

Abstract
Bioluminescence resonance energy transfer (BRET) is a valuable tool to detect protein-protein interactions. BRET utilizes bioluminescent and fluorescent protein tags with compatible emission and excitation properties, making it possible to examine resonance energy transfer when the tags are in close proximity (<10 nm) as a typical result of protein-protein interactions. Here we describe a protocol for detecting BRET from two known protein binding partners (Gαi1 and RGS14) in HEK 293 cells using Renilla luciferase and yellow fluorescent protein tags. We discuss the calculation of the acceptor/donor ratio as well as net BRET and demonstrate that BRET can be used as a platform to investigate the regulation of protein-protein interactions in live cells in real time.
Keywords: Bioluminescence resonance energy transfer (BRET), Renilla luciferase (RLuc), Yellow fluorescent protein (YFP), RGS14, G protein regulatory (GPR) motif, G protein
1 Introduction
Bioluminescence Resonance Energy Transfer (BRET) is a method of studying protein-protein interactions in live cells [1]. BRET utilizes non-radiative energy transfer between energy donor and energy acceptor protein tags. The energy transfer occurs when the protein tags are in close proximity, as described by the Förster distance [2]. As shown in Fig. 1, BRET serves as a molecular ruler, detecting protein-protein interactions under 10 nm (see Note 1). For a comprehensive review, see refs. [3, 4].
Fig. 1.

BRET is dependent on the distance between the donor luciferase and the acceptor fluorophore. Addition of the cell permeant Renilla luciferase substrate coelenterazine (ctz) results in oxidation of the substrate to coelenteramide, which produces blue light at 482 nm. When protein-protein interactions between Protein X and Protein Y bring the donor luciferase (RLuc) and acceptor fluorophore (YFP) in close proximity (<10 nm), the energy from the donor can be transferred to the acceptor and light is produced at 527 nm. When the BRET tags are not in close enough proximity, light is only emitted at 482 nm
BRET makes use of a bioluminescent energy donor while the energy acceptor is a fluorophore. The choice of BRET pair is based on the overlap of the bioluminescent protein (donor) emission spectrum with the excitation spectrum of the fluorescent protein (acceptor). For BRET experiments, the most commonly chosen bioluminescent donor is luciferase from the sea pansy Renilla reniformis. Renilla luciferase (RLuc) catalyzes the oxidation of its substrate, coelenterazine, to produce blue light at 482 nm. The emission spectrum of RLuc overlaps well with the excitation spectra of the yellow fluorescent protein (YFP) family of proteins including the mutant YFP variants enhanced YFP (EYFP) and Venus [5] which emit light at ~527 nm (Fig. 2). For more information on BRET pairs, see Note 2.
Fig. 2.
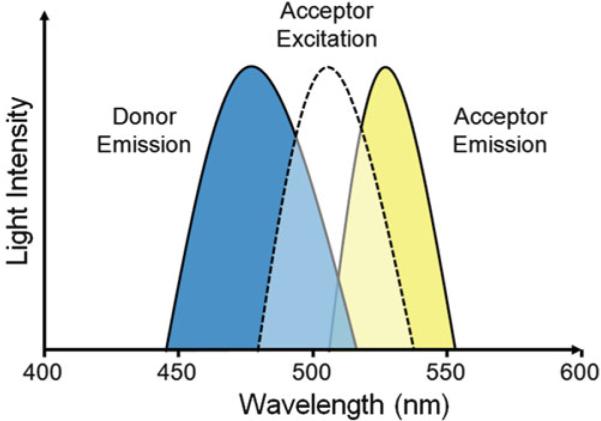
The energy transfer between BRET pairs depends on the overlap of the donor emission spectrum with the excitation spectrum of the acceptor. For Renilla luciferase, oxidation of coelenterazine results in an emission peak at 482 nm. This emission overlaps well with the excitation spectrum of yellow fluorescent protein (excitation peak: 514 nm). The resulting energy transfer yields yellow light with an emission peak of 527 nm
BRET has distinct advantages over other techniques to detect protein-protein interactions. First, BRET is amenable to detecting interactions in live cells, thus proteins retain posttranslational modifications and cellular trafficking regulations that may be important for protein-protein interactions. BRET is readily adaptable to almost any cell type that allows expression of the donor and acceptor proteins. In live cells, protein-protein interactions can be monitored in real time over a time-course or for a fixed time interval in response to cellular treatments such as exposure to GPCR agonists, growth factors, or other drugs as an approach to define the regulation of protein complexes [6–12]. Additionally, fusion of BRET pairs to the same recombinant protein can be used to develop small molecule biosensors [13]. Methods have also been developed using BRET as a reporter for movement and subcellular location of target proteins [14]. Moreover, unlike the similar technique fluorescence resonance energy transfer (FRET), BRET does not require external excitation but instead relies on the addition of the cell permeant substrate coelenterazine to initiate the assay, thereby endowing the experimenter with temporal control over the assay and preventing unintentional activation of the acceptor fluorophore. Given these many advantages, BRET can be readily adapted for high throughput screening for small molecule modulators of protein-protein interactions. For review, see refs. [9, 15].
