

Summary
We utilized induced pluripotent stem cells (iPSCs) derived from Huntington’s disease (HD) patients as a human model of HD and determined that the disease phenotypes only manifest in the differentiated neural stem cell (NSC) stage, not in iPSCs. To understand the molecular basis for the CAG repeat expansion-dependent disease phenotypes in NSCs, we performed transcriptomic analysis of HD iPSCs and HD NSCs compared to isogenic controls. Differential gene expression and pathway analysis pointed to transforming growth factor β (TGF-β) and netrin-1 as the top dysregulated pathways. Using data-driven gene coexpression network analysis, we identified seven distinct coexpression modules and focused on two that were correlated with changes in gene expression due to the CAG expansion. Our HD NSC model revealed the dysregulation of genes involved in neuronal development and the formation of the dorsal striatum. The striatal and neuronal networks disrupted could be modulated to correct HD phenotypes and provide therapeutic targets.
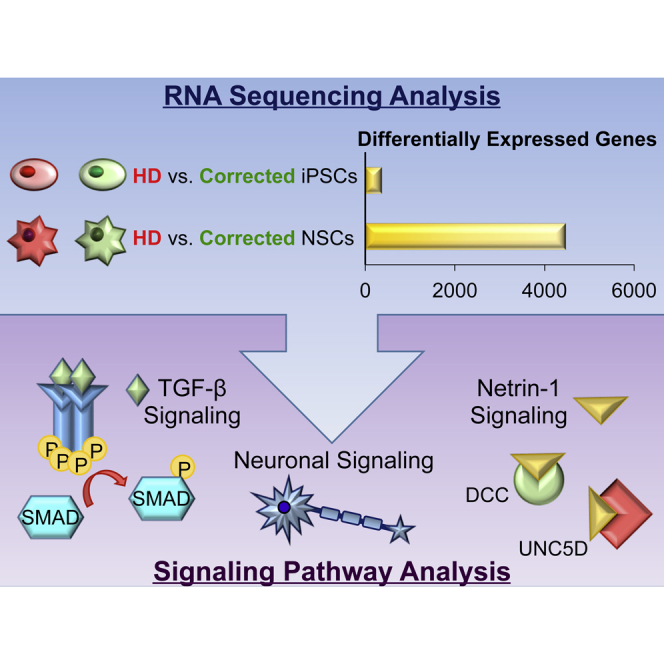
Graphical Abstract
Highlights
-
•
HD stem cell models have phenotypes in the differentiated NSC and not the IPSC state
-
•
Bioinformatic analysis of RNA-seq data identifies TGF-β and netrin-1 in HD phenotypes
-
•
WGCNA analysis in the HD-NSC linked module to development of the dorsal striatum
-
•
Modulation of TGF-β and netrin-1 in HD-NSCs is neuroprotective
Ellerby and colleagues use expression profiling and bioinformatic analysis to identify critical pathways mediating Huntington’s disease (HD) phenotypes in neural stem cells. They identify and show modulation of TFG-β and netrin-1 signaling is relevant to HD phenotypes found in neural stem cells and not iPSCs. Further, HD phenotypes are linked to genes in the dorsal striatum.
Introduction
Huntington’s disease (HD) is a debilitating, inherited neurological disorder characterized by chorea, psychological changes, and cognitive decline leading to dementia (Victorson et al., 2014). These symptoms are correlated with loss of striatal and cortical neurons in the brain (Ehrlich, 2012). HD is caused by a CAG expansion mutation coding for the polyglutamine tract located in the N-terminal region of the huntingtin protein (HTT) (The Huntington’s Disease Collaborative Research Group., 1993). At this time, there is no cure for HD or treatments to delay its onset and progression (Videnovic, 2013).
Standard HD models include transgenic animal models, immortalized rodent and human cell lines, and post-mortem tissue from HD patients (Bard et al., 2014, Lee et al., 2013). These models have been very useful in understanding some mechanisms behind HD pathogenesis; however, they do not fully represent human HD pathology (Kaye and Finkbeiner, 2013). Particularly important is the field’s reliance on HD mouse models, which do not account for the potential to miss key drug targets, the effects of polymorphisms on human protein toxicity, human-specific cell subtypes, and transcription factor binding sites specific to humans. A promising complementary approach for modeling HD is the use of human induced pluripotent stem cells (iPSCs) derived from HD patient somatic cells (An et al., 2012, Zhang et al., 2010). HD iPSCs harboring mutant HTT protein (mHTT) have the potential to model the disease more accurately, as they are untransformed and capable of differentiating into multiple types of neural tissue. Human iPSCs also provide the advantage of following the progress of the disease during neural development and detecting early pathological changes—the presymptomatic stage. iPSCs provide a platform for systemic genomic profiling and drug screening and are a promising tool for cellular replacement therapy in HD patients.
We have previously established HD-patient-derived iPSCs and corrected their genetic defect through the use of homologous recombination-based gene targeting methods (An et al., 2012). Characterization of these isogenic lines and derivative neural precursors showed that correction of the expanded polyglutamine region to a non-disease causing length resulted in a normalization of cellular phenotypes consistent with several well-established and reproducible aspects of the disease—cell death, loss of brain-derived neurotrophic factor (BDNF) expression, and reduction of mitochondrial respiratory capacity, among other cellular phenotypes (An et al., 2012, Zhang et al., 2010). These phenotypes were apparent in differentiated neural stem cells (NSCs) but not the pluripotent stem cell fate. In a separate study, HD iPSCs displayed elevated lysosomal activity indicating a disruption in cellular maintenance and protein degradation (Camnasio et al., 2012). Finally, a study identified the key functional differences in striatal medium spiny neurons (MSNs) generated from HD and control patient iPSCs (HD iPSC Consortium, 2012). HD MSNs display altered electrophysiological properties including differences in their ability to fire spontaneous and evoked action potentials and to regulate intracellular calcium signaling.
Our initial endeavors in characterizing our isogenic iPSCs lines included a microarray-based large-scale gene expression analysis comparing the HD iPSCs with the corrected line C116 iPSCs (An et al., 2012). These studies were restricted to a comparison of only the iPSCs lines but yielded several useful insights regarding the biology of these established cell models. Specifically, we found that global gene expression remained essentially unchanged at the iPSC state upon analysis of isogenic pairs (HD iPSCs versus corrected iPSCs). We identify an order of magnitude fewer significantly differentially expressed (DE) genes when compared to a separate experiment, evaluating a non-isogenic pair of HD iPSCs versus normal iPSCs derived from an unrelated healthy individual, likely due to differences in genetic background as found in other studies (Fogel et al., 2014). The low degree of DE genes between corrected and uncorrected isogenic iPSCs supports several important points regarding the biological characterization of this HD model: (1) isogenic gene modification does not dramatically alter the expression profile of these cells, (2) HTT gene correction does not markedly alter gene expression at the iPSCs state—consistent with disease biology, (3) gene expression analysis in controlled isogenic cell line studies may represent a cleaner approach to discovery of disease-relevant pathway effects, and (4) further analysis in disease-affected cell types may allow the ability to resolve disease-specific coexpression traits unique to those cell types.
