

Abstract
Background
Triple-Negative Breast Cancer (TNBC) is an aggressive disease with a poor prognosis. Clinically, TNBC patients have limited treatment options besides chemotherapy. The goal of this study was to determine the kinase dependency in TNBC cell lines and to predict compounds that could inhibit these kinases using integrative bioinformatics analysis.
Results
We integrated publicly available gene expression data, high-throughput pharmacological profiling data, and quantitative in vitro kinase binding data to determine the kinase dependency in 12 TNBC cell lines. We employed Kinase Addiction Ranker (KAR), a novel bioinformatics approach, which integrated these data sources to dissect kinase dependency in TNBC cell lines. We then used the kinase dependency predicted by KAR for each TNBC cell line to query K-Map for compounds targeting these kinases. Wevalidated our predictions using published and new experimental data.
Conclusions
In summary, we implemented an integrative bioinformatics analysis that determines kinase dependency in TNBC. Our analysis revealed candidate kinases as potential targets in TNBC for further pharmacological and biological studies.
Keywords: Kinase dependency, Triple-Negative Breast Cancer, high-throughput screening, bioinformatics
Background
Triple-negative breast cancer (TNBC) is a subtype of breast cancer that is lacking the expression ofestrogen receptor (ER), progesterone receptor (PR) and HER2 (ERBB2)[1]. TNBC, also known as basal-like breast cancer, is an aggressive disease with a poor prognosis. Unlike ER-positive, PR-positive, and HER2-amplified breast cancer subtype patients, chemotherapy is the only treatment option for TNBC patients. Advances in the treatment of TNBC have been hampered by the lack of novel effective targeted therapies due to the poor understanding of the underlying molecular characteristics of this disease. Recent large-scale molecular characterization studies in breast cancer have revealed some frequently mutated genes and altered pathways in TNBC[2,3]. These genes and pathways include TP53, BRCA1/2, PIK3CA, and PTEN mutations and activation of PI3K/AKT and RAS/RAF/MEK signaling pathways. Many of these genes and pathways are regulated by kinases (e.g. PIK3CA, RAS, MAPKs); therefore providing an opportunity to identify potential druggable targets by small moleculesfor TNBC therapy.
Protein kinases represent one of the largest “druggable” and well-studied protein families in the human genome[4]. This class of proteins (kinome) plays key role in regulating various signaling pathways in cells. There are>500 members of the human kinome which can be classified into seven different kinase families based on their conserved catalytic domain sequences[5]. In cancer cells, some kinases are mutated and have acquired oncogenic properties to drive tumorgenesis. Small molecules that inhibit these oncogenic kinases can effectively kill cancer cells. Targeted cancer therapies have exploited this “oncogene addiction” concept[6]; this has lead to several successful clinical applications of targeted therapies: BCR-ABL tyrosine kinase inhibition in chronic myeloid leukemia by imatinib[7], inhibition of EGFR in EGFR-mutated non-small cell lung cancers (NSCLC) by erlotinib or gefitinib[8-10], inhibition of BRAF in BRAF-mutated melanoma by vemurafenib[11]and inhibition of ALK in EML4-ALK NSCLC by crizotinib[12]. Furthermore, many of the small molecules inhibit multiple kinases and could be repositioned or repurposedfor other applications. For example, imatinib has been repositioned to inhibit KIT and PDGFRA in gastrointestinal stromal tumors[13] and crizotinib has been repositioned to inhibit ROS1 in ROS1-fusion NSCLC patients[14]. Large-scale quantitative in vitro kinase binding assays have been developed to capture the complex interactions between inhibitors and kinases[15-17].
High-throughput screening (HTS) provides a different perspective to interrogate biological systems using chemical biology. Large-scale HTS studies such as Cancer Cell Line Encyclopedia (CCLE)[18], Genomics of Drug Sensitivity in Cancer (GDSC)[19,20], Cancer Therapeutics Response Portal (CTRP)[21], and NCI-60 Developmental Therapeutic Program Screen[22]represent examples of the HTS pharmacological profiling data sources. One recent study has performed HTS of 180 kinase inhibitors in 12 TNBC cell lines[23]. Typically, HTS was performed on a panel of cancer cell lines screened with multiple compounds to generate pharmacological profiling data. From the pharmacological profiling data, one can correlate the compound sensitivity with other molecular genomics data to derive drug sensitivity signatures[18-21]. Another application of HTS pharmacological data is to correlate with in vitro kinase binding assays to deconvolute kinase dependency in biological systems[24]. However, no efforts have been made to integrate HTS pharmacological profiling data, in vitro kinase binding data, and genomics data for dissecting kinase dependency in cancer cells.
The goal of this study was to determine the kinase dependency in TNBC cell lines and to predict compounds that could inhibit these kinases using integrative bioinformatics analysis. In this study, we used publicly available gene expression data, HTS pharmacological profiling data, and quantitative in vitro kinase binding data. We employed our recently developed Kinase Addiction Ranker (KAR) to integrate these data sourcesto dissect kinase dependency in TNBC cell lines[25]. We then used the kinase dependency predicted by KAR to query K-Map [26,27]for connecting compounds with kinases for individual TNBC lines. For validation, we performed literature search on published experimental data and tested K-Map predictionsin cell lines. Our research strategy for this study is illustrated in Figure 1.
Figure 1.

Integrative bioinformatics research strategyto dissect kinase dependency in TNBC. The Kinase Addiction Ranker (KAR) algorithm integrates gene expression, drug screen, and quantitative kinase-drug binding data to predict kinase dependence in TNBC cell lines. The top ranking kinases by KAR are then queried by K-Map to predict effective compounds. These predictions are then validated experimentally or in literature.