Below we describe a BRET experiment to explore the interactions between Regulator of G protein Signaling 14 (RGS14) and its binding partner Gαi1. RGS14 has previously been shown to interact with Gαi1 through its G protein regulatory (GPR) motif by traditional biochemical methods [16, 17]. We detail transfection of a C-terminal luciferase tagged RGS14 (RGS14-Luc) donor and internal YFP tagged Gαi1 (Gαi1-YFP) acceptor. We demonstrate a robust BRET signal between wild type RGS14 and Gαi1 that is disrupted with a mutant RGS14 (Q515A/R516A) that can no longer bind Gαi1. In our example, we show how to vary the acceptor protein expression level to achieve optimal net BRET signal. We describe how to calculate net BRET, acceptor/donor ratio, and fit the data using graphing software.
2 Materials
2.1 Cell Lines
Maintain HEK 293 cells in 1× Dulbecco's Modified Eagle Medium (DMEM) without phenol red indicator, supplemented with 2 mM l-glutamine, 100 U/mL penicillin, 100 mg/mL streptomycin, and 10 % fetal bovine serum (5 % for transfection). Grow cells in a humidified incubator with 5 % CO2 at 37 °C.
2.2 Buffer Compositions/Stock Solutions
BRET buffer (Tyrode's solution): 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 0.37 mM NaH2PO4, 24 mM NaHCO3, 10 mM HEPES, pH 7.4, 0.1 % glucose.
Polyethylenimine (PEI) transfection reagent stock solution: Dissolve PEI (1 mg/mL) in dH2O at 80 °C while stirring, cool and adjust to pH 7.2 using 0.1 N HCl and filter-sterilize. Aliquot and store at −80 °C. Use each aliquot only once.
Luciferase substrate stock solution: 2 mM benzyl coelenterazine H in 100 % ethanol containing 60 mM HCl, aliquot and store at −80 °C.
2.3 Instrumentation
2.4 Plasmids
Donor plasmids can be constructed by inserting your gene of interest into a vector containing the humanized RLuc gene. For construction of RGS14-Luc constructs presented below, rat RGS14 cDNA was inserted into the phRLuc-N2 vector, which places the RLuc tag at the C-terminus of RGS14. Determining the optimal location of the Rluc tag relative to the protein of interest is an important parameter and must be determined empirically (see Note 1).
For many BRET experiments, acceptor plasmids can be constructed by inserting your gene of interest into a commercially available vector encoding YFP, EYFP or Venus. For construction of Gαi1-YFP used below, insertion of the YFP tag at either terminus compromised the function of Gαi1. Thus, the Gαi1-YFP construct was engineered by inserting the YFP coding sequence between the b and c helices of the helical domain of Gαi1 which was then expressed in a pcDNA3.1 vector [18].