Here, we present transcriptomic and bioinformatic analysis of disease-relevant and non-relevant cell types in tandem with an isogenic human stem cell model of HD. DE analysis and weighted coexpression analyses confirmed the cell-specific nature of gene expression changes due to mHTT. We used weighted gene coexpression network analysis (WGCNA) to analyze the non-disease and disease states of our isogenic HD stem cell model and determine clusters of co-regulated genes, known as modules, that define each state (Langfelder and Horvath, 2008). The modules were analyzed by Genemania, functionally annotated by DAVID, analyzed by Enrichr, and the top, most connected transcripts (hubs) were studied in detail. Inclusion of HD iPSCs, which do not show a phenotype, enabled the isolation of coexpression traits specific to the HD NSCs and the identification of pathways involved in HD pathogenesis. Further modulation of key members of these signaling pathways rescued some HD disease phenotypes.
Results
Analysis of Differential Gene Expression in Undifferentiated and Differentiated HD Induced Pluripotent Stem Cell Models
Previously, we characterized the genetically corrected lines of HD patient-derived iPSCs and found that a phenotype was apparent in the differentiated NSC stage but absent at the pluripotent stem cell state (Figure 1A) (An et al., 2012). We aimed to use this neural-specific correlation between disease genotype and disease phenotype to inform our global analysis of transcriptional changes that occur as a result of mHTT in our established human HD isogenic stem cell model (Figure 1A). Both iPSCs and NSCs were grown in tandem for three passages and eight replicates of each line were plated. Libraries were made from all 32 samples and sequenced using a HiSeq2000 sequencer to an average depth of 23.7 million 50 bp, paired-end reads per sample, 87% uniquely mapping to the human reference genome. Multidimensional scaling (MDS) showed distinct clustering of sample groups (Figure 1B). All iPSCs samples fall within a distinct cluster from NSCs samples. HD and corrected NSCs samples fall within distinct clusters dependent on the CAG length. By contrast, HD and corrected iPSC samples do not clearly delineate from each other. This is consistent with the distinct phenotypes found in HD NSCs and not HD iPSCs.
Figure 1.

RNA-Seq Analysis of HD Isogenic Stem Cell Model
(A) Schematic illustration of experimental design and analysis. iPSCs reprogrammed from HD patient fibroblasts were genetically corrected for the HTT mutation using traditional homologous recombination via a targeting construct. Resulting isogenic iPSC pairs and differentiated isogenic NSC pairs were grown in tandem and plated as replicates for RNA isolation and RNA-seq analysis. There are eight biological replicates (BR) of each-corrected iPSCs, HD iPSCs, corrected NSCs, and HD NSCs: a total of 32 samples.
(B) Multi-dimensional scaling based on VSD-normalized expression levels of the top 5,000 most variable genes.
(C) Number of DE genes across comparisons at <0.01 false discover rate (FDR) threshold. Green bars represent the numbers of downregulated genes and red upregulated for each comparison.
(D) Heatmap representing DE genes in any of the comparisons of HD versus control. Samples (in columns noted H1–H16) and genes (in rows) are clustered by similarity. Shades of red represent upregulation, shades of green downregulation.
(E) Heatmap representing relative expression levels of the DE genes when comparing HD NSCs and corrected NSCs at an adjusted p value of <0.01. Gene names are listed on the right. The nomenclature is the NSC corrected line C116 and the sample number (C116_NSC_#) and HD-NSC and its corresponding sample number (HD_NSC_#).
(F) Table summary of top upstream regulators in HD versus corrected NSC samples via IPA analysis.
EdgeR was used to identify DE genes in each of the two paradigms (NSCs, iPSCs). We assessed the effect of HD mutation in both groups (HD iPSCs versus corrected iPSCs; HD NSCs versus corrected NSCs, false discovery rate threshold: 1%). We identified 4,466 DE genes when comparing HD NSCs and corrected NSCs, of which 2,367 were upregulated and 2,099 were downregulated in the disease state (Figure 1C; Table S1). In contrast, only 370 genes were DE when comparing HD iPSCs to corrected iPSCs (Figure 1C; Table S1). This greater than 10-fold difference in globally DE genes in the disease/corrected NSC pair compared to the same cell pairs in the iPSC state correlates with the neural specific disease phenotypes in HD. Clustering of these DE genes across all samples shows discrete clusters of genes with distinct expression patterns between NSC and iPSC samples (Figure 1D). Functional annotation of the DE genes between HD NSCs and corrected NSCs groups are listed, representing the most significant changes in gene expression at the NSC state due to CAG repeat length (Figure 1E; Table S2). Functional annotation of the DE genes when comparing HD NSCs and corrected NSCs highlighted several gene ontology (GO) categories, including extracellular matrix organization (p = 0.000027), regulation of synapse assembly (p = 0.000097), mammalian phenotype-abnormal survival or perinatal lethality (p = 2.8e-7), and numerous genes involved in axonal guidance such as netrin-1, semaphorin 3D (SEMA3D), BDNF, and cadherin family members. Targets of the transcription factor SMAD3 were overrepresented (p = 0.0018), and the top WikiPathway was BDNF signaling (p = 0.0053).
Ingenuity Pathway Analysis (IPA) of the genes that are DE between HD NSCs and corrected NSCs highlighted, among the top upstream regulators, transforming growth factor β (TGF-β), β-estradiol, and tumor necrosis factor alpha (TNF-α) (Figure 1F). TGF-β is particularly interesting as it plays a critical role in temporal neurogenesis where the onset of TGF-β signaling determines the overall lifespan of the temporal lineage (Dias et al., 2014). HTT is required for neurogenesis and mHTT disrupts striatal and cortical neuronal development (Molina-Calavita et al., 2014, Molero et al., 2009). The role of TGF-β has not been investigated extensively in HD but has been in other neurological diseases, such as Alzheimer’s disease and Parkinson’s disease, where levels of this factor are elevated (Vawter et al., 1996, Zetterberg et al., 2004). Levels of TGF-β are altered in cortical neurons and in peripheral blood of HD patients, as well as in mouse models of HD (Battaglia et al., 2011, Kandasamy et al., 2011). Microarray and bioinformatics analysis of a HD neuronal Caenorhabditis elegans model revealed Wnt/TGF-β as relevant to pathogenesis (Tourette et al., 2014). Similarly, a link with β-estradiol has not been well studied, but it has been shown that male HD transgenic rats display decreased levels of 17β-estradiol at older ages (Bode et al., 2008). Lastly, HD is associated with increased levels of proinflammatory cytokines such as TNF-α, suggesting a role in inflammatory response, which may contribute to HD progression (Björkqvist et al., 2008, Hsiao et al., 2014).
