

Abstract
Lynch syndrome is a hereditary cancer syndrome caused by a constitutional mutation in one of the mismatch repair genes. The implementation of predictive testing and targeted preventative surveillance is hindered by the frequent finding of sequence variants of uncertain significance in these genes. We aimed to determine the pathogenicity of previously reported variants (c.-28A>G and c.-7C>T) within the MLH1 5′untranslated region (UTR) in two individuals from unrelated suspected Lynch syndrome families. We investigated whether these variants were associated with other pathogenic alterations using targeted high-throughput sequencing of the MLH1 locus. We also determined their relationship to gene expression and epigenetic alterations at the promoter. Sequencing revealed that the c.-28A>G and c.-7C>T variants were the only potentially pathogenic alterations within the MLH1 gene. In both individuals, the levels of transcription from the variant allele were reduced to 50% compared with the wild-type allele. Partial loss of expression occurred in the absence of constitutional epigenetic alterations within the MLH1 promoter. We propose that these variants may be pathogenic due to constitutional partial loss of MLH1 expression, and that this may be associated with intermediate penetrance of a Lynch syndrome phenotype. Our findings provide further evidence of the potential importance of noncoding variants in the MLH1 5′UTR in the pathogenesis of Lynch syndrome.
Keywords: Lynch syndrome, MLH1, colorectal cancer, promoter variants
Introduction
Lynch syndrome (MIM #120435) predisposes to the development of colorectal, endometrial, and other cancers [Lynch and de la Chapelle, 2003]. It is most commonly caused by constitutional heterozygous loss-of-function mutations in the DNA mismatch repair (MMR) genes, usually MLH1 (MIM #120436) or MSH2 (MIM #609309) [Lynch et al., 2009]. The reported mutations in Lynch syndrome families are genetically heterogeneous and include gross structural alterations such as deletions or inversions, [Wagner et al., 2002] missense, nonsense or frameshift mutations, [Tavtigian et al., 2008] splice site mutations, [Thompson et al., 2014] or variants within MMR gene regulatory regions such as promoter regions [Green et al., 2003]. In many cases, the functional and hence clinical significance of the sequence alterations, in particular single-nucleotide variants (SNVs) outside coding regions, is uncertain. For these families, predictive testing and targeted preventative surveillance of family members cannot be offered. A recent study reported the classification of 2,360 constitutional MMR gene variants into class 1 (not pathogenic), class 2 (likely not pathogenic), class 3 (uncertain), class 4 (likely pathogenic), or class 5 (pathogenic) [Thompson et al., 2014]. The finding that a large proportion (32%) of variants belonged to class 3 demonstrates the importance of collecting further evidence about these variants.
Several class 3 variants within the MLH1 promoter have been found in cases of suspected Lynch syndrome, namely, c.-411_-413del, c.-432_-435del, c.-64G>T, c.-53G>T, c.-42C>T, c.-28A>G, c.-28A>T, c.-27C>A, c.-11C>T, and c.-7C>T [Green et al., 2003; Hitchins et al., 2011; Ward et al., 2013; Kwok et al., 2014; Thompson et al., 2014]. However, for most of these variants, it is unclear whether they abrogate MLH1 function directly or whether they are linked to another genetic defect within MLH1. Studies of the c.-27C>A variant provide the most compelling evidence that MLH1 promoter variants can directly affect the regulation of MLH1. This variant segregates with colorectal cancer in multiple cancer-affected families [Raevaara et al., 2005; Hitchins et al., 2011; Ward et al., 2013; Kwok et al., 2014] and has been associated with reduced transcriptional activity and the dominant inheritance of a mosaic constitutional MLH1 epimutation [Hitchins et al., 2011]. These studies suggest that MLH1 promoter variants may cause unbalanced constitutional expression of MLH1. In support of this, the c.-411_-413del, c.-42C>T, c.-27C>A, and c.-11C>T variants also significantly reduce the activity of the MLH1 promoter in driving the expression of a reporter gene [Green et al., 2003; Hitchins et al., 2011; Ward et al., 2013]. To date, however, no analysis of the potential pathogenicity of the c.-28A>G or c.-7C>T variants has been undertaken, despite the fact that these variants have been described in at least two unrelated individuals with suspected Lynch syndrome [Lee et al., 2005; Muller-Koch et al., 2001].
In this study, we aimed to determine the pathogenicity of the c.-28A>G and c.-7C>T variants within the MLH1 5′ untranslated region (UTR) in two individuals from suspected Lynch syndrome families. We investigated whether these variants were linked to other constitutional pathogenic alterations within or around the MLH1 gene and whether they were associated with epigenetic alterations at the gene promoter or changes in gene expression.