3 Methods
3.1 Experimental Setup
Below we describe an experiment where the donor expression level is set and the acceptor expression level is varied. This experimental setup will allow the acceptor to saturate the donor and provide a maximal BRET signal. The expression level should be empirically determined; however, we typically use donor plasmid amounts that yield relative luminescence units (RLU) of 100,000–350,000 in our microplate reader (TriStar LB 941). In our example, 5 ng of phRLuc-N2:: RGS14 yields an RLU of ~100,000–300,000 but this will vary for other donor constructs depending on transfection efficiency, the ability of the transfected cells to express the donor protein and the type of instrument used for detection. Additionally, on our microplate platform, we typically use acceptor plasmid amounts that yield relative fluorescence units (RFUs) of 30,000–200,000. In our example experiment, pcDNA3.1::Gαi1-YFP typically yields ~30,000–60,000 RFUs.
For the present experiment, 5 ng of the donor plasmid (RGS14-WT-Luc or RGS14-Q515A/R516A-Luc) was transfected with increasing amounts of acceptor (0, 10, 50, 100, 250, and 500 ng pcDNA3.1::Gαi1-YFP) (see Note 4).
3.2 Transient Transfection with Polyethylenimine (PEI) (See Note 5)
Seed 8 × 105 cells per well in six-well plates in 2 mL medium per well, grow in a humidified incubator at 37 °C overnight with 5 % CO2.
Prior to transfection, change medium to 1× DMEM containing 2 mM l-glutamine, 100 U/mL penicillin, 100 mg/mL streptomycin, and 5 % fetal bovine serum, 2 mL per well.
Generate solution A by adding 8 μL of 1 mg/mL PEI from stock to 92 μL of serum-free medium for each well, allow this solution to incubate for 3 min.
Generate solution B by adding up to 1.5 μg of DNA to 100 μL of serum-free medium for each condition in 1.5 mL microcentrifuge tubes. DNA amount is adjusted to a final concentration of 1.5 μg by adding empty pcDNA3.1 plasmid.
Add 100 μL of solution A to microcentrifuge tubes containing solution B to create solution C.
Cap the 1.5 mL microcentrifuge tube and immediately vortex for 3 s.
Incubate solution C at room temperature for 15 min.
Add solution C (~200 μL) dropwise to the appropriate well of cells in the six-well plates.
Allow cells to grow for 1–2 days (the medium does not need to be changed for PEI transfection).
3.3 BRET
Immediately prior to beginning the BRET experiment, prepare coelenterazine by diluting the stock solution to 50 μM in room temperature BRET buffer (see Note 6).
Aspirate the transfection medium from the six-well plates.
To each well, add 750 μL of room temperature BRET buffer, using a pipette to gently remove the cells from the plate.
Plate 90 μL of the cells in triplicate into white-bottomed 96-well plates.
Load plate into plate reader and detect fluorescence levels (excitation: 485 nm, emission: 530 nm) using microplate reader software (see Note 7).
Add 10 μL of coelenterazine solution to each well (5 μM final concentration of coelenterazine per well).
Incubate the cells with coelenterazine for 2 min at room temperature.
Take BRET readings by measuring luminescence at 485 ± 20 nm and fluorescence at 530 ± 20 nm (see Note 8).
3.4 Analysis
Export fluorescence and BRET data into spreadsheet software.
The BRET ratio can be determined by dividing fluorescence by luminescence (BRET readings at 530/485 nm).
Calculate net BRET by subtracting out background luminescence (BRET readings in cells expressing the donor without any acceptor).
Calculate the acceptor/donor ratio by dividing the initial fluorescence measurements (530 nm) by the luminescence measurements (485 nm).
Using graphing software, plot the acceptor/donor ratio against the net BRET as in Fig. 3. The data can then be fit using a nonlinear regression, (typically a one-site binding [hyperbola] is the most appropriate) to observe BRET saturation as a key indicator of signal specificity.
Fig. 3.