Network Analysis Reveals Modules of Genes Associated with the Pathogenic CAG Repeat
We chose to take advantage of the multi-differentiated state of our experimental design by implementing a coexpression network analysis that could further inform our understanding of the biology of HD. WGCNA was performed on the full RNA sequencing (RNA-seq) expression data across the four distinct cellular states (32 samples) (Figure 1A). WGCNA identifies modules of coexpressed genes that reflect shared functions or cellular components. In addition, identification of highly connected genes (hubs) allows us to pinpoint genes and pathways relevant to further analysis. WGCNA revealed seven distinct modules representing highly co-expressed networks of genes (Figures 2A and 2B; Table S3). A module first principal component (eigengene) can be used to visualize the expression levels for each module across the 32 samples. This analysis showed that some modules (blue, turquoise) represented differences across cell types, whereas for others (red, black), the eigengene was correlated primarily with genotype (Figures 2C–2J).
Figure 2.

WGCNA Analysis of HD Isogenic Stem Cell Model Reveals Drivers of HD Phenotypes in NSCs
(A) Dendogram of genes clustered based on topological overlap. Identified modules are defined by colors.
(B) Dendrogram representing the seven modules identified by WGCNA. The gray module represents genes that are not included in any of the modules.
(C–I) Heatmap (top) and first principal component (eigengene, bottom) of expression data for genes in WGCNA modules across 32 samples. Samples are labeled by group, and the number of genes in each module is in parentheses. Of seven distinct modules defined, turquoise and blue modules are characterized by coexpression traits consistent with expression differences between the iPSC and NSC state (C and D), whereas other individual modules define expression profiles consistent with changes in gene expression in NSCs due to CAG expansion (E and F) y axis is first principal component for (C)–(I).
(J) Summary of all modules by sample group.
We used DAVID to assess the GO annotation of each module (Table S4). The two largest modules by size, blue and turquoise, represent expression changes that clearly delineate between NSC and iPSC sample groups, strongly reflecting expression differences between the iPSC and the differentiated NSC states (Figures 2C, 2D, and 2J). Top biological processes associated with the blue module (downregulated in NSCs relative to iPSCs) include translation, metabolic process, ribosome biogenesis, and proteasomal catabolic process, while conversely, processes associated with the turquoise module (upregulated in NSCs relative to iPSCs) include regulation of transcription, DNA-dependent chromatin modification, chromosome and chromatin organization, cell cycle, and response to DNA damage (Table S4).
We further identified two coexpression modules with features highly relevant to the HD phenotype (Figures 2E and 2F). We hypothesize that the genes in these modules would be more closely relevant to HD as we observe a relevant phenotype in neuronal states and less difference in pluripotent stem cell states. The black (59 genes) and red (73 genes) modules represent genes that are highly upregulated in HD NSCs, while expression differences between HD and control are less pronounced in the iPSC state (Figures 2E, 2F, and 2J). Functional annotation highlighted differences in the biological processes (GO terms) associated with gene members of the red and black modules. Most significant biological processes in the red module include enriched terms neuron differentiation, neuron development, morphogenesis, axonogenesis and axon guidance, whereas no terms were significantly associated with the black module after multiple testing correction (Table S5). The remaining three modules, brown, yellow, and green, (Figures 2G–2I) showed modest correlation with either the cell type or the genotype.
Identifying the Top Genes within the Consensus Modules Based on Significance and Fold Change
Having identified two modules of interest with regard to cell-type-specific gene expression changes due to mHTT, we further focused on the members of these two groups and the gene expression fold changes at the NSC state (HD versus corrected). At a p value cutoff of <0.01, all members of the black module, and all but one in the red module are below that threshold (Table S6). A volcano plot of red and black module members allows further visualization of the highly significant genes in these two groups, while also identifying large magnitude fold changes from these groups (Figure 3A). A log2 fold change cutoff of 1 is shown (Figure 3A; Table S6). Genes that we found upregulated in HD NSCs compared to controls were NTN1, CDH2, DPYSL5, CTNND2, YWHAE, C7orf29, LRRC61, LRRC4B, SEMA3D, PCDHB15, PCDHB18, UQCC, SPOCK3, and GRIP1.
Figure 3.

Volcano and Hub Connectivity of Module Genes, Visualized for the Red and Black Modules
(A) Volcano plot of genes in the red and black WGCNA modules, at p < 0.1. Individual top fold change genes are labeled.
(B and C) Network plot depicting the top connections for each module. Most highly connected module members (hub genes) are shown for each group.
(D and E) Genemania network analysis diagram of red and black modules. All module members in black, with genes of interest highlighted in red.
See also Figure S1.
Identification of Hub Genes in the Modules
Next, we focused on the genes with the highest intramodular connectivity (hub genes), as these are the best representative of the expression of the entire module (Langfelder et al., 2013) and may represent points of biological interest in defining the HD phenotypes. Network plots for the red and black modules, highlighting the top most connected genes, are represented in Figures 3B and 3C (Figures S1A–S1E show the other modules).
For the red module, four hub genes were identified: SEMA3D, CDH2, IRS1, and APBB2 (Figure 3B). Semaphorins are a class of secreted and membrane proteins that act as growth cone guidance molecules through the receptors plexins and are coupled to RRAS. Previously, we identified RRAS as a therapeutic target for HD using a siRNA screen to the druggable genome (Miller et al., 2012). Semaphorin 4D inhibition was shown to ameliorate neuropathology and some cognitive deficits in HD mouse models (Southwell et al., 2015). Cadherin has been linked directly to the function of the HTT protein (Lo Sardo et al., 2012). The HTT protein evolved to acquire a unique regulatory activity for controlling neural adhesion via ADAM10-N-cadherin. Mouse striatal neurons expressing mHTT have altered N-cadherin levels and neuritogenesis (Reis et al., 2011). Insulin growth receptor substrate 1 (IRS1) has not been investigated in HD, but mice lacking this protein are long-lived and lack many of the diseases of aging (Selman et al., 2008). Finally, the amyloid-β precursor protein-binding, family B, member 2 (APPB2) is involved in APP processing and levels of phosphorylation (Sakuma et al., 2009).
For the black module, eight hub genes were identified: HSPB8, DOK5, SH3BGRL2, VGLL3, LYN, GRIP1, FAM131B, and EFEMP1 (Figure 3C). A few of these hub genes particularly stand out with respect to HD. AMPA receptors generate intracellular signals from the cell surface to the nucleus through the Lyn-MAPK pathway, which may contribute to synaptic plasticity by regulating the expression of BDNF (Hayashi et al., 1999). SH3BGL2 (intersectin 2) is involved in clathrin-mediated endocytosis and is a partner of dynamin (Evergren et al., 2007). DOK5 is a substrate for TrkB and TrkC receptors (Shi et al., 2006). Genemania network analysis of black and red modules are shown in Figures 3D and 3E highlighting the genes interconnections.
Enrichment Analysis of the Modules
As an alternative approach to rank and visualize enriched terms, we used the web-based gene list enrichment analysis tool Enrichr. Enrichr tools were applied to all seven modules (Table S6). We found that the genes of the black module are statistically enriched for genes associated with the mouse gene atlas term dorsal striatum (Figure 4A). We used GeneMANIA, a gene function tool that allows visualization between these genes with respect to coexpression, co-localization, and protein interaction to generate an annotated Cytoscape network for the black module (Figure 4B). We found DARPP-32 to be associated with a number of these enriched networks. PPP1R1B (DARPP-32) is downregulated in NSCs but not iPSCs in our RNA-seq data, and differential expression of DARPP-32 has been confirmed by qPCR analysis (Figure 4C) in distinct NSC cultures. DARPP-32 is an abundant protein expressed in striatal MSNs and its expression levels decrease in HD mouse models and human postmortem tissue.