Methods
Pharmacological profiling data
We obtained the HTS pharmacological profiling data of 12 TNBC cell lines from a recently published paper [23]. The 12 TNBC cell lines are: BT20, BT549, CAL148, HCC38, HCC70, HCC1143, HCC1187, HCC1806, Hs578T, MDA-MB231, MDA-MB468 and MFM223. These cell lines were screened with 180 kinase inhibitors. The drug sensitivity read out from this dataset is half maximal effective concentration values (EC50).
Quantitative kinase inhibition data
We obtained comprehensive quantitative kinase inhibition data for 72 of the 180 screened kinase inhibitors from literature. Drug sensitivity data from these 72 drugs were used in our algorithm. This comprehensive inhibition data allows for better interpretation of high-throughput screening results as most kinase inhibitors interact with far more kinases than the ones that are most commonly reported[15].
Microarray gene expression data
We obtained the TNBC microarray gene expression data from the Cancer Cell Line Encyclopedia (GSE36133). These cell lines were profiled using Affymetrix HG-U133 Plus 2.0 microarrays. Raw CEL files for these cell lines were normalized using Robust Multiarray Average (RMA) approach in Affymetrix Power Tools (APT).
Kinase Addiction Ranker (KAR)
We have recently developed KAR (Kinase Addiction Ranker), a novel computational method that integrates high-throughput drug screening data, quantitative kinase binding data, and transcriptomics data to define kinase dependency for individual cancer cell lines [25]. For each cell line, KAR first assigns compounds in the high-throughput drug screen to1 of 5 bins based on drug sensitivity. The bin number determines how many points each kinase target of the drug receives by the scoring algorithm. Targets of compounds in Bin 1 receive 20 points, Bin 2 targets receive 10 points, Bin 3 targets receive 5 points, Bin 4 targets receive 0 points, and Bin 5 targets receive -10 points. Bin 4 and 5 therefore contain drugs that do not meet the threshold for drug sensitivity in the sample, with compounds in Bin 5 receiving a negative penalty.
Next, quantitative kinase binding data is dichotomized as inhibited or not inhibited for each compound based on user-defined threshold (default: a kinase is considered as inhibited by the compound if IC50/Kd< 1 μM or percent of inhibition > 85%). Transcriptomics data is used to filter out low expressed kinases. Kinases are scored by adding or subtracting points based on the sensitivity bin of each drug that inhibits the kinase. Finally, p-values are computed using chi-square and Fisher's exact tests to determine if there is a significant association between a kinase being inhibited and the drug being sensitive (Sensitivity Bins 1-3) in the cell line. KAR returns the ranked list of kinases based on p-values and scaled scores. Kinases with p < 0.05 will be deemed as significant and dependent by the cancer cell line. We have implemented KAR in two programming languages: python and Matlab. KAR is freely available at: http://tanlab.ucdenver.edu/KAR.
Kinase Connectivity Map (K-Map) Analysis
We recently developed and implemented K-Map that systematically connects a kinase profile with a reference kinase inhibitor database and predicts the most effective inhibitor for a queried kinase profile [26,27]. The K-Map consists of three key components: (1) a reference database that contains a set of kinase inhibitors profiles; (2) a query signature; and (3) a pattern matching algorithm or similarity metric defined between a query signature and a reference kinase inhibitor profile to quantify the connection (or similarity) between the interactions of kinases and inhibitors.
Reference Database
The current K-Map reference database was builtbased on two recently published comprehensive analyses of kinase inhibitor selectivity [15,16]. The first study systematically interrogated 178 commercially available inhibitors against a panel of 300 protein kinases using a radiometric phospho-transfer method to assess the percent kinase inhibition (IC50)[15]. The second study measured the selectivity and potency of 72 inhibitors against 442 kinases using direct binding affinities between inhibitors and kinases (Kd)[16]. These datasets were converted into rank-ordered lists according to the inhibitors' potencies against the kinases and used as the K-Map reference profiles for matching query kinases.
Query Signature
For each TNBC line, the top five kinases ranked by KAR were used as the query kinase profile and connected through the K-Map in this study.
Pattern Matching Algorithm
K-Map implementsthe pattern matching strategy based on the Kolmogorov-Smirnov (KS) statistics. The KS-test is a non-parametric, rank-based pattern-matching approach implemented in the connectivity map[28]. The algorithm aims to correlate kinase inhibitors, based on kinase inhibition profiles in the reference database, with a given query (i.e. top five kinases ranked by KAR). For every inhibitor in the reference database, the KS statistic is computed and a “connectivity score” is defined where it ranges from 1 (maximal efficacy) to 0 (minimal efficacy). K-Map then returnsthe ranked list of kinase inhibitors that best inhibit the list of queried kinases sorted by their “connectivity scores”. We used K-Map to connect the top five kinases for 12 TNBC cell lines with drugs in this study. K-Map is freely available at: http://tanlab.ucdenver.edu/kMap.
Cell lines and culture
HCC1806 was obtained from the American Type Culture Collection (ATCC). The cell line has been authenticated as previously described[29,30]. Cell cultures were prepared as previously described[29,30].
Drugs
Erlotinib and bosutinib were obtained commercially (Selleck Chemicals) and prepared according to the manufacture's guidelines.