Materials and Methods
Patients and Tissue Samples
Two probands (Proband 32 and Proband N; Fig. 1) with the c.-28A>G and c.-7C>T variants (all nucleotide numbering used throughout the manuscript is based on cDNA sequence) were enrolled in this study in Australia (HREC/09/SVH/63 and HREC 14/169) and Norway (HREC 2014/493/REK midt). The c.-28A>G and c.-7C>T variants studied in this manuscript have been submitted to the LOVD database at http://www.lovd.nl/MLH1. At the age of 38 years, Proband 32 had microsatellite-unstable colorectal cancer with wild-type BRAF (MIM #164757) and loss of MLH1 and PMS2 expression (as determined by immunohistochemistry). Proband N was a 54-year-old woman who sought advice due to a family history of colorectal neoplasia (Fig. 1). Multiplex ligation-dependent PCR amplification (MLPA) was performed using a commercially available kit (MRC Holland, Amsterdam, The Netherlands). No copy-number alterations were identified in any exons of the MLH1, PMS2 (MIM #600259), MSH2, and MSH6 (MIM #600678) genes in either proband. Sequence alterations across the MLH1 and PMS2 genes were further assessed using long-range PCR and Sanger sequencing. No sequence alterations were detected in the MLH1 or PMS2 genes except for the c.-28A>G and c.-7C>T heterozygous variants in the MLH1 5′UTR. The following samples were available from Proband 32; fresh frozen peripheral blood mononuclear cells (PBMCs), DNA from PBMCs, buccal, saliva, normal colon, and formalin-fixed paraffin-embedded tumor tissue, and RNA from PBMCs. DNA from saliva was also available from a first-degree relative of Proband 32. The following samples were available from Proband N: DNA from PBMCs and buccal, and RNA from PBMCs. DNA from paraffin tissue was available from a colonic adenoma resected from the father of Proband N and used to obtain the MLH1 haplotype of this individual. RNA was available from the PBMCs of 43 deidentified samples from healthy individuals who had donated to an institutional biobank (HREC 11/160 and HC12060). These individuals were <50 years of age (20 male) with no history of diabetes or cancer. Fresh frozen PBMCs were available from three of these healthy donors. The colorectal cancer cell line SW620 was obtained from the American Type Culture Collection (ATCC) after cell authentication testing using microsatellite and mutation analysis. Cells were maintained in Dulbecco’s modification of Eagle’s medium supplemented with 25 mM glucose, 10% (v/v) fetal bovine serum, 100 units penicillin, 100 μg/ml streptomycin and 2 mM glutamate (Life Technologies, Carlsbad, CA) and grown at 37°C in 5% CO2.
Figure 1.
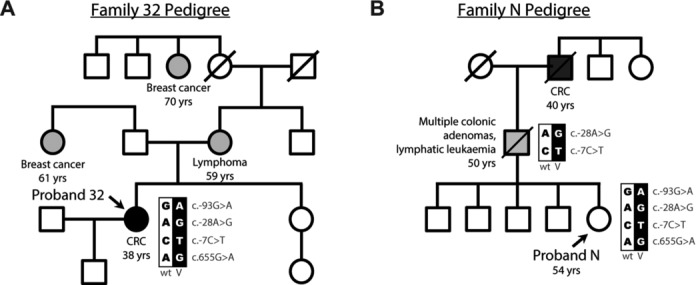
Identification of two probands with the MLH1 c.-28A>G and c.-7C>T variants from two unrelated cancer-affected families. A and B: The pedigrees of Family 32 and Family N, respectively. Shown are the genotypes with respect to the c.-93, c.-28, c.-7, and c.655 sites, the age of cancer onset and other family members affected by cancer. Family N showed cosegregation of the c.-28A>G and c.-7C>T variants across two generations. See also Supp. Figure S1. Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence.
DNA Methylation Analysis
Bisulfite pyrosequencing of five CpG sites immediately upstream of the MLH1 transcription initiation site was performed as described previously [Goel et al., 2011]. Samples were analyzed in quadruplicate. Single molecule bisulfite sequencing was performed as described previously [Hesson et al., 2013]. All primers used in this study are listed in Supp. Table S1.
Targeted DNA Enrichment and High-Throughput Sequencing
We used the HaloplexTM targeted enrichment system (Agilent Technologies, Santa Clara, CA) to sequence a 1.7 Mb region encompassing 15 genes (chr3: 36422675-38128073) across the MLH1 locus. Briefly, 200 ng of genomic DNA was digested with 16 different restriction enzymes at 37°C for 30 min. Digested DNA was then hybridized with specific biotinylated nucleotide adapters using an initial denaturing step of 95°C for 10 min followed by hybridization at 54°C for 16 hr. DNA-adaptor hybrids were captured using streptavidin beads and circularized using DNA ligase. Herculase II Fusing DNA polymerase was used to amplify the captured target library using the following conditions: denaturing at 98°C for 2 min followed by 25 cycles of 98°C for 30 sec, 60°C for 30 sec, 72°C for 1 min, and a final extension of 10 min at 72°C. The amplified target library was purified using AMPure XP Kit (Beckman Coulter Genomics, Danvers, MA) and sequenced on a HiSeq 1500 using paired-end 2 × 100 bp TruSeq chemistry. A total of 20,709,684 sequence reads were obtained. Alignment of reads was performed using Bowtie2 [Langmead and Salzberg, 2012] against the human reference genome (hg19) using default parameters. This provided an average depth of 140, with >10x coverage across 89% of the target region. Coverage across the MLH1 gene was >10x across 91.8% and 82.4% of exonic and intronic DNA, respectively. Variant calling was performed using GATK [McKenna et al., 2010] and Pindel [Ye et al., 2009]. Variants identified by Pindel that had variable allele ratios >0.2 and <0.8 were considered as heterozygous. Only variants predicted by reads with phred quality scores >30, which have a 1 in 1,000 chance that the base is incorrectly called [Ewing and Green, 1998], were considered as variants. Novel variants were defined as those not present in the 1000 genomes or SNP137 databases.