Figure 4.

The Black Module Is Linked to Expression of Genes in the Striatum
(A) Gene enrichment of black module gene members shows significant enrichment of genes associated with dorsal striatum based on the Mouse Gene Atlas list using the Enrichr tool.
(B) Subsequent visualization of networks associated with these five genes using the Genemania tool.
(C) qPCR analysis of DARPP-32 mRNA levels in HD and C116 NSC samples (n = 5 BR). Five independent BR are displayed as mean ± SD.
(D) Both the red and black modules have genes enriched for human striatum. The matches for the red module are ACTB, C17orf58, CORO2B, CTNND2, FAM20C, GNAZ, GPRIN3, HBEGF, KIF5C, MTSS1, MURC, NEFM, NRXN1, NTN1, PCDHB12, PCDHB18, PCP4, RAB1F, RND3, SDK1, STK32B, TMEM88, TPM1, TUBB3, and ZN597.
See also Figures S2 and S3.
Given the black module’s enrichment to the mouse striatum, we next evaluated if this module is associated with the human striatum during development. We evaluated if any of the HD-relevant modules were enriched for striatum-specific marker genes identified from a genome-wide adult brain mouse atlas (Lein et al., 2007). While the top 100 genes with the most striatum-specific expression were selected, only 49 of these were actually expressed in our dataset. From the 50 mouse homologous genes in the black module, only Dach1 is among the most striatum-specific marker genes, while there was no overlap with the 61 mouse homologous genes in red module. We also evaluated if the black or red modules were enriched for genes associated with human striatum during development, identified from gene expression data in prenatal human striatum (from 2 to 20 weeks of age) (Onorati et al., 2014). We found that the red module was statistically enriched for DE genes found to be upregulated in the developing human striatum (ELOVL4, KIF5C, MTSS1, NEFM, NRXN1, NTN1, PCP4, STK32B, and TMEM88; Z score = 4.18). Using WGCNA analysis, the group also identified a module, named M25, whose members are highly expressed in the developing human striatum (Onorati et al., 2014). We found that the black and red modules were statistically enriched for genes found in the M25 module (Figure 4D). The genes matching the highly expressed genes in the striatum were BEX2, C7orf13, FAM131B, GREB1L, GRM2, LPPR1, LYN, MAP2, MMP11, MVD, NUAK1, POU3F1, PPARGC1A, SH3BGRL2, SRCIN1, TNFRSF21, and UQCC. In addition to the black module, we also evaluated other key striatal-specific genes identified in the Onorati study using our RNA-seq expression dataset (Table S1). Well known genes in human striatal development CTIP2, DARPP-32, ISL1, TBR1, FOXP1, and PAX6 are all downregulated in HD NSCs compared to control NSCs (Figure S2). Interestingly, FOXP2 is upregulated in HD NSCs compared to control NSCs, and its expression becomes more restricted in the human striatum during development (Onorati et al., 2014).
Because degeneration of MSNs is the pathological hallmark of HD, we used three independently generated lists of MSN-enriched gene sets, by laser-captured HD patient MSNs (Hodges et al., 2006), tagged ribosome profiling in adult mice (Heiman et al., 2008) and by FACS-sorting of D1/D2 MSNs (Lobo et al., 2006) for analysis. We found that in the red module, 16 of the 61 mouse homolog genes were MSN-specific marker genes, though the enrichment was not significant (Z score = 2.03). Moreover, the red module also included NRXN1, one of the nine genes differentially enriched in striatonigral (D1) neurons (Lobo et al., 2006).
We note that our coexpression modules identified highly upregulated genes at the HD NSC state in the black and red modules compared to control NSCs. Coexpression modules representing highly downregulated genes at the HD NSC state were not identified by the WGCNA analysis (Figure 2J). In particular, reduced expression of BDNF is a well-characterized phenotype in HD models and in brains of HD patients. BDNF is downregulated in HD NSCs in our RNA-seq analysis, and as noted above, BDNF signaling is the top Wikipathway identified among the 100 top DE genes (p = 0.0053). Using a hierarchical clustering approach, we identified genes whose expression pattern follows that of BDNF (Figure S3A). This cluster contains genes that, while unchanged in iPSCs, are downregulated in HD-NSCs compared to control NSCs and may therefore represent genes that are dysregulated in the NSC lineage of HD patients. It included several genes known to be associated with HD and striatal development, such as BDNF, TGM2, and DARPP-32, as well as five cadherins (CDH15, CHD6, PCDHGA3, PCDHGB7, and PCDHGC4) (Figure S3B). Genemania shows the coexpression network for this cluster of genes (Figure S3C). Enrichr links this module to dorsal striatum as well, and genes are enriched for the M25 module (Onorati et al., 2014). Further, this group contains a key constituent of the TNF upstream regulatory pathway that was identified in our IPA analysis of DE genes (Figure 1F): TNFRSF1B (TNFR-II or p75), whose expression highly correlates with BDNF. Based on these findings, we evaluated if the black or red modules were enriched for genes-associated BDNF depletion, identified from gene expression striatal profiles from BDNF knockout mice (Strand et al., 2007). We found that the red module was statistically enriched for genes upregulated (five genes: CSRP1, NTN1, PCYT2, PTPRK, and SEMA3A; Z score = 4.2) and downregulated (five genes: ACTN1, GNAZ, HBEGF, PCP4, and TPM1; Z score = 5.03) in the BDNF-KO mice.
We next tested whether genes in HD-associated modules were enriched for transcription factor binding sites using the ENCODE chromatin immunoprecipitation sequencing (ChIP-seq) significance tool (Auerbach et al., 2013). We found that both HD modules were enriched for the Ezh2 (Enhancer of zeste homolog 2) factor, with 21 (out of the 58 valid) black module genes (Q value = 3.204e-2) and 28 (out of 65 valid) red module genes (Q value = 5.905e-5) having a binding site for this particular transcription factor. EZH2, is the functional enzymatic component of the polycomb repressive complex 2 (PRC2), which is involved in various biological processes, including differentiation, maintenance of cell identity and proliferation, and stem-cell plasticity (Margueron and Reinberg, 2011). HTT facilitates PRC2 function and this is at least partially mediated by the physical interaction of full-length wild-type HTT protein with the Ezh2 and Suz12 PRC2 core component (Seong et al., 2010).
Therapeutic Targets Can Be Identified from Bioinformatic Analysis of Expression Data from HD NSCs
Bioinformatics analysis revealed key signaling molecules in neurogenesis, striatal development and axonal guidance. Consistent with this, two pathways identified relevant to HD phenotypes include TGF-β (top altered RNA-seq pathway) and netrin-1 (black module). Both of these pathways are required for neurogenesis and are linked to development of the striatum. Therefore, we evaluated whether these pathways were altered in our HD NSC model and if modulation was neuroprotective.