Cell proliferation assay
To evaluate the drug effects in TNBC cells, we used the CellTiter-Glo assay. In brief, cell viability assayswere performed using CellTiter-Glo (Promega) according to manufacturer's instructions. TNBC cells were seeded at 4000 cells/well in a 96-well plate, and exposed to increasing concentrations of erlotinib or bosutinib from 0 - 10 μmol/L for 96 hours. CellTiter-Glo measurements were obtained for these different concentrations to determine cell viability. Cell viability curves were derived from the data and IC50 values calculated from a minimum of three experiments.
Results and discussion
Kinase Addiction Ranker for ranking kinase dependency in TNBC cell lines
To identify kinase dependency in TNBC, we first analyzed a HTS pharmacological profiling data set of 180 kinase inhibitors profiled across 12 TNBC cell lines[23]. We selected 72 of the 180 profiled drugs based on availability of a published quantitative in vitrokinase inhibition profile and inhibition of at least one kinase above threshold (Kd/EC50<1 μM or >85% inhibition). We used KAR (Kinase Addiction Ranker), a novel bioinformatics approach, which integrates gene expression, drug sensitivity, and kinase inhibition data to generate a ranked list of kinase dependency in these TNBC cell lines. As described in the methods section, KAR integrates three data sources (pharmacological profiling data, kinase inhibition data and gene expression data) to delineate kinase dependency in individual cell lines. On average, KAR identified 24 kinases with a significant association with drug sensitivity in each cell line (range: 9 - 46) (Table 1).
Table 1.
Top 5 kinases ranked by KAR for the 12 TNBC lines.
| TNBC Cell Line | Top 5 Kinases (Score, p-value) ranked by KAR | ||||
|---|---|---|---|---|---|
| (Number of significant kinases, p < 0.05) | Rank 1 | Rank 2 | Rank 3 | Rank 4 | Rank 5 |
| BT20 | EGFR | MAP4K2 | MAP4K3 | MAP3K9 | MARK4 |
| (9) | (160, 0.0095) | (150, 0.0095) | (155, 0.016) | (160, 0.0257) | (90, 0.039) |
| BT549 | MAP4K4 | MINK1 | MAP3K11 | MAP4K5 | NUAK1 |
| (42) | (115, 1.1 × 10-5) | (80, 7.1 × 10-5) | (95, 0.0001) | (120, 0.0002) | (110, 0.0002) |
| CAL148 | MAP4K2 | CDK5 / p25 | CDK2 / cyclin A | IRAK1 | AURKB |
| (14) | (75, 0.0029) | (65, 0.0073) | (60, 0.0073) | (40, 0.0084) | (95, 0.0108) |
| HCC38 | MAP3K9 | MAP4K5 | MARK4 | MAP4K4 | MINK1 |
| (43) | (85, 1.79 × 10-5) | (95, 5.73 × 10-5) | (85, 5.73 × 10-5) | (100, 8.05 × 10-5) | (85, 9,47 × 10-5) |
| HCC70 | PRKD3 | CDK5 / p25 | STK3 | MAP3K9 | AURKA |
| (24) | (90, 0.0015) | (100, 0.0031) | (75, 0.0115) | (65, 0.0127) | (45, 0.0129) |
| HCC1143 | CDK5 / p25 | MST4 | CDK1/cyclin A | CDK3/cyclin E | RAF1 |
| (46) | (45, 0.0005) | (40, 0.0005) | (30, 0.0041) | (30, 0.0041) | (10, 0.0041) |
| HCC1187 | CLK4 | ROCK2 | PIK3CA | GSK3B | MAP3K9 |
| (13) | (60, 0.0182) | (50, 0.0182) | (45, 0.0182) | (85, 0.0197) | (85, 0.0229) |
| HCC1806 | YES1 | TNK2 | EGFR | MAP4K4 | LYN |
| (23) | (195, 0.0004) | (180, 0.0004) | (125, 0.0041) | (170, 0.0058) | (130, 0.0072) |
| HS578T | PDGFRB | MAP4K4 | LYN | MTOR | CLK4 |
| (22) | (60, 0.0005) | (50, 0.0022) | (70, 0.0024) | (60, 0.0024) | (30, 0.0037) |
| MDA-MB-231 | MAP3K7 | CDK1 / cyclin A | IGF1R | CDK5 / p25 | CDK6 / cyclin D1 |
| (32) | (40, 0.0007) | (40, 0.0007) | (25, 0.0007) | (35, 0.0013) | (25, 0.0039) |
| MDA-MB-468 | CDK2 / cyclin A | CDK5 / p25 | MAP3K9 | STK4 | PRKD3 |
| (10) | (95, 0.0047) | (95, 0.0047) | (155, 0.0062) | (155, 0.0062) | (90, 0.0164) |
| MFM-223 | ROCK2 | CLK4 | AURKA | RET | MAP3K9 |
| (13) | (50, 0.0043) | (45, 0.0043) | (35, 0.0043) | (40, 0.0126) | (25, 0.0171) |
The kinases most commonly associated with drug sensitivity in the TNBC cell lines were MAP4K4 and PRKD3, which were each significant in 10/12 TNBC cell lines. MAP4K4 (also known as HGK or NIK) activates MAPK8/JNK signaling. MAP4K4 is involved in cell migration and invasion in melanoma, ovarian, breast and prostate cancers[31]. Moreover, overexpression of MAP4K4 correlated with larger tumor size, increased lymph node involvement, and recurrence in pancreatic ductal carcinoma[32]. PRKD3 has been shown to promote proliferation and chemoresistance in TNBC [33,34]. Since MAP4K4 and PRKD3 are so frequently associated with drug sensitivity in this dataset, they may represent targets that could benefit larger populations of TNBC patients. MAP4K4 and PRKD3 are simultaneously inhibited by CDK1/2 inhibitor III and PKR inhibitor, which were sensitive in nearly all of the 12 cell lines.
