolabeling with 89Zr [28]. Investigations in this direction are ongoing.
Conflict of Interest
The authors have declared that there is no conflict of interest.
Acknowledgements
This work was supported by the Austrian Science Foundation (FWF) grant P 25899‐B23 and the China Scholarship Council.
Zhai, C. , Summer, D. , Rangger, C. , Haas, H. , Haubner, R. , and Decristoforo, C. (2015), Fusarinine C, a novel siderophore‐based bifunctional chelator for radiolabeling with Gallium‐68. J. Label Compd. Radiopharm, 58, 209–214. doi: 10.1002/jlcr.3286.
References
- 1. Decristoforo C., Pickett R. D., Verbruggen A., Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 31–40. [DOI] [PubMed] [Google Scholar]
- 2. Velikyan I., J Labelled Comp Radiopharm. 2015. doi: 10.1002/jlcr.3250. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 3. Shannon R. D., Acta Cryst. 1976, A32, 751–767. [Google Scholar]
- 4. Eisenwiener K. P., Prata M. I., Buschmann I., Zhang H. W., Santos A. C., Wenger S., et al. Bioconjug. Chem. 2002, 13, 530–541. [DOI] [PubMed] [Google Scholar]
- 5. Notni J., Šimeček J., Hermann P., Wester H. J., Chemistry 2011, 17, 14718–14722. [DOI] [PubMed] [Google Scholar]
- 6. Šimeček J., Zemek O., Hermann P., Notni J., Wester H. J., Mol. Pharmaceutics. 2014, 11, 3893–3903. [DOI] [PubMed] [Google Scholar]
- 7. Šimeček J., Notni J., Kapp T. G., Kessler H., Wester H. J., Mol. Pharmaceutics. 2014, 11, 1687–1695. [DOI] [PubMed] [Google Scholar]
- 8. Neilands J. B., J. Biol. Chem. 1995, 270, 26723–26726. [DOI] [PubMed] [Google Scholar]
- 9. Yokoyama A., Ohmomo Y., Horiuchi K., Saji H., Tanaka H., Yamamoto K., et al. J. Nucl. Med. 1982, 23, 909–914. [PubMed] [Google Scholar]
- 10. Maecke H. R., Heppeler A., Nock B. A., Samatostatin analogues labeled with different radionuclides In Technetium, Rhenium and other Metals in Chemistry and Nuclear Medicine, (Eds: Nicolini M., Mazzi U.). SGE Ditoriali: Padua, 1999, 77–91. [Google Scholar]
- 11. Petrik M., Haas H., Schrettl M., Helbok A., Blatzer M., Decristoforo C.. Nucl. Med. Biol. 2012, 39, 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Petrik M., Franssen G. M., Haas H., Laverman P., Hörtnagl C., Schrettl M., et al. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1175–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu S., Mol. Pharmaceutics. 2006, 3, 472–487. [DOI] [PubMed] [Google Scholar]
- 14. Knetsch P. A., Zhai C., Rangger C., Blatzer M., Haas H., Kaeopookum P., et al. Nucl. Med. Biol. 2015, 42, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Knetsch P. A., Petrik M., Griessinger C. M., Rangger C., Fani M., Kesenheimer C., et al. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1303–1312. [DOI] [PubMed] [Google Scholar]
- 16. Velikyan I., Beyer G. J., and Långström B., Bioconjug. Chem. 2004, 15, 554–560. [DOI] [PubMed] [Google Scholar]
- 17. Notni J., Pohle K., Wester H. J., EJNMMI research. 2012, 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meyer G. J., Macke H., Schuhmacher J., Knapp W. H., Hofmann M., Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 1097–1104. [DOI] [PubMed] [Google Scholar]
- 19. Mas‐Moruno C., Dorfner P. M., Manzenrieder F., Neubauer S., Reuning U., Burgkart R., et al. J. Biomed. Mater. Res. A 2013, 101, 87–97. [DOI] [PubMed] [Google Scholar]
- 20. Šimeček J., Hermann P., Wester H. J., Notni J., ChemMedChem 2013, 8, 95–103. [DOI] [PubMed] [Google Scholar]
- 21. Caraco C., Aloj L., Eckelman W., Appl. Radiat. Isot. 1998, 49, 1477–1479. [DOI] [PubMed] [Google Scholar]