TGF-β is the top signaling pathway that is altered between HD and corrected NSCs based on our RNA-seq results. Therefore, we evaluated TGF-β signaling in our human HD stem cell model and in zQ175 knockin HD mice. First, we determined basal levels of TGF-β expression in HD and corrected NSCs. TGF-β is part of a larger complex and appears in multiple forms including its precursor form (∼60 kDa), its active dimer form (∼24 kDa), and its inactive monomer form (∼12 kDa) (Annes et al., 2003). Using a pan-TGF-β antibody that recognizes all three TGF-β isoforms, western blot analysis of HD and corrected NSC lysates revealed that HD NSCs significantly express more TGF-β in its precursor, dimer, and monomer forms (Figures 5A and 5B). TGF-β ligands signal through TGF-β receptors and activate SMAD proteins through phosphorylation of SMAD2/3 (Dennler et al., 2002). Because TGF-β1 expression is increased in the brain in response to injury (Krupinski et al., 1996), we determined whether recombinant TGF-β1 could modulate TGF-β signaling in HD NSCs and ameliorate HD phenotypes. To test for activation of SMAD signaling, HD and corrected NSCs were treated with or without TGF-β1. Both corrected and HD NSCs showed an increase in pSMAD2 levels upon TGF-β1 treatment compared to untreated NSCs (Figures 5C and 5D). In basal conditions without TGF-β1 treatment, HD NSCs expressed more pSMAD2 than corrected NSCs. Furthermore, TGF-β1 treatment resulted in higher pSMAD2 phosphorylation expression in HD NSCs compared to corrected NSCs. Thus, both HD and corrected NSCs can activate SMAD signaling upon exposure to TGF-β1, however HD NSCs have more pSMAD2 overall. This could be due to HD NSCs having more TGF-β or to a compensatory mechanism to combat the HD mutation. To confirm that expression of TGF-β changes are observed in multiple patient HD iPSC lines (nonisogenic), we also analyzed HD iPSC lines ND41656 (CAG57) and ND42222 (CAG109) and compared them to a control (Figures 5E and 5F). We found that the levels of TGF-β were higher in NSCs derived from the HD lines compared to the control.
Figure 5.

TGF-β Expression and SMAD Signaling Is Altered in HD NSCs
(A) TGF-β protein levels in HD (n = 5 BR) and corrected C116 NSCs (n = 5 BR) were determined by western blot and normalization to α-tubulin.
(B) HD NSCs had elevated TGF-β protein in multiple forms including the TGF-β precursor complex, TGF-β dimer and monomer, compared to C116 NSCs.
(C) SMAD activation, indicated by phosphorylation of SMAD2 (pSMAD2), was determined in HD and C116 NSCs via western blot by normalization to SMAD2.
(D) Both corrected and HD NSCs activated the SMAD signaling pathway upon treatment with recombinant human TGF-β1 (10 ng/ml) for 1 hr. Under normal basal conditions, HD NSCs have higher levels of activated pSMAD2 compared to corrected NSCs. Similarly, HD NSCs treated with TGF-β1 had higher levels of pSMAD2 compared to C116 NSCs. All experiments had five biological replicates per genotype, and each experiment was repeated three times.
(E) TGF-β protein levels in HD lines (n = 2 BR) and control NSCs (n = 2 BR) were determined by western blot and normalization to α-tubulin.
(F) Densitometry of the western blot shown in (E).
Elevated levels of TGF-β protein and changes in TGF-β signaling in HD suggest a potential neuroprotective role for TGF-β in the brain. Therefore, we determined whether TGF-β1 could rescue HD-related phenotypes that we identified in our earlier studies (An et al., 2012, Zhang et al., 2010). We previously found that HD NSCs had elevated caspase-3/7 activity compared to corrected NSCs upon removing serum from cell culture medium. To determine if TGF-β is protective in a toxicity assay, we cultured corrected or HD NSCs in either (1) their regular NSC media supplemented with growth factors (complete), or (2) in basic medium without growth factor but supplemented either with recombinant TGF-β1 or TGF-β2 for 48 hr. Both TGF-β1 and TGF-β2 significantly reduced caspase-3/7 activity in basic conditions (Figure 6A). The reduction in caspase activity occurred in a dose-responsive manner (Figure 6B), as higher concentrations of TGF-β1 were more effective at reducing caspase activity in HD NSCs in basic conditions. We did not see a significant reduction in caspase activity in corrected NSCs in basic conditions with addition of either TGF-β1 or TGF-β2. We also evaluated whether the deficit in maximal respiratory capacity in HD NSCs was normalized by TGF-β1 treatment. HD NSCs have deficits in mitochondrial function when compared to corrected NSCs as measured by Seahorse respiration (An et al., 2012). TGF-β1 treatment rescued the respiratory deficit in HD NSCs (Figure 6C).
Figure 6.

TGF-β Expression Has Neuroprotective Effects In Vitro and Is Elevated In Vivo
(A) HD NSCs have elevated caspase-3/7 activity upon growth factor withdrawal. This phenotype is partially rescued by treatment with either recombinant human TGF-β1 or TGF-β2 (10 ng/ml) (n = 5 BR per genotype, each experiment repeated three times).
(B) TGF-β1 ameliorates elevated caspase-3/7 activity in HD NSCs in a dose response manner (n = 5 BR per genotype).
(C) Maximal respiratory capacity of HD NSCs and C116 NSCs was measured in the presence and absence of TGF-β (N = 6 BR per genotype, each experiment repeated three times).
(D) TGF-β expression in the cortex of zQ175 mice at 6 months was higher in the precursor and dimer forms and higher in the dimer form at 12 months of age (n = 3 BR per genotype, each experiment repeated three times).
(E) In the striatum, TGF-β monomer was elevated in zQ175 compared to wild-type at 12 months, but no significance was seen at 6 months (n = 3 BR per genotype, each experiment repeated four times).
After confirming that TGF-β protein levels and SMAD signaling are altered in HD NSCs, we next asked whether TGF-β levels are also altered in vivo. We analyzed the cortex and striatum of zQ175 HD knockin mice for TGF-β expression at 6 and 12 months of age. In the cortex, zQ175 homozygous mice had elevated levels of multiple TGF-β protein forms compared to wild-type littermate controls. Specifically, zQ175 cortex at 6 months had significantly higher levels of the TGF-β precursor and dimer and exhibited a trend in having more TGF-β monomer compared to controls. At 12 months, zQ175 cortex had higher levels of TGF-β dimer and showed a trend toward having more TGF-β monomer when compared to controls (Figure 6D). Results were not as conclusive for the striatum (Figure 6E). We only observed a significant increase in TGF-β monomer in zQ175 mice when compared to controls at 12 months of age (Figure 6D). Our zQ175 results support our in vitro findings and suggest that TGF-β expression is elevated in HD brains.