Next, we performed hierarchical clustering on the scaled KAR scores to reveal relationships between the 12 TNBC cell lines and kinases (Figure 2). Clustering included the 89 kinases that had a significant association with drug sensitivity in at least one of the 12 cell lines analyzed. No correlation of TNBC subtypes[1] were found in these clusters, this is similar to the previous published pharmacological profiling data. This suggests the heterogeneity of the molecular subtypes of TNBC andthat understanding the kinase dependency could provide better treatment strategy for this disease.
Figure 2.
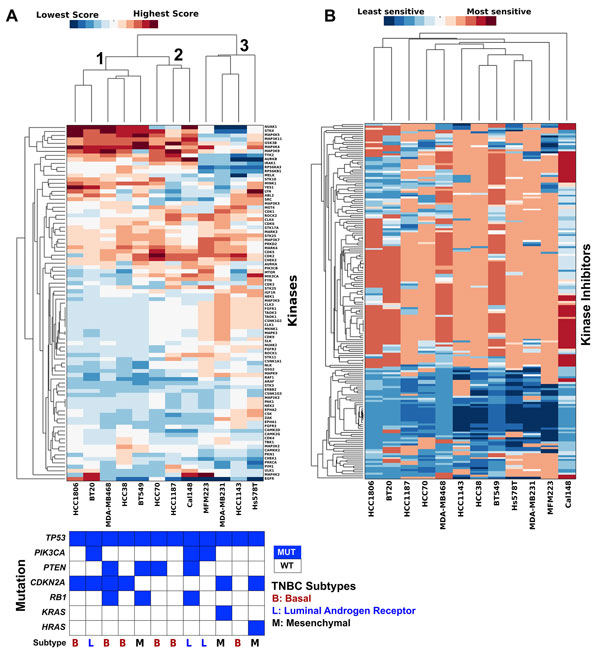
KAR identifies relationships in kinase dependency in TNBC. A. Hierarchical clustering ofTNBC cell lines and kinases based on scaled kinase dependency scores. Each column was normalized to give a mean of 0 and a standard definition of 1. Red indicates that a cell line has a high dependence on a given kinase and blue indicates low dependence. Mutation and subtype information are also provided [1]. B. Hierarchical clustering of TNBC cell lines based on kinase inhibitor sensitivity data (from [23]). Clustering based on kinase inhibitor sensitivity resulted in different groupings compared to kinase dependency score. Red indicates that a cell line has higher sensitivity to a particular kinase inhibitor (lower IC50) and blue indicates lower sensitivity (higher IC50).
From Figure 2, the cluster analysis reveals three main groups of TNBC cell lines (Figure 2A). The first group contains HCC1806, BT20, MDA-MB-468, HCC38 and BT549, the second group contains HCC70, HCC1187 and CAL148 and the third group contains MFM223, MDA-MB-231, HCC1143 and Hs578T. Within the first group, HCC1806 and BT20 show a unique dependence on EGFR when compared to the other three lines (MDA-MB-468, HCC38 and BT549). Interestingly, EGFR also does not cluster with any of the other kinases analyzed, indicating EGFR dependence is a fairly unique marker in a cell line compared to other kinases.
We also clustered the pharmacological profiling data (EC50) from all 180 drugs[23](Figure 2B). As with the kinase score, EGFR-dependent BT20 and HCC1806 grouped together. The other cell lines show less distinct groupings with many cell lines being paired with different cell lines than when clustered based on kinase score. This suggests that the kinase dependency relationships derived from KAR are different from the relationships derived from clustering pharmacological profiling data.
Validating kinase dependency in TNBC cell lines
Here, we validate the kinases with high KAR rankings (Table 1) in a subset of the TNBC cell lines studied based on previously published studies.
BT20
KAR ranks Epidermal Growth Factor Receptor (EGFR), as the top kinase for this cell line. Indeed, previously published papers have verified that this cell line expressed high levels of EGFR[35,36], however, this cell line is not sensitive to EGFR inhibitors such as erlotinib or gefitinib[35-37]. This indicates that there may be some other kinases driving the proliferation of this cell line.
HCC1806
For this cell line, KAR ranks YES1 and LYN in the top 5 kinases. Both of these kinases are SRC kinase family members. Indeed, previous studies have demonstrated that this cell line is highly sensitive to dasatinib (FDA approved SRC inhibitor)[38]. Interestingly, KAR also ranks EGFR as one of the kinase dependent in this cell line. From the clustering of kinase dependency score (Figure 2), BT20 and HCC1806 clustered together.
HCC70
KAR ranks Aurora Kinase A (AURKA) as one of the top five kinases in this cell line. Previously, we have tested two different Aurora Kinase inhibitors across a large panel of TNBC cell lines, and found that HCC70 is very sensitive to MLN8237 (IC50 = 0.1 μM)[30] and ENMD2076 (IC50 = 0.549 μM)[29]. This supports AURKA dependence in this cell line. In fact, there is an ongoing Phase II clinical trial of treating TNBC patients with ENMD2076 (http://ClinicalTrials.gov ID: NCT01639248).
MDA-MB-231
KAR ranks MAP3K7, which is commonly known as the Transforming growth factor beta-activated kinase 1 (TAK1), as the top kinase for this cell line. Interestingly, this cell line is the only TNBC cell line analyzed that is a KRAS mutant (p.G13D) and is highly dependent on KRAS ("KRAS-dependent" cell line)[39]. Indeed, previous studies have demonstrated that this cell line has high MAP3K7 expression, and is sensitive to the TAK1 kinase inhibitor 5Z-7-oxozeaenol [40]. Previous studies in colorectal cancer cell lines have suggested that TAK1 could be a therapeutic target in KRAS-dependent lines [41]. This confirms that KAR could identify relevant kinases for individual cell lines.