Zygosity Assessment
Quantitative DNA fragment analysis with or without prior restriction enzyme digest or pyrosequencing were used to identify genotype, as indicated (Supp. Table S2). Heterozygosity at the c.1164del1 site within the VILL gene was assessed using pyrosequencing (primers labeled as “VILL [exon 10]”) in Supp. Table S1.
5′ Rapid Amplification of cDNA Ends
Transcription initiation sites were identified using a 5’/3’ rapid amplification of cDNA ends (RACE) Kit (2nd generation; Roche, Basel, Canton of Basel-Stadt, Switzerland) as described previously [Hesson et al., 2009]. One microgram of total RNA was treated with DNaseI (Fermentas, Waltham, MA) and cDNA synthesized using an antisense primer specific to MLH1 exon 11 (Supp. Table S1). Nested PCR was performed using a second antisense primer specific to MLH1 exon 10 in combination with a polyT-anchor primer. Duplicate samples in which reverse transcriptase was omitted were assayed to control for DNA contamination. Transcripts were characterized by gel extraction of PCR products and single molecule sequencing. Sequences were aligned against the human genome (hg19) and allele specificity was determined using the c.655G>A site.
MLH1 Expression
RNA was extracted using AllPrep DNA/RNA/miRNA Universal Kit (Qiagen, Venlo Limburg, The Netherlands) as per the manufacturer’s instructions. RNA quality was determined by Agilent 2100 electrophoresis bioanalyzer. cDNA was synthesized from 250 ng of high-quality RNA (RNA integrity number [RIN] >8) using the QuantiTect Reverse Transcription Kit (Qiagen). We excluded RNA samples with a RIN <8.0 and generated all cDNA libraries simultaneously in a single batch. This approach eliminated variables such as RNA quality and differences in the efficiency of cDNA synthesis associated with batch effects. Allelic representation (i.e., the proportion of MLH1 transcripts that originate from either allele) was determined using PCR amplification of cDNA using primers complimentary to exons 1a and 9 followed by pyrosequencing across the c.655G>A site [Kwok et al., 2010].
Nucleosome Occupancy and Methylome Sequencing
We designed a nucleosome occupancy and methylome sequencing (NOMe-Seq) assay encompassing the transcription initiation sites identified by 5′RACE, as well as the annotated initiation sites of the EPM2AIP1 [NM_041805.3] and MLH1 [NM_000249.3] genes. This 393 bp product was located c.-335 to c.58 relative to MLH1 allowing the detection of multiple nucleosomes across this promoter. NOMe-Seq was performed as described previously [Kelly et al., 2010; Taberlay et al., 2011; You et al., 2011]. This involved harvesting intact nuclei and treating with 200 U GpC methyltransferase M.CviPl (New England Biolabs, Ipswich, MA) for 15 min at 37°C followed by termination of the reaction with an equal volume of 20 mM Tris HCl pH 7.9, 600 mM NaCl, 1% (w/v) SDS, and 10 mM EDTA and overnight digestion with 200 μg/ml Proteinase K (Ambion, Austin, TX). DNA was subsequently isolated and bisulfite converted using EZ DNA Methylation-Gold kit (Zymo Research, Irvine, CA). PCR amplicons were cloned using the TOPO TA Cloning kit (Invitrogen, Carlsbad, CA) and individual molecules isolated by colony PCR for sequencing as described previously [Hesson and Ward, 2014]. M.CviPI enzyme methylates accessible DNA at GpC sites, whereas nucleosome-bound DNA is inaccessible and remains refractory to GpC methylation. The promoter of the HSPA5 (MIM #138120) gene, known to be nucleosome free and accessible, was used as a control for GpC methyltransferase M.CviPl in each sample examined. GpCpG sites were excluded from analysis. Nucleosome occupancy was defined as a region ≥150 bp that was inaccessible to M.CviPI. At the extreme ends of each molecule, nucleosome-occupied DNA was identified as M.CviPI inaccessibility >75 bp (half the size of a nucleosome occupied region of DNA), as described previously [Hesson et al., 2014]. Duplicate molecules (those containing identical patterns of GpC methylation) were removed from further analysis to prevent data misinterpretation due to cloning or PCR bias. Molecules containing non-CpG methylation (except in the context of GpC sites) were also discarded to eliminate amplicons derived from incompletely converted DNA. The term occupancy refers to the proportion of molecules bearing a nucleosome at a specific location, as described previously [Hesson et al., 2014].