HEK 293 cells were transfected with increasing amounts of Gαi1-YFP (0, 10, 50, 100, 250, and 500 ng) and either 5 ng RGS14-WT-Luc or RGS14-Q515A/R516A-Luc. Wild type RGS14 shows a robust BRET signal with Gαi1. Conversely, the RGS14 mutant (Q515A/R516A) that can no longer bind Gαi1 shows a drastically reduced maximal BRET signal indicating a disruption in the protein-protein interaction. The above data is representative of three independent experiments. Curves were generated with GraphPad Prism 5 using the one-site binding curve fitting function. Additionally, Gαi1-YFP expression levels were verified by immunoblot analysis
Footnotes
BRET efficiency (donor energy transfer to acceptor) is sensitive to the donor-acceptor proximity and is inversely proportional to the sixth power of distance between them. Thus, BRET signals generally reflect direct protein association; however, non-robust BRET signals can be detected due to close proximity without the occurrence of direct binding. For example, when a third intermediate protein brings the donor and acceptor into close proximity [10–12]. In addition to proximity, BRET also depends on the orientation of the protein tag dipoles. Inefficient dipole coupling can prevent energy transfer, despite protein-protein interactions. Thus, it is advantageous to use linkers (typically four or more Gly residues) inserted between the proteins of interest and the BRET tags to allow sufficient movement of the tags. Moreover, placement of tags must be considered when engineering recombinant proteins with BRET tags. Placement of the BRET tag at the N-terminus, internally, or at the C-terminus of the protein can have a profound impact on the observed BRET signal and whether protein function is compromised. For example, placement of the luciferase tag at the N-terminus of RGS14 rather than the C-terminus results in a dramatic reduction in BRET signal with Gαi1-YFP. As another example, the acceptor YFP tag in Gαi1 cannot be placed at either termini without affecting protein function, and was placed internally between the b and c helices of the alpha helical domain with minimal consequences to Gαi1 function [18]. Due to these considerations, two interacting proteins may not always be detected by BRET due to the distance between the tags or abnormal protein function or localization through improper tag placement.
Additional BRET pairs and BRET substrates have been developed. Many of these BRET pairs can be used with the method described above. For a comprehensive review of other BRET pairs and substrates, see ref. [19].
We use the TriStar LB 941 Multimode Microplate Reader (Berthold Technologies) for our BRET experiments; however, other plate readers can be used with similar results. The plate reader must detect light signals at two distinct wavelengths either simultaneously or sequentially. In our assay, we use Berthold Technologies filters at 485 nm to measure luminescence and at 530 nm to measure fluorescence though similar filters can be purchased from other vendors. To collect BRET data, we use the MikroWin 2000 software (Mikrotek). MikroWin is specialized for microplate experiments and optimized to run with a variety of instruments from various manufacturers. Additionally, data collected in MikroWin can be exported and further analyzed in spreadsheet software such as Microsoft Excel. Data can then be graphed in graphing software such as GraphPad Prism.
In order to calculate net BRET, it is necessary to include a donor-only control (donor transfected without any acceptor). The donor-only control is used to assess any background BRET observed in the absence of acceptor. BRET from donor-only controls are subtracted from observed BRET values to calculate the net BRET.
Other transfection reagents can be used with similar results. We choose to use PEI as it yields high transfection efficiency and reproducibility at very affordable cost.
Coelenterazine is light sensitive and should be kept away from light exposure until ready to use. Dilute coelenterazine stock immediately before performing BRET to prevent breakdown of the substrate.
Initial fluorescence levels are taken to determine the acceptor/donor ratio as well as an internal control to verify expression of the fluorescently tagged protein. For experiments where the amount of donor is held constant and the amount of acceptor is increased, corresponding increases of the acceptor should be observed in the fluorescence measurement.
As stated above, for detection on the TriStar platform, ideal luminescence should be about 100,000–350,000 relative luminescence units (RLUs). Ideal fluorescence should range between 30,000 and 200,000 relative fluorescence units (RFUs). As a corollary, typical acceptor/donor ratios range between 0 and 15; however, this number will vary depending on transfection efficiency and the expression level of the acceptor. In addition, lowering the level of donor expression will also inflate the acceptor/donor ratio.