KAR results indicate that the other TNBC cell lines seem highly dependent on MAPKs (e.g. MAP4K2, MAP4K3) and CDK kinases (e.g. CDK1, CDK2, CDK3, CDK5, CDK6). A previous study evaluating kinase expression in Estrogen Receptor (ER) positive vs. negative breast cancer samples identified a subgroup in the ER-negative samples also enriched with MAPKs[42].
Predicting compounds for individual TNBC cell lines by using K-Map
For each TNBC cell lines, we used the top five ranking kinases (lowest chi-square p-values) as the query to K-Map for predicting effective compounds. Compounds with p < 0.05 are selected and sorted by connectivity scores. Table 2 lists the top five compounds predicted by K-map based on the top five kinases for each TNBC cell lines.
Table 2.
To 5 kinase inhibitors predicted by K-Map for the 12 TNBC lines.
| Top 5 Drugs (Score) ranked by K-Map | |||||
|---|---|---|---|---|---|
| TNBC Cell Line | Rank 1 | Rank 2 | Rank 3 | Rank 4 | Rank 5 |
| BT20 | Staurosporine | Bosutinib | Go 6976 | TWS119 | PKR Inhibitor |
| (1.00) | (0.976) | (0.972) | (0.963) | (0.953) | |
| BT549 | Staurosporine | K-252a | SB 218078 | CDK1/2 Inhibitor III | Go 6976 |
| (1.00) | (1.00) | (0.997) | (0.995) | (0.995) | |
| CAL148 | Lestaurtinib | K-252a | Staurosporine | JAK3 Inhibitor VI | CDK1/2 Inhibitor III |
| (1.00) | (1.00) | (1.00) | (0.996) | (0.994) | |
| HCC38 | Staurosporine | K-252a | CDK1/2 Inhibitor III | SB 218078 | Go 6976 |
| (1.00) | (1.00) | (0.995) | (0.991) | (0.989) | |
| HCC70 | Lestaurtinib | Staurosporine | CDK1/2 Inhibitor III | K-252a | SB 218078 |
| (1.00) | (1.00) | (0.998) | (0.998) | (0.983) | |
| HCC1143 | JNK Inhibitor II | CDK4 Inhibitor III | CDK4 Inhibitor II | VEGFR Receptor Inhibitor II | CHK2 Inhibitor II |
| (0.977) | (0.943) | (0.938) | (0.936) | (0.929) | |
| HCC1187 | Staurosporine | CDK1/2 Inhibitor III | JAK3 Inhibitor VI | JNJ-7706621 | PKR Inhibitor |
| (1.00) | (0.998) | (0.990) | (0.973) | (0.971) | |
| HCC1806 | Bosutinib | TWS119 | Staurosporine | Dasatinib | WHI-P154 |
| (1.00) | (0.990) | (0.98) | (0.966) | (0.956) | |
| HS578T | Staurosporine | SU11652 | K-252a | Sunitinib | Dorsomorphin |
| (1.00) | (0.992) | (0.986) | (0.984) | (0.982) | |
| MDA-MB-231 | CDK1/2 Inhibitor III | Indirubin Derivative E804 | Sunitinib | Aminopurvalanol A | CDK2 Inhibitor IV |
| (1.00) | (0.975) | (0.956) | (0.949) | (0.94) | |
| MDA-MB-468 | Lestaurtinib | Staurosporine | CDK1/2 Inhibitor III | K-252a | JAK3 Inhibitor VI |
| (1.00) | (1.00) | (0.998) | (0.998) | (0.99) | |
| MFM-223 | Staurosporine | CDK1/2 Inhibitor III | K-252a | JAK3 Inhibitor VI | SYK Inhibitor |
| (1.00) | (1.00) | (0.99) | (0.974) | (0.972) | |
Staurosporine, a multi-kinase inhibitor used as the positive control in the K-Map, was predicted as an effective compound for ten TNBC cell lines (Table 2). This is likely because a highly non-specific compound like Staurosporine can inhibit > 400 kinases by itself.
Validating compounds predicted by K-Map in TNBC cell lines
From Table 2 K-Map predicts bosutinib as one of the compounds that targets the top five ranking kinases of BT20 and HCC1806. Both cell lines have EGFR dependency as determined by KAR, and one of the targets of bosutinib is EGFR. In addition to EGFR, bosutinib also inhibits top ranking kinases YES1, TNK2, MAP4K4, and LYN (Table 1). Therefore, bosutinib inhibits each top ranking kinase for HCC1806 while erlotinib only inhibits one of the top five (EGFR). We would therefore predict that HCC1806 would be more sensitive to bosutinib than erlotinib. To validate this prediction, we tested HCC1806 in vitro with bosutinib and erlotinib using a CellTiter-Glo assay. As depicted in Figure 3, the IC50 of bosutinib(3 μM) is lower than erlotinib (>10 μM) in HCC1806. This validates the K-Map prediction that this cell line is more sensitive to bosutinibthan erlotinib. We also validated BT20 with bosutinib and erlotinib, and found that bosutinib also exhibited lower IC50 than erlotinib. (data not shown).
Figure 3.