Statistics
Differences in allelic balance were assessed using a two-sample t-test.
Results
Two MLH1 5′UTR Variants Associated with Early-Onset CRC
To confirm the presence of the c.-28A>G and c.-7C>T variants, we performed sequencing of a 1,067-bp region across the MLH1 promoter (c.-960 to c.107 relative to NM_000249.3) in constitutional DNA from both probands and identified three heterozygous variants, namely, c.-93G>A, c.-28A>G, and c.-7C>T (Supp. Fig. S1A). No further sequence variants were identified in this region. While the c.-93G>A is a common and benign variant, the c.-28A>G and c.-7C>T are described as Class 3 variants of uncertain pathogenicity in the InSiGHT database [Fokkema et al., 2011]. Data from 6,515 control exomes [Fu et al., 2013] showed both variants were present in 4/13,006 of chromosomes tested (minor allele frequency [MAF] = 0.0003). Sequencing of individual promoter molecules determined that these variants were present on the same allele (Supp. Fig. S1B). Sequencing of long-range amplicons generated using cDNA derived from PBMCs showed that in both probands the c.-93A/-28G/-7T haplotype also contained the expressed c.655G variant in exon 8 of the MLH1 gene, whereas the c.-93G/-28A/-7C haplotype contained the c.655A variant.
Targeted Sequencing and Microsatellite Analysis Across the MLH1 Locus Confirms the Lack of Deleterious Sequence or Structural Alterations in MLH1
To determine whether the c.-28A>G and c.-7C>T variants were in cis with another pathogenic sequence alteration within MLH1, we employed targeted, high-throughput sequencing of constitutional DNA from Proband 32. High-coverage sequencing data were obtained over a 1.7-Mb region that consisted of exonic, intronic, and intergenic regions of MLH1 and flanking genes (see Materials and Methods and Fig. 2A). We detected the c.-93A/-28G/-7T and c.-93G/-28A/-7C haplotypes within the MLH1 promoter (Fig. 2B; Supp. Table S3), which were each confined to single 100 bp reads, as expected. We also detected three novel intronic SNVs within MLH1 (Supp. Fig. S2; Supp. Table S3). These novel intronic variants were located 736, 869, and 1,216 bp from the nearest MLH1 exon and it is highly unlikely that they would contribute to splicing defects. Therefore, no potentially pathogenic sequence alterations were detected across the MLH1 gene other than the c.-28A>G and c.-7C>T variants.
Figure 2.

Targeted sequencing confirms the lack of potentially pathogenic sequence alterations within the MLH1 gene other than the c.-28A>G and c.-7C>T variants. A: The locations of the 15 genes within the region targeted for enrichment and sequencing, the locations and number of all variants identified (black vertical bars), the locations and number of novel variants identified (red vertical bars), and aligned read depth across the region (blue histogram). Shown is the region from chr3:36395082-38171759 (hg19, February 2009). B: The three variants in the MLH1 5′UTR that were detected by targeted sequencing. No other potentially pathogenic sequence alterations were detected in the MLH1 gene. C: The locations of sequence tagged site (STS) and SNV markers within chromosome 3 (Chr3) that were assayed for zygosity. See also Supp. Table S2. D: A schematic of EPM2AIP1 and MLH1, indicating the locations and number of variants used to assess zygosity. Shown is the region from chr3:37026918-37093479 (hg19, February 2009). E: The location of the VILL (villin-like) c.1164del1 variant within the target region that was sequenced. See also Supp. Figures S3 and S4. B and E: The type of dbSNP137 polymorphisms is indicated by different colors with red representing nonsynonymous polymorphisms within the coding region (including splice-site mutations); black representing intronic polymorphisms; green representing synonymous polymorphisms within the coding region; blue representing 5′ UTR variants.
To investigate the possibility of large deletions in or flanking the MLH1 gene, we determined zygosity at five microsatellite markers and six SNPs within and flanking both sides of the MLH1 gene (Supp. Table S2). These markers encompassed ∼20.2 Mb of DNA across the 3p22.3-p14.3 region (Fig. 2C). All but one microsatellite marker showed heterozygosity in constitutional DNA. Identical results were obtained from tumor DNA. We extended this assessment of zygosity using the variants identified across the MLH1 and EPM2AIP1 genes by targeted sequencing. A total of 52 sequence variants across these genes were heterozygous, whereas only one was homozygous (Fig. 2D). These results confirmed the presence of two intact alleles of the MLH1 gene, and therefore the absence of MLH1 deletion, in constitutional and tumor DNA.