References
- 1.Xu Y, Piston DW, Johnson CH. A bioluminescence resonance energy transfer (BRET) system: application to interacting circadian clock proteins. Proc Natl Acad Sci U S A. 1999;96:151–156. doi: 10.1073/pnas.96.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu P, Brand L. Resonance energy transfer: methods and applications. Anal Biochem. 1994;218:1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- 3.Xu Y, Kanauchi A, von Arnim AG, et al. Bioluminescence resonance energy transfer: monitoring protein-protein interactions in living cells. Methods Enzymol. 2003;360:289–301. doi: 10.1016/s0076-6879(03)60116-3. [DOI] [PubMed] [Google Scholar]
- 4.Lohse MJ, Nuber S, Hoffmann C. Fluorescence/bioluminescence resonance energy transfer techniques to study G-protein-coupled receptor activation and signaling. Pharmacol Rev. 2012;64:299–336. doi: 10.1124/pr.110.004309. [DOI] [PubMed] [Google Scholar]
- 5.Nagai T, Ibata K, Park ES, et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 6.Romero-Fernandez W, Borroto-Escuela DO, Tarakanov AO, et al. Agonist-induced formation of FGFR1 homodimers and signaling differ among members of the FGF family. Biochem Biophys Res Commun. 2011;409:764–768. doi: 10.1016/j.bbrc.2011.05.085. [DOI] [PubMed] [Google Scholar]
- 7.Gales C, Rebois RV, Hogue M, et al. Real-time monitoring of receptor and G-protein interactions in living cells. Nat Methods. 2005;2:177–184. doi: 10.1038/nmeth743. [DOI] [PubMed] [Google Scholar]
- 8.Angers S, Salahpour A, Joly E, et al. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc Natl Acad Sci U S A. 2000;97:3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamdan FF, Audet M, Garneau P, et al. High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J Biomol Screen. 2005;10:463–475. doi: 10.1177/1087057105275344. [DOI] [PubMed] [Google Scholar]
- 10.Vellano CP, Maher EM, Hepler JR, et al. G protein-coupled receptors and resistance to inhibitors of cholinesterase-8A (Ric-8A) both regulate the regulator of g protein signaling 14 RGS14.Galphai1 complex in live cells. J Biol Chem. 2011;286:38659–38669. doi: 10.1074/jbc.M111.274928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oner SS, Maher EM, Breton B, et al. Receptor-regulated interaction of activator of G-protein signaling-4 and Galphai. J Biol Chem. 2010;285:20588–20594. doi: 10.1074/jbc.C109.088070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oner SS, An N, Vural A, et al. Regulation of the AGS3.G{alpha}i signaling complex by a seven-transmembrane span receptor. J Biol Chem. 2010;285:33949–33958. doi: 10.1074/jbc.M110.138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang LI, Collins J, Davis R, et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J Biol Chem. 2007;282:10576–10584. doi: 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lan TH, Liu Q, Li C, et al. Sensitive and high resolution localization and tracking of membrane proteins in live cells with BRET. Traffic. 2012;13:1450–1456. doi: 10.1111/j.1600-0854.2012.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couturier C, Deprez B. Setting Up a Bioluminescence Resonance Energy Transfer High throughput Screening Assay to Search for Protein/Protein Interaction Inhibitors in Mammalian Cells. Front Endocrinol. 2012;3:100. doi: 10.3389/fendo.2012.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollinger S, Taylor JB, Goldman EH, et al. RGS14 is a bifunctional regulator of Galphai/o activity that exists in multiple populations in brain. J Neurochem. 2001;79:941–949. doi: 10.1046/j.1471-4159.2001.00629.x. [DOI] [PubMed] [Google Scholar]
- 17.Kimple RJ, De Vries L, Tronchere H, et al. RGS12 and RGS14 GoLoco motifs are G alpha(i) interaction sites with guanine nucleotide dissociation inhibitor Activity. J Biol Chem. 2001;276:29275–29281. doi: 10.1074/jbc.M103208200. [DOI] [PubMed] [Google Scholar]
- 18.Gibson SK, Gilman AG. Gialpha and Gbeta subunits both define selectivity of G protein activation by alpha2-adrenergic receptors. Proc Natl Acad Sci U S A. 2006;103:212–217. doi: 10.1073/pnas.0509763102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bacart J, Corbel C, Jockers R, et al. The BRET technology and its application to screening assays. Biotechnol J. 2008;3:311–324. doi: 10.1002/biot.200700222. [DOI] [PubMed] [Google Scholar]