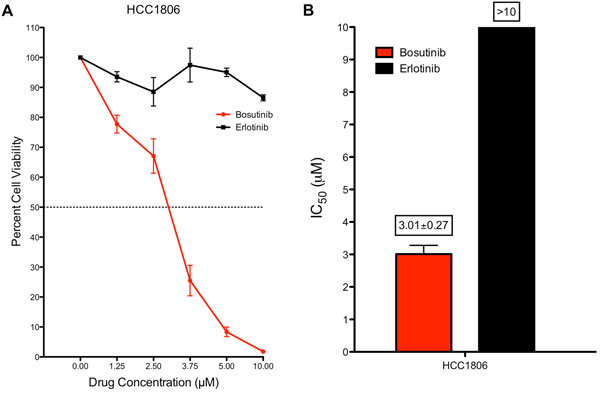
Validation of bosutinib and erlotinib in TNBC cell line HCC1806. A. Cell viability (mean +/- SE) dose response for bosutinib and erlotinib in HCC1806. B. Estimation of the IC50 for bosutinib and erlotinib in HCC1806. HCC1806 was much more sensitive to bosutinib than erlotinib. While both compounds target EGFR (KAR rank = 3), bosutinib targets other top ranking kinases by KAR: YES1, TNK2, MAP4K4, and LYN.
K-Map predicts SU11652 and sunitinib as potential compounds to be effective against Hs578T. Both compounds are PDGFR inhibitors, where PDGFRB is the top dependent kinase predicted by KAR for this cell line. Indeed, previous studies have demonstrated that Hs578T has high expression of PDGFRB (both at mRNA and protein levels), and this cell line is more sensitive to sunitinib[43].
CDK 1/2 inhibitor was predicted by K-Map as one of the compounds that inhibits the top five kinases in nine TNBC cell lines (BT549, CAL148, HCC38, HCC70, HCC1143, HCC1187, MDA-MB-231, MDA-MB-468 and MFM-223). Recent studies have suggested that MYC-dependence is synthetic lethal with CDK inhibitor in TNBC cell lines[44]. Indeed, six of these cell lines (BT549, HCC38, HCC70, HCC1143, MDA-MB-231 and MDA-MB-468) were MYC-dependent [45]. This supports that the K-Map prediction of CDK1/2 inhibitor could be a potential therapeutic for these TNBC cell lines.
Similar to Fink et al.'s analysis of the pharmacological profiling data [23], we observed heterogeneity of kinase dependence among the 12 TNBC cell lines and no co-clustering of cell lines of the same molecular subtype. We also showed EGFR dependence for BT-20 and HCC1806, but our experiments showed much lower sensitivity to Erlotinibthan Fink et al. (IC50 0.2 μM, our experiments: >10 μM). Fink et al.'s clustering of the drug sensitivity datarevealed co-clustering of HCC70, BT549, and MDA-MD468, and reported increased sensitivity of this group to PI3K pathway inhibitors[23]. Here, KAR revealed significant association between PIK3CB inhibition and drug sensitivity in HCC70 and BT549, but much higher correlations with drug sensitivity for other kinases (Table 1). Fink et al. also report that another group of cell lines which co-cluster (HCC38, HCC1143, HCC1187, HS578T, MDA-MB231, and MFM-223), are generally resistant to kinase inhibition with no kinase target being selectively toxic to this group [23]. Our approach incorporating more comprehensive target lists for each drug, however, was able to find kinases with significant associations with drug sensitivity for each cell line in this group. Moreover, MAPK4K4, which was one of the kinases most commonly associated with drug sensitivity in the 12 TNBC cell lines, is significant in all but HCC1187in this group of cell lines.
Here, we presented examples of how the KAR algorithm and K-Map research tool can be integrated to determine kinase dependency and predict effective cancer drugs for TNBC. KAR aids greatly in preventing misinterpretation of HTS data, as kinase inhibitors typically inhibit many more targets than are commonly reported. KAR therefore helps uncover kinase dependency that may be overlooked if only focusing on the commonly reported drug targets. Moreover, incorporation of gene expression data can help ensure that high-ranking kinases will be translationally applicable. Compared to previous approaches[24,46-48], KAR benefits from producing scores and p-values that can be easily interpreted by biologists without computational backgrounds, incorporation of transcriptomics data, increased accessibility (MATLAB and python functions available at http://tanlab.ucdenver.edu/KAR), and does not require preliminary optimization of the drug screening list. K-Map allows for quick connection of essential kinases revealed by KAR to compounds for experimental testing. K-Map can help reveal drugs that may not have been part of the original screening set. While we used this approach with TNBC cell lines, a similar strategy can be used with patient samples to predict effective kinase inhibitor therapies and drug combinations.
Conclusions
We presented an integrative bioinformatics analysis to determine kinase dependency in TNBC. We integrated three different high-throughput data sources with the KAR algorithm: HTS pharmacological profiling data, quantitative in vitro kinase binding data, and gene expression data. We then queried the top five kinases from each TNBC cell lines to K-Map to predict compounds that could inhibit these sets of kinases. We validated the KAR and K-Map predictions using experiments and published studies. Using the integrative bioinformatics analysis, we have discovered kinase dependency in these TNBC cell lines. The data provide candidate kinases and drugs for further pharmacological and biological studies.
List of abbreviations used
AURKA: Aurora Kinase A; EGFR: Epidermal Growth Factor Receptor; HTS: High-throughput Screening; KAR: Kinase Addiction Ranker; NSCLC: Non-small cell lung cancer; TNBC: Triple Negative Breast Cancer.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
KAR and ACT conceived the study, coordinated the experiments, performed acquisition of data, participated in data analysis, and drafted the manuscript. LEH and JK contributed to the study design. KAR, JK, JS, MY and ACT conducted bioinformatics analysis. PK, AI, TMP, JTT, JRD and SGE conducted experimental validation. All authors read and approved the final manuscript.