Next, we examined variants in other genes and intergenic sites across the 1.7-Mb region that was sequenced. We identified a total of 1,452 sequence variants, comprising 1,374 SNVs and indels previously described in the 1000 genomes or SNP 137 databases and 78 novel sequence variants (Supp. Table S3). Only one of these 78 novel variants was associated with an amino acid alteration within the coding region of a gene. This frameshift variant was a single-nucleotide deletion (c.1164del1) within exon 10 of the VILL (villin-like) gene that was predicted to cause the truncation of VILL protein to p.Asp388Glu.fs.*64 (Fig. 2E). The presence of this variant in constitutional DNA from Proband 32 was confirmed by single molecule sequencing across exon 10 (Supp. Fig. S3). Analysis of DNA from a saliva sample obtained from a first-degree relative of Proband 32 revealed the c.1164del1 variant in VILL, and the c.-93A/-28G/-7T variants in MLH1 were also present confirming they cosegregation on the same chromosome (Supp. Fig. S4). Data from control exomes [Fu et al., 2013] showed that the c.1164del1 variant was present in 1/12,520 chromosomes (MAF = 0.00008). Given the predicted pathogenicity of this variant, we determined whether it was also present in Proband N. Pyrosequencing across the c.1164del1 site showed this variant was not present in constitutional DNA from this individual (Supp. Fig. S5A). To further define the variant MLH1 haplotype, we determined whether the novel intronic variants described in Supp. Figure S2 cosegregated with the c.-93G>A, c.-28A>G, c.-7C>T, and VILL c.1164del variants in the first-degree relative of Proband 32. Neither variant was found in this individual showing that they were located on the wild-type MLH1 allele. Both intronic variants were also absent in Proband N. This shows that the two probands in our study share the same ancestral variant MLH1 haplotype, and that the VILL variant in Proband 32 most likely arose as a later event within this haplotype.
Reduced Constitutional MLH1 Expression from the Variant Allele
The absence of potentially pathogenic sequence alterations within or flanking the MLH1 gene other than the c.-28A>G and c.-7C>T variants prompted us to investigate their role in regulating MLH1. Given the location of these variants within the MLH1 5′UTR, we investigated their influence on gene expression. Allelic representation of expression was performed by using primers complementary to exons 1a and 9 followed by pyrosequencing across the c.655G>A site (Fig. 3A). In Proband 32, the proportion of transcripts originating from the variant allele was half that observed from the wild-type allele, at 32.3%:67.7% (G:A; Fig. 3B and D). This showed there was a ∼50% reduction in expression from the variant allele relative to the wild-type allele. Similar levels of allelic imbalance were observed in the PBMCs of Proband N with the proportion of transcripts from each allele detected at 33.7%:66.3% (G:A; Fig. 3C and D), again indicating a reduction in expression from the variant allele to ∼50% of the levels of the wild-type allele. These levels of allelic imbalance were significantly different to the allelic balance observed in 12 control PBMCs that were also heterozygous at the c.655G>A site (Fig. 3D; mean normal allelic balance = 45.2%:54.8% G:A, range = 49.8%:50.2%–41.6%:58.4% G:A; P < 0.0001, paired t-test). These data show that the c.-28A>G and c.-7C>T variants are associated with partial loss of constitutional MLH1 expression to ∼50% the levels observed from the wild-type allele and that this is consistent in two individuals.
Figure 3.
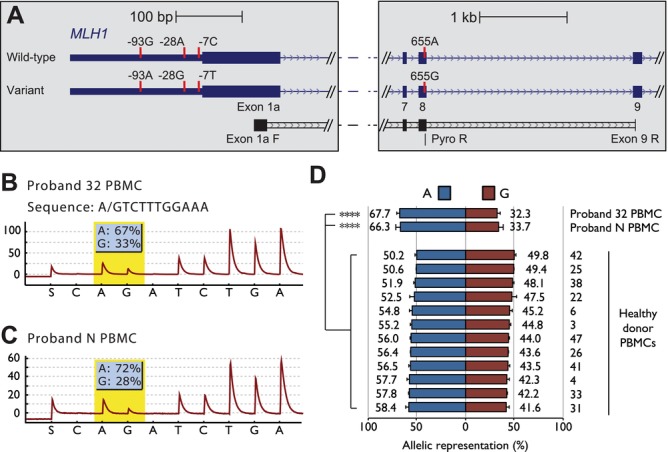
Partial loss of MLH1 expression from the variant allele. A: The locations of primers used to assess allele-specific expression of MLH1 transcripts from exon 1a. Indicated are the locations of sequence variants associated with the wild-type and variant MLH1 alleles (vertical red bars), the locations of PCR primers within exon 1a and 9 (exon 1a F and exon 9 R, respectively), and the pyrosequencing primer used to assess allelic balance across the c.655A>G site (Pyro R). B and C: Representative pyrograms indicating reduced expression of the variant MLH1 allele in Proband 32 and Proband N PBMCs, respectively. Partial loss of expression from the variant allele is indicated by the reduced representation of transcripts containing the c.655G variant. D: Compares the allelic balance of MLH1 expression in Proband 32 and Proband N with that in 12 PBMCs from healthy donors that were informative at the c.655A>G site. ****P < 0.0001 (paired t-test). Allelic balance was determined with a minimum of three technical replicates for each sample. Data are represented as mean ± SD.