Next, we investigated if modulation of the genes in the red module would have a neuroprotective effect in our HD NSC model. We focused on the axonal guidance molecule netrin-1 (Figures 3A and 3D) since it was one of the most significantly altered genes in this module that has defined function in the axonal guidance pathway. Furthermore, netrin-1 is expressed in the striatal ventricular zone during development, guides the large influx of striatal matrix neurons into the striatum, and is critical to the formation of fundamental striatal structures (Hamasaki et al., 2001). As shown in Figures 7A and 7B, the receptors for netrin-1, such as DCC and UNC5D, have increased expression in HD NSCs as determined by qPCR (Figure 7B). Elevated levels of netrin-1 and changes in this signaling pathway in HD may suggest a potential neuroprotective role for netrin-1 in the brain. To test this, we treated HD NSCs with recombinant netrin-1. We found that netrin-1 treatment reduced caspase-3/7 activity in growth factor starved HD NSCs and is potentially neuroprotective (Figure 7C). We also evaluated whether the deficit in maximal respiratory capacity in HD NSCs was normalized by netrin-1 treatment. Netrin-1 treatment rescued the respiratory deficit in HD NSCs (Figure 7D). The netrin protein levels were not significantly altered in the zQ175 mice at 12 month of age compared to controls as measured by western blot analysis (data not shown). Perhaps the alteration occurs early in development and is normalized in adults.
Figure 7.

Netrin-1 Is Neuroprotective in HD Stem Cell Model
(A) The expression of netrin-1 mRNA in increased in HD NSCs (n = 8 BR per genotype, repeated three times).
(B) The receptors for netrin signaling have altered expression in HD NSCs (n = 8 BR per genotype).
(C) HD NSCs have elevated caspase-3/7 activity upon growth factor withdrawal. This phenotype is rescued by treatment with netrin-1 (100 ng/ml) (n = 5 BR per condition, repeated three times).
(D) Maximal respiratory capacity of HD NSCs and C116 NSCs was measured in the presence and absence of netrin-1 (n = 6 BR per genotype, repeated three times).
Discussion
In our previous work, we found cellular and molecular phenotypes associated with HD in NSCs derived from HD patient-specific iPSCs when compared to isogenic controls. The phenotypes we reported included susceptibility to cellular death as measured by caspase activity and TUNEL staining, mitochondrial deficits, lower levels of BDNF, altered cadherin and TGF signaling (An et al., 2012). The cellular and molecular phenotypes were only associated with the differentiated HD NSC state and were not detected in HD iPSCs. This finding suggests that the first disease phenotypes in HD manifest during early development at the NSC stage. This is interesting because we may be detecting HD developmental changes that occur early in disease progression, in our human HD NSC models.
To examine the molecular basis of the phenotypes found in HD NSCs, we used RNA-seq to measure expression changes in these cellular states. Inclusion of HD iPSCs, which do not show a distinct disease-associated phenotype, enabled specific isolation of coexpression traits specific to the HD NSCs and the identification of pathways involved in HD pathogenesis. WGCNA analysis revealed the effects of HTT CAG expansion on NSCs and identified two modules driving HD phenotypes—the black and red modules. By studying gene expression changes in prenatal human striatum during development, using WGCNA analysis, another group identified a module they named M25 whose members are highly expressed in the developing human striatum (Onorati et al., 2014). We found that the black and red modules were enriched for genes found in the M25 module, which is associated with human striatal tissue. In addition to the black and red module, we also evaluated other key striatal-specific and well known genes in human striatal development including CTIP2 (BCL11B), DARPP-32 (PPP1R1B), ISL1, TBR1, FOXP1, and PAX6, which were all downregulated in HD NSCs compared to control NSCs. This is consistent with a number of studies showing mHTT alters cortical and striatal neurogenesis in mouse models of HD (Molero et al., 2009), and the normal function of HTT is implicated in neurogenesis. Indeed, knockin HD mice with Q111 repeats exhibited delayed striatal cytoarchitecture with altered expression of markers of MSNs (Molero et al., 2009). The role of development in neurodegeneration has been addressed in a related polyglutamine expansion disease, SCA1 (Serra et al., 2006). When ATXN1[82Q] expression in SCA1 mice was delayed during postnatal development, the disease was less severe. This correlation of genes found in our NSC analysis and in previous mouse studies in the field support the concept that NSCs can be a valuable tool for modeling a disease in a dish.
A question arising from this work is whether development sets the stage for the early pathophysiological abnormalities in HD and whether we can identify candidate pathways or genes that will restore neuronal homeostasis and protect striatal MSNs halting disease progression. We believe the black and red modules defined in our studies represent critical cellular pathways that are disrupted in early HD pathogenesis and that normalization will restore neuronal homeostasis and prevent neuronal degeneration. Our bioinformatics analysis revealed key signaling molecules in neurogenesis, striatal development, and axonal guidance for these modules. To test whether these pathways were altered in HD and represented therapeutic targets for HD, we focused on two signaling pathways: TGF-β and netrin. During development the striatum forms from the ventral telencephalon and is dependent upon BMP/TGF-β inhibition (Kuang et al., 2006) as well as netrin-1 signaling (Hamasaki et al., 2001). Our studies of TGF-β signaling in HD reveal that this signaling pathway is altered in HD. Both HD NSCs and HD knockin mouse brains express more TGF-β than wild-type controls. Furthermore, TGF-β1 is neuroprotective in our NSC models and can ameliorate some HD phenotypes including reducing caspase activity and improving respiratory capacity in HD NSCs. These results suggest a possible compensatory mechanism in which HD NSCs express higher levels of TGF-β in order to compensate for the negative effects caused by the HD mutation. Our findings show that netrin-1 levels and receptors were altered in HD NSCs. The addition of axonal guidance molecule was neuroprotective consistent with the neuroprotective role of netrin-1 after stroke (Liao et al., 2013). In addition to these two signaling pathways, our analysis also revealed that the top WikiPathway was BDNF signaling for the top 100 genes. This agrees with the strong correlation of expression changes of BDNF knockout mice with human HD brain (Kuhn et al., 2007, Strand et al., 2007) and numerous studies showing that BDNF is neuroprotective in HD (Crane et al., 2014). All of these findings support the use of NSCs for studying HD in its developmental phases.
In conclusion, we report here that a HD NSC model provides evidence for early disruption of signaling pathways that are critical to establishing striatal lineage, and restoring these pathways either early or late in the disease process may provide disease-modifying therapies. Future work will focus on modulating these critical pathways in vivo to provide therapeutics treatments for HD.
Experimental Procedures
Culture of iPSCs
All lines of iPSCs were grown on Matrigel (BD Biosciences) in mTeSR1 defined medium (STEMCELL). Optimal passaging occurred at 70%–80% confluency and was performed by treatment for 1 hr with 1 mg/ml collagenase. For RNA-seq sample preparation, comparison lines (HD iPSCs, C116 iPSCs) were grown and expanded in tandem for three passages before collection of total RNA. The isogenic lines (HD iPSCs and C116 iPSCs) were described in An et al. (2012) and Zhang et al. (2010) as well as further control lines. The non-isogenic HD iPSCs lines ND41656 (CAG57) and ND42222 (CAG109) were obtained from Coriell repository. The methods for differentiation to NSCs, RNA isolation, RNA-seq, qPCR, enrichment and network analysis, western blot of NSCs and zQ175 mice, caspase activity, and seahorse measurements are found in the Supplemental Experimental Procedures and previous publications (An et al., 2012, Zhang et al., 2010).