Contributor Information
Karen A Ryall, Email: karen.ryall@ucdenver.edu.
Jihye Kim, Email: jihye.kim@ucdenver.edu.
Peter J Klauck, Email: peter.klauck@ucdenver.edu.
Jimin Shin, Email: jimin.shin@ucdenver.edu.
Minjae Yoo, Email: minjae.yoo@ucdenver.edu.
Anastasia Ionkina, Email: anastasia.ionkina@ucdenver.edu.
Todd M Pitts, Email: todd.pitts@ucdenver.edu.
John J Tentler, Email: john.tentler@ucdenver.edu.
Jennifer R Diamond, Email: jennifer.r.diamond@ucdenver.edu.
S Gail Eckhardt, Email: gail.eckhardt@ucdenver.edu.
Lynn E Heasley, Email: lynn.heasley@ucdenver.edu.
Jaewoo Kang, Email: kangj@korea.ac.kr.
Aik Choon Tan, Email: aikchoon.tan@ucdenver.edu.
Acknowledgements
We thank the PETT lab members for constructive comments on the manuscript.
This work is partly supported by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award T32CA17468 (K.A.R.), the National Institutes of Health P30CA046934, Cancer League of Colorado, the David F. and Margaret T. Grohne Family Foundation. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the funders.
Declaration
Publication charges for this article have been funded bythe David F. and Margaret T. Grohne Family Foundation.
This article has been published as part of BMC Genomics Volume 16 Supplement 12, 2015: Joint 26th Genome Informatics Workshop and 14th International Conference on Bioinformatics: Genomics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/16/S12.
References
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK. et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2013;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER, Wilson RK, Ally A, Balasundaram M, Butterfield YSN, Carlsen R, Carter C, Chu A, Chuah E, Chun H-JE, Coope RJN, Dhalla N, Guin R, Hirst C, Hirst M, Holt RA, Lee D, Li HI, Mayo M, Moore RA, Mungall AJ. et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature Reviews Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–297. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, Cornelissen JJ, Fischer T, Hochhaus A, Hughes T, Lechner K, Nielsen JL, Rousselot P, Reiffers J, Saglio G, Shepherd J, Simonsson B, Gratwohl A, Goldman JM, Kantarjian H, Taylor K, Verhoef G, Bolton AE, Capdeville R, Druker BJ. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak EL, Bang Y-J, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou S-HI, Dezube BJ, Jänne PA, Costa DB, Varella-Garcia M, Kim W-H, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber DA, Wilner K, Salgia R, Shapiro GI, Clark JW. et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetri GD, Mehren von M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Bin Peng, Dimitrijevic S, Druker BJ, Corless C, Fletcher CDM, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- Shaw AT, Ou S-HI, Bang Y-J, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, Doebele RC, Le LP, Zheng Z, Tan W, Stephenson P, Shreeve SM, Tye LM, Christensen JG, Wilner KD, Clark JW, Iafrate AJ. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–1971. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nature Biotechnology. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Pietro Ciceri, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nature Biotechnology. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Yoo M, Shin J, Kim J, Ryall KA, Lee K, Lee S, Kang J, Tan AC. DSigDB: drug signatures database for gene set analysis. Bioinformatics. 2015;31:3069–3071. doi: 10.1093/bioinformatics/btv313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, de Silva M. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR, Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F. et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, Ramaswamy S, Futreal PA, Haber DA, Stratton MR, Benes C, McDermott U, Garnett MJ. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Research. 2012;41:D955–D961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, Bracha AL, Liefeld T, Wawer M, Gilbert JC, Wilson AJ, Stransky N, Kryukov GV, Dancik V, Barretina J, Garraway LA, Hon CS-Y, Munoz B, Bittker JA, Stockwell BR, Khabele D, Stern AM, Clemons PA, Shamji AF, Schreiber SL. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–1161. doi: 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nature Reviews Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Fink LS, Beatty A, Devarajan K, Peri S, Peterson JR. Pharmacological profiling of kinase dependency in cell lines across triple-negative breast cancer subtypes. Molecular Cancer Therapeutics. 2015;14:298–306. doi: 10.1158/1535-7163.MCT-14-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujral TS, Peshkin L, Kirschner MW. Exploiting polypharmacology for drug target deconvolution. Proc Natl Acad Sci USA. 2014;111:5048–5053. doi: 10.1073/pnas.1403080111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall KA, Shin J, Yoo M, Hinz TK, Kim J, Kang J, Heasley LE, Tan AC. Identifying kinase dependency in cancer cells by integrating high-throughput drug screening and kinase inhibition data. Bioinformatics. 2015;31:3799–3806. doi: 10.1093/bioinformatics/btv427. doi:10.1093/bioinformatics/btv427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Yoo M, Kang J, Tan AC. K-Map: connecting kinases with therapeutics for drug repurposing and development. Human Genomics. 2013;7:20. doi: 10.1186/1479-7364-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Vasu VT, Mishra R, Singleton KR, Yoo M, Leach SM, Farias-Hesson E, Mason RJ, Kang J, Ramamoorthy P, Kern JA, Heasley LE, Finigan JH, Tan AC. Bioinformatics-driven discovery of rational combination for over- coming EGFR-mutant lung cancer resistance to EGFR therapy. Bioinformatics. 