The MLH1 CpG Island Promoter Within the Variant Allele Is Epigenetically Unaltered
To investigate the potential causes of reduced expression from the variant allele, we investigated the MLH1 promoter for evidence of constitutional epigenetic changes. Routine diagnostic tests using methylation-sensitive MLPA (MS-MLPA) had suggested that the DNA from Proband 32 was 60% methylated at one CpG site within the MLH1 promoter, whereas six other CpG sites were unmethylated. However, we deduced that the CpG site identified as hypermethylated was within a HhaI restriction enzyme site (used in the MS-MLPA assay) that was abolished by the c.-7C>T variant (Fig. 4A). Bisulfite sequencing of individual promoter molecules across the c.-93G>A variant confirmed the lack of methylation on both alleles of the MLH1 promoter throughout all normal and tumor tissues from Proband 32 (Fig. 4B). The lack of methylation in constitutional DNA from both probands was also confirmed using bisulfite pyrosequencing across five CpG dinucleotides that are associated with the transcriptional silencing of MLH1 when methylated (Fig. 4C and D) [Deng et al., 1999].
Figure 4.
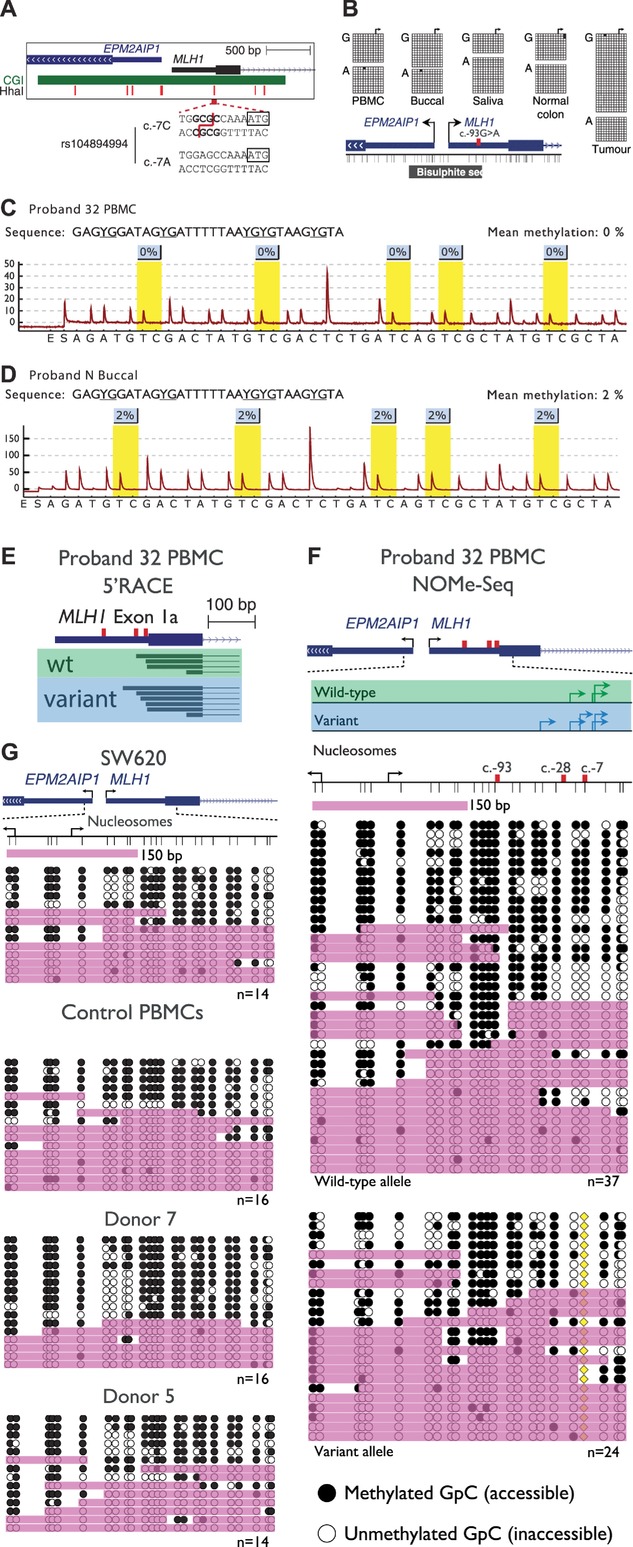
The variant allele is not epigenetically altered at the MLH1 promoter. A: A schematic of the MLH1 and EPM2AIP1 bidirectional promoter indicating the location of the CpG island (green bar), seven HhaI sites used to detect methylation by MS-MLPA (red vertical bars), and the sequence encompassing the c.-7C>T site. The presence of the c.-7C>T variant abolishes a HhaI restriction site. B: Single molecule bisulfite sequencing data of various tissues from Proband 32. The c.-93G>A site was used to distinguish between wild-type (c.-93G) and variant (c.-93A) MLH1 alleles. The black horizontal bar labeled “Bisulfite seq” indicates the region analyzed. Both alleles were unmethylated in all tissues examined including tumor tissue. C and D: Representative pyrograms indicating methylation levels at five CpG sites within the MLH1 promoter in Proband 32 PBMCs and Proband N buccal DNA, respectively. The nominal limit of quantification for this assay is 5%. E: The locations of unique transcription initiation sites in exon 1a of the wild-type (green box) and variant (blue box) MLH1 alleles. The c.-93, c.-28, and c.-7 sites are indicated by the red vertical bars. F: Nucleosome occupancy across individual promoter molecules separated according to allele of origin, as determined by the c.-7C>T variant (yellow diamond). Black arrows indicate the annotated MLH1 [NM_000249.3] or EPM2AIP1 [NM_014805.3] transcription initiation sites, whereas green and blue arrows indicate the locations of sites identified by 5′RACE in wild-type and variant alleles, respectively. Thin vertical black lines represent the positions of GpC dinucleotides. Black circles = GpC dinucleotides methylated/accessible to the GpC methyltransferase M.CviPI. White circles = GpC dinucleotides unmethylated/inaccessible to GpC methyltransferase. Pink shading indicates regions of accessibility ≥150 or >75 bp at the extreme ends of amplicons. G: Nucleosome occupancy across the same region in MLH1-expressing colorectal carcinoma cells and PBMCs from healthy donors. The number of molecules sequenced is indicated at the bottom right of each panel.