Author Contributions
K.R., M.C.A., and N.Z. carried out experiments. K.R., M.C.A., N.Z., S.M., and L.M.E. designed experiments. K.R., M.C.A., N.Z, R.N.O., F.G., E.M.R., R.A., B.B., S.M., S.D.M., G.C., and L.M.E. performed analysis of the data sets. M.C.A., K.R., and L.M.E. wrote the paper. All authors edited and contributed to the manuscript preparation.
Acknowledgments
The funding for this research was provided by the CHDI, Inc. (L.M.E.), NIH T32 AG000266 (L.M.E., R.N.O., and K.R.), and F32 NS080551 (R.N.O.) training grants. We acknowledge the support of the NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691).
Published: December 8, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, three figures, and six tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.11.005.
Accession Numbers
The accession number for the RNA-seq data reported in this paper is GEO: GSE74201.
Supplemental Information
References
- An M.C., Zhang N., Scott G., Montoro D., Wittkop T., Mooney S., Melov S., Ellerby L.M. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. 2012;11:253–263. doi: 10.1016/j.stem.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes J.P., Munger J.S., Rifkin D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Auerbach R.K., Chen B., Butte A.J. Relating genes to function: identifying enriched transcription factors using the ENCODE ChIP-Seq significance tool. Bioinformatics. 2013;29:1922–1924. doi: 10.1093/bioinformatics/btt316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard J., Wall M.D., Lazari O., Arjomand J., Munoz-Sanjuan I. Advances in huntington disease drug discovery: novel approaches to model disease phenotypes. J. Biomol. Screen. 2014;19:191–204. doi: 10.1177/1087057113510320. [DOI] [PubMed] [Google Scholar]
- Battaglia G., Cannella M., Riozzi B., Orobello S., Maat-Schieman M.L., Aronica E., Busceti C.L., Ciarmiello A., Alberti S., Amico E. Early defect of transforming growth factor β1 formation in Huntington’s disease. J. Cell. Mol. Med. 2011;15:555–571. doi: 10.1111/j.1582-4934.2010.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkqvist M., Wild E.J., Thiele J., Silvestroni A., Andre R., Lahiri N., Raibon E., Lee R.V., Benn C.L., Soulet D. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008;205:1869–1877. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode F.J., Stephan M., Suhling H., Pabst R., Straub R.H., Raber K.A., Bonin M., Nguyen H.P., Riess O., Bauer A. Sex differences in a transgenic rat model of Huntington’s disease: decreased 17β-estradiol levels correlate with reduced numbers of DARPP32+ neurons in males. Hum. Mol. Genet. 2008;17:2595–2609. doi: 10.1093/hmg/ddn159. [DOI] [PubMed] [Google Scholar]
- Camnasio S., Delli Carri A., Lombardo A., Grad I., Mariotti C., Castucci A., Rozell B., Lo Riso P., Castiglioni V., Zuccato C. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis. 2012;46:41–51. doi: 10.1016/j.nbd.2011.12.042. [DOI] [PubMed] [Google Scholar]
- Crane A.T., Rossignol J., Dunbar G.L. Use of genetically altered stem cells for the treatment of Huntington’s disease. Brain Sci. 2014;4:202–219. doi: 10.3390/brainsci4010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S., Goumans M.J., ten Dijke P. Transforming growth factor beta signal transduction. J. Leukoc. Biol. 2002;71:731–740. [PubMed] [Google Scholar]
- Dias J.M., Alekseenko Z., Applequist J.M., Ericson J. Tgfβ signaling regulates temporal neurogenesis and potency of neural stem cells in the CNS. Neuron. 2014;84:927–939. doi: 10.1016/j.neuron.2014.10.033. [DOI] [PubMed] [Google Scholar]
- Ehrlich M.E. Huntington’s disease and the striatal medium spiny neuron: cell-autonomous and non-cell-autonomous mechanisms of disease. Neurotherapeutics. 2012;9:270–284. doi: 10.1007/s13311-012-0112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evergren E., Gad H., Walther K., Sundborger A., Tomilin N., Shupliakov O. Intersectin is a negative regulator of dynamin recruitment to the synaptic endocytic zone in the central synapse. J. Neurosci. 2007;27:379–390. doi: 10.1523/JNEUROSCI.4683-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel B.L., Cho E., Wahnich A., Gao F., Becherel O.J., Wang X., Fike F., Chen L., Criscuolo C., De Michele G. Mutation of senataxin alters disease-specific transcriptional networks in patients with ataxia with oculomotor apraxia type 2. Hum. Mol. Genet. 2014;23:4758–4769. doi: 10.1093/hmg/ddu190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki T., Goto S., Nishikawa S., Ushio Y. A role of netrin-1 in the formation of the subcortical structure striatum: repulsive action on the migration of late-born striatal neurons. J. Neurosci. 2001;21:4272–4280. doi: 10.1523/JNEUROSCI.21-12-04272.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T., Umemori H., Mishina M., Yamamoto T. The AMPA receptor interacts with and signals through the protein tyrosine kinase Lyn. Nature. 1999;397:72–76. doi: 10.1038/16269. [DOI] [PubMed] [Google Scholar]
- HD iPSC Consortium Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012;11:264–278. doi: 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M., Schaefer A., Gong S., Peterson J.D., Day M., Ramsey K.E., Suárez-Fariñas M., Schwarz C., Stephan D.A., Surmeier D.J. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A., Strand A.D., Aragaki A.K., Kuhn A., Sengstag T., Hughes G., Elliston L.A., Hartog C., Goldstein D.R., Thu D. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Hsiao H.Y., Chiu F.L., Chen C.M., Wu Y.R., Chen H.M., Chen Y.C., Kuo H.C., Chern Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 2014;23:4328–4344. doi: 10.1093/hmg/ddu151. [DOI] [PubMed] [Google Scholar]
- Kandasamy M., Reilmann R., Winkler J., Bogdahn U., Aigner L. Transforming growth factor-beta signaling in the neural stem cell niche: a therapeutic target for Huntington’s disease. Neurol. Res. Int. 2011;2011:124256. doi: 10.1155/2011/124256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye J.A., Finkbeiner S. Modeling Huntington’s disease with induced pluripotent stem cells. Mol. Cell. Neurosci. 2013;56:50–64. doi: 10.1016/j.mcn.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupinski J., Kumar P., Kumar S., Kaluza J. Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans. Stroke. 1996;27:852–857. doi: 10.1161/01.str.27.5.852. [DOI] [PubMed] [Google Scholar]
- Kuang C., Xiao Y., Yang L., Chen Q., Wang Z., Conway S.J., Chen Y. Intragenic deletion of Tgif causes defectsin brain development. Hum. Mol. Genet. 2006;15:3508–3519. doi: 10.1093/hmg/ddl427. [DOI] [PubMed] [Google Scholar]