2014;30:2393–2398. doi: 10.1093/bioinformatics/btu323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J-P, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Diamond JR, Eckhardt SG, Tan AC, Newton TP, Selby HM, Brunkow KL, Kachaeva MI, Varella-Garcia M, Pitts TM, Bray MR, Fletcher GC, Tentler JJ. Predictive biomarkers of sensitivity to the aurora and angiogenic kinase inhibitor ENMD-2076 in preclinical breast cancer models. Clinical Cancer Research. 2013;19:291–303. doi: 10.1158/1078-0432.CCR-12-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentler JJ, Ionkina A, Tan AC, Newton TP, Pitts TM, Glogowska MJ, Kabos P, Sartorius CA, Sullivan KD, Espinosa JM, Eckhardt SG, Diamond JR. p53 Family Members Regulate Phenotypic Response to Aurora Kinase A Inhibition in Triple-Negative Breast Cancer. Molecular Cancer Therapeutics. 2015;14:1117–1129. doi: 10.1158/1535-7163.MCT-14-0538-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CS, Hong J, Sapinoso L, Zhou Y, Liu Z, Micklash K, Schultz PG, Hampton GM. A small interfering RNA screen for modulators of tumor cell motility identifies MAP4K4 as a promigratory kinase. Proc Natl Acad Sci USA. 2006;103:3775–3780. doi: 10.1073/pnas.0600040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JJ, Wang H, Rashid A, Tan T-H, Hwang RF, Hamilton SR, Abbruzzese JL, Evans DB, Wang H. Expression of MAP4K4 is associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Clinical Cancer Research. 2008;14:7043–7049. doi: 10.1158/1078-0432.CCR-08-0381. [DOI] [PubMed] [Google Scholar]
- Borges S, Storz P. Protein kinase D isoforms: new targets for therapy in invasive breast cancers? Expert Review of Anticancer Therapy. 2013;13:895–898. doi: 10.1586/14737140.2013.816460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huck B, Duss S, Hausser A, Olayioye MA. Elevated protein kinase D3 (PKD3) expression supports proliferation of triple-negative breast cancer cells and contributes to mTORC1-S6K1 pathway activation. J Biol Chem. 2014;289:3138–3147. doi: 10.1074/jbc.M113.502633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkery B, Crown J, Clynes M, O'Donovan N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann Oncol. 2009;20:862–867. doi: 10.1093/annonc/mdn710. [DOI] [PubMed] [Google Scholar]
- Yamasaki F, Zhang D, Bartholomeusz C, Sudo T, Hortobagyi GN, Kurisu K, Ueno NT. Sensitivity of breast cancer cells to erlotinib depends on cyclin-dependent kinase 2 activity. Molecular Cancer Therapeutics. 2007;6:2168–2177. doi: 10.1158/1535-7163.MCT-06-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Sequential Application of Anticancer Drugs Enhances Cell Death by Rewiring Apoptotic Signaling Networks. Cell. 2012;149:780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Dering J, Ginther C, Wilson CA, Glaspy P, Tchekmedyian N, Slamon DJ. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/"triple-negative" breast cancer cell lines growing in vitro. Breast Cancer Res Treat. 2007;105:319–326. doi: 10.1007/s10549-006-9463-x. [DOI] [PubMed] [Google Scholar]
- Scholl C, Fröhling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S, Döhner K, Bullinger L, Sandy P, Boehm JS, Root DE, Jacks T, Hahn WC, Gilliland DG. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- Huang H-L, Chiang C-H, Hung W-C, Hou M-F. Targeting of TGF-β-activated protein kinase 1 inhibits chemokine (C-C motif) receptor 7 expression, tumor growth and metastasis in breast cancer. Oncotarget. 2015;6:995–1007. doi: 10.18632/oncotarget.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, Haber DA, Settleman J. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 2012;148:639–650. doi: 10.1016/j.cell.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers C, Tsimelzon A, Sexton K, Herrick AM, Gutierrez C, Culhane A, Quackenbush J, Hilsenbeck S, Chang J, Brown P. Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clinical Cancer Research. 2009;15:6327–6340. doi: 10.1158/1078-0432.CCR-09-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, Sphyris N, Shariati M, Kumar SV, Battula VL, Herschkowitz JI, Guerra R, Chang JT, Miura N, Rosen JM, Mani SA. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Research. 2013;73:1981–1992. doi: 10.1158/0008-5472.CAN-12-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, Creasman KJ, Bazarov AV, Smyth JW, Davis SE, Yaswen P, Mills GB, Esserman LJ, Goga A. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med. 2012;209:679–696. doi: 10.1084/jem.20111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Sergio CM, Sutherland RL, Musgrove EA. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC Cancer. 2014;14:32. doi: 10.1186/1471-2407-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlow N, Davis LE, Cantor EL, Séguin B, Keller C, Pal R. A new approach for prediction of tumor sensitivity to targeted drugs based on functional data. BMC Bioinformatics. 2013;14:239. doi: 10.1186/1471-2105-14-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TP, Ong E, Hodges AP, Paternostro G, Piermarocchi C. Prediction of kinase inhibitor response using activity profiling, in vitro screening, and elastic net regression. 2014. p. 74. [DOI] [PMC free article] [PubMed]
- Tyner JW, Yang WF, Bankhead A, Fan G, Fletcher LB, Bryant J, Glover JM, Chang BH, Spurgeon SE, Fleming WH, Kovacsovics T, Gotlib JR, Oh ST, Deininger MW, Zwaan CM, Boer Den ML, van den Heuvel-Eibrink MM, O'Hare T, Druker BJ, Loriaux MM. Kinase Pathway Dependence in Primary Human Leukemias Determined by Rapid Inhibitor Screening. Cancer Research. 2013;73:285–296. doi: 10.1158/0008-5472.CAN-12-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]