Altered nucleosome occupancy, specifically around the transcription initiation site of a gene, can correlate with gene silencing in the absence of hypermethylation [Hesson et al., 2014]. We performed 5′RACE to map the positions of transcription initiation sites within the MLH1 promoter in PBMCs from Proband 32. Because transcription may be initiated downstream of the informative c.-93, c.-28 or c.-7 sites, we sequenced across the c.655A>G variant to determine the allele of transcript origin. A total of 20 wild-type and 15 variant transcript molecules were sequenced, which collectively identified 10 separate sites of transcription initiation within exon 1a. Interestingly, 8/10 clustered within the region c.-27 to c.-3. These initiation sites overlapped with those identified in 975 human primary cells, tissues, and cancer cell lines (Supp. Fig. S6) [Consortium et al., 2014]. Therefore, the predominant transcription initiation sites within the MLH1 promoter were located in the immediate vicinity of the c.-28A>G and c.-7C>T variants. However, comparison of the initiation sites between the variant and wild-type alleles showed that the presence of these variants did not alter the precise sites of transcription initiation (Fig. 4E). Next, we examined nucleosome occupancy across these initiation sites using the informative c.-7C>T site to distinguish between the wild-type and variant alleles. One or more of the initiation sites were accessible on 59% (22/37) of wild-type promoter molecules (Fig. 4F). By comparison, 50% (12/24) of variant promoter molecules were accessible across the same sites (Fig. 4F) suggesting a slight increase in nucleosome occupancy. However, in PBMCs from three healthy donors and a carcinoma cell line, all of which express high levels of MLH1, 50%–69% of molecules were accessible across the same sites showing that the slight increase in nucleosome occupancy observed in the variant allele was within the normal range (Fig. 4G). These data showed there were no marked differences in nucleosome occupancy across transcription initiation sites between the wild-type and variant alleles or with the same sites in normal control cells. Therefore, despite reduced expression of the variant allele, the MLH1 promoter is epigenetically unaltered.
Discussion
We show that two SNVs in the MLH1 promoter are associated with partial loss of MLH1 expression in two individuals from suspected Lynch syndrome families. Specifically, the c.-28A>G and c.-7C>T variants were associated with ∼50% reduction in the abundance of transcripts initiated from the variant allele when compared with the wild-type allele. This loss of expression occurred in the context of an intact MLH1 promoter and in the absence of any other potentially pathogenic sequence alterations in or around the MLH1 gene. Though these variants are located in the immediate vicinity of transcription initiation sites, we show that they do not alter the precise location of these initiation sites. Finally, despite partial loss of expression from the variant allele, we show that the c.-28A>G and c.-7C>T variants are not associated with promoter hypermethylation or alterations in nucleosome occupancy. These findings suggest that the c.-28A>G and c.-7C>T variants may be pathogenic due to allelic imbalance of MLH1 expression.