- Kuhn A., Goldstein D.R., Hodges A., Strand A.D., Sengstag T., Kooperberg C., Becanovic K., Pouladi M.A., Sathasivam K., Cha J.H. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Mischel P.S., Horvath S. When is hub gene selection better than standard meta-analysis? PLoS ONE. 2013;8:e61505. doi: 10.1371/journal.pone.0061505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.Y., Cantle J.P., Yang X.W. Genetic manipulations of mutant huntingtin in mice: new insights into Huntington’s disease pathogenesis. FEBS J. 2013;280:4382–4394. doi: 10.1111/febs.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein E.S., Hawrylycz M.J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A.F., Boguski M.S., Brockway K.S., Byrnes E.J. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Liao S.J., Gong Q., Chen X.R., Ye L.X., Ding Q., Zeng J.S., Yu J. Netrin-1 rescues neuron loss by attenuating secondary apoptosis in ipsilateral thalamic nucleus following focal cerebral infarction in hypertensive rats. Neuroscience. 2013;231:225–232. doi: 10.1016/j.neuroscience.2012.11.059. [DOI] [PubMed] [Google Scholar]
- Lo Sardo V., Zuccato C., Gaudenzi G., Vitali B., Ramos C., Tartari M., Myre M.A., Walker J.A., Pistocchi A., Conti L. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat. Neurosci. 2012;15:713–721. doi: 10.1038/nn.3080. [DOI] [PubMed] [Google Scholar]
- Lobo M.K., Karsten S.L., Gray M., Geschwind D.H., Yang X.W. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat. Neurosci. 2006;9:443–452. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- Margueron R., Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.P., Yates B.E., Al-Ramahi I., Berman A.E., Sanhueza M., Kim E., de Haro M., DeGiacomo F., Torcassi C., Holcomb J. A genome-scale RNA-interference screen identifies RRAS signaling as a pathologic feature of Huntington’s disease. PLoS Genet. 2012;8:e1003042. doi: 10.1371/journal.pgen.1003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero A.E., Gokhan S., Gonzalez S., Feig J.L., Alexandre L.C., Mehler M.F. Impairment of developmental stem cell-mediated striatal neurogenesis and pluripotency genes in a knock-in model of Huntington’s disease. Proc. Natl. Acad. Sci. USA. 2009;106:21900–21905. doi: 10.1073/pnas.0912171106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Calavita M., Barnat M., Elias S., Aparicio E., Piel M., Humbert S. Mutant huntingtin affects cortical progenitor cell division and development of the mouse neocortex. J. Neurosci. 2014;34:10034–10040. doi: 10.1523/JNEUROSCI.0715-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onorati M., Castiglioni V., Biasci D., Cesana E., Menon R., Vuono R., Talpo F., Goya R.L., Lyons P.A., Bulfamante G.P. Molecular and functional definition of the developing human striatum. Nat. Neurosci. 2014;17:1804–1815. doi: 10.1038/nn.3860. [DOI] [PubMed] [Google Scholar]
- Reis S.A., Thompson M.N., Lee J.M., Fossale E., Kim H.H., Liao J.K., Moskowitz M.A., Shaw S.Y., Dong L., Haggarty S.J. Striatal neurons expressing full-length mutant huntingtin exhibit decreased N-cadherin and altered neuritogenesis. Hum. Mol. Genet. 2011;20:2344–2355. doi: 10.1093/hmg/ddr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma M., Tanaka E., Taru H., Tomita S., Gandy S., Nairn A.C., Nakaya T., Yamamoto T., Suzuki T. Phosphorylation of the amino-terminal region of X11L regulates its interaction with APP. J. Neurochem. 2009;109:465–475. doi: 10.1111/j.1471-4159.2009.05988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C., Lingard S., Choudhury A.I., Batterham R.L., Claret M., Clements M., Ramadani F., Okkenhaug K., Schuster E., Blanc E. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2008;22:807–818. doi: 10.1096/fj.07-9261com. [DOI] [PubMed] [Google Scholar]
- Seong I.S., Woda J.M., Song J.-J., Lloret A., Abeyrathne P.D., Woo C.J., Gregory G., Lee J.-M., Wheeler V.C., Walz T. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2010;19:573–583. doi: 10.1093/hmg/ddp524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra H.G., Duvick L., Zu T., Carlson K., Stevens S., Jorgensen N., Lysholm A., Burright E., Zoghbi H.Y., Clark H.B. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 2006;127:697–708. doi: 10.1016/j.cell.2006.09.036. [DOI] [PubMed] [Google Scholar]
- Shi L., Yue J., You Y., Yin B., Gong Y., Xu C., Qiang B., Yuan J., Liu Y., Peng X. Dok5 is substrate of TrkB and TrkC receptors and involved in neurotrophin induced MAPK activation. Cell. Signal. 2006;18:1995–2003. doi: 10.1016/j.cellsig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Southwell A.L., Franciosi S., Villanueva E.B., Xie Y., Winter L.A., Veeraraghavan J., Jonason A., Felczak B., Zhang W., Kovalik V. Anti-semaphorin 4D immunotherapy ameliorates neuropathology and some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 2015;76:46–56. doi: 10.1016/j.nbd.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Strand A.D., Baquet Z.C., Aragaki A.K., Holmans P., Yang L., Cleren C., Beal M.F., Jones L., Kooperberg C., Olson J.M., Jones K.R. Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J. Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Tourette C., Farina F., Vazquez-Manrique R.P., Orfila A.M., Voisin J., Hernandez S., Offner N., Parker J.A., Menet S., Kim J. The Wnt receptor Ryk reduces neuronal and cell survival capacity by repressing FOXO activity during the early phases of mutant huntingtin pathogenicity. PLoS Biol. 2014;12:e1001895. doi: 10.1371/journal.pbio.1001895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vawter M.P., Dillon-Carter O., Tourtellotte W.W., Carvey P., Freed W.J. TGFbeta1 and TGFbeta2 concentrations are elevated in Parkinson’s disease in ventricular cerebrospinal fluid. Exp. Neurol. 1996;142:313–322. doi: 10.1006/exnr.1996.0200. [DOI] [PubMed] [Google Scholar]
- Victorson D., Carlozzi N.E., Frank S., Beaumont J.L., Cheng W., Gorin B., Duh M.S., Samuelson D., Tulsky D., Gutierrez S. Identifying motor, emotional-behavioral, and cognitive deficits that comprise the triad of HD symptoms from patient, caregiver, and provider perspectives. Tremor Other Hyperkinet. Mov. (N. Y.) 2014;4:224. doi: 10.7916/D8JW8BWS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videnovic A. Treatment of huntington disease. Curr. Treat. Options Neurol. 2013;15:424–438. doi: 10.1007/s11940-013-0219-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg H., Andreasen N., Blennow K. Increased cerebrospinal fluid levels of transforming growth factor-beta1 in Alzheimer’s disease. Neurosci. Lett. 2004;367:194–196. doi: 10.1016/j.neulet.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Zhang N., An M.C., Montoro D., Ellerby L.M. Characterization of human Huntington’s disease cell model from induced pluripotent stem cells. PLoS Curr. 2010;2:RRN1193. doi: 10.1371/currents.RRN1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.