There are now several reports of Lynch syndrome associated with partial loss of MLH1 expression [Curia et al., 1999; Green et al., 2003; Hinrichsen et al., 2013]. Curia et al. (1999) described Lynch syndrome associated with a silent variant within exon 9 of the MLH1 gene and 50% reduction in constitutional levels of expression compared with the wild-type allele. Green et al. (2003) described a c.-42C>T variant within the MLH1 gene associated with the reduction of promoter activity to ∼37% that of wild-type promoter sequence. Furthermore, several constitutional MLH1 missense mutations that lead to ∼50% or more reduction in protein levels were recently reclassified as pathogenic [Hinrichsen et al., 2013]. In each case, loss of the wild-type allele in tumor cells would likely lead to impaired MMR activity due to insufficient MLH1 expression from the variant allele. According to a recently described standardized classification system, [Thompson et al., 2014] MMR gene variants that abrogate gene function include those that cause-defective transcription. Our observation that the c.-28A>G and c.-7C>T variants are associated with partial constitutional loss of MLH1 expression provides strong evidence that they may be pathogenic. It is possible that reduced but not abolished MMR functionality caused by the partial loss of MLH1 expression may give rise to an intermediate mutation rate and the onset of cancer due to increased occurrence of somatic mutations. A potential limitation of our study is that we assessed MLH1 expression in PBMCs, which may not necessarily reflect the levels of expression in colorectal mucosa. Should samples become available, further studies could aim to determine the transcriptional activity associated with the c.-28A>G and c.-7C>T variants in other tissues, including colorectal mucosa. Our findings show that these variants are associated with abrogated MLH1 function (partial loss of expression) but this alone is insufficient to reclassify as Class 4 variants (likely pathogenic) and both remain Class 3 variants (uncertain). Additional evidence, such as the presence of one or both of these variants on a different background MLH1 haplotype, ≥2 tumors with a Lynch syndrome molecular phenotype in a carrier or the cosegregation of these variants with Lynch syndrome will be required to reclassify these variants as Class 4. If however, these variants are indeed associated with intermediate penetrance, segregation with Lynch syndrome is unlikely to be observed.
The variable age of onset of cancer associated with the c.-28A>G and c.-7C>T variants was evident from the two families we investigated. While Proband 32 presented with CRC aged 38 years, Proband N was asymptomatic at 54 years. However, the father and grandfather of Proband N presented with colonic adenomas and colorectal cancer at ages 50 and 40 years, respectively, though we were unable to ascertain whether the grandfather had the c.-28A>G and c.-7C>T variants. This variable penetrance may be explained by incomplete inactivation of MLH1 expression, as described previously for the c.-42C>T variant, which was associated with drastically different ages of cancer onset between 35 and 76 years (average 62 years) in a single Lynch syndrome family [Green et al., 2003]. Interestingly, in this study, loss of heterozygosity analyses of the tumor from an individual with the c.-42C>T variant revealed deletion of the MLH1 allele with the variant and not of the wild-type allele, as one would expect. In our analyses, both MLH1 alleles were retained in the tumor of Proband 32. These results are most likely explained by the presence of somatic MLH1 mutations in the tumor.
Further evidence that the c.-28A>G and c.-7C>T variants are the most likely cause of cancer predisposition was provided by our detailed genetic analysis of the MLH1 locus. We combined targeted high-throughput sequencing of the wider MLH1 locus with microsatellite marker analysis to provide high-resolution sequence and zygosity information across the 3p14.3-p22.3 region. Collectively, this data confirmed the lack of potentially pathogenic sequence alterations within or flanking the MLH1 gene, other than the c.-28A>G and c.-7C>T variants. This approach identified a frameshift variant within the neighboring VILL gene. The cosegregation of the c.1164del1, c.-28A>G, and c.-7C>T variants in a first-degree relative of Proband 32 showed that they were located on the same chromosome. Furthermore, their precise frequencies in control exomes provided clues to the genetic history of this haplotype. For example, the c.-28A>G and c.-7C>T variants have an identical MAF of 0.0003, showing that they represent part of an ancestral haplotype. Moreover, the absence of any identifiable sequence differences on the variant MLH1 haplotype between the two probands investigated in our study supports the conclusion that this MLH1 haplotype originates from a common ancestor. The VILL c.1164del1 frameshift variant has a MAF of only 0.00008 and is therefore a more recent genetic event within this haplotype. The function of VILL is unknown but its homology with the VIL1 (villin [MIM #193040]) gene suggests it may play a role in regulating the actin cytoskeleton and the formation of microvilli at the epithelial surface of the gut. The presence of a constitutional frameshift variant in VILL in Proband 32 raised the possibility that it might explain early-onset cancer. However, this variant is unlikely to be the cause of cancer predisposition because it was absent in Family N in which the c.-28A>G and c.-7C>T variants cosegregated. Therefore, the c.-28A>G and c.-7C>T variants are the most likely cause of predisposition to CRC with MMR deficiency and loss of MLH1.
The InSiGHT database [Thompson et al., 2014] describes two suspected individuals with this haplotype in addition to two cases described previously [Muller-Koch et al., 2001; Lee et al., 2005]. One was diagnosed with CRC aged 46 years and had a sibling who was diagnosed with CRC aged 28 years. The proband’s tumor was MLH1 and PMS2 negative and BRAF wild-type. Separate from the c.-7C>T and c.-28A>G variants, no constitutional mutations in MLH1, EPCAM, or MSH2 were found by MLPA and no expression analyses were undertaken. The other individual with this haplotype was also diagnosed with MLH1 negative CRC; however, no further information is provided. Therefore, the rarity of the c.-28A>G and c.-7C>T haplotype at the population level, but its presence in several suspected Lynch syndrome individuals to date suggests it is associated with a Lynch syndrome phenotype.
The cosegregation of these two variants makes it difficult to ascertain whether both or only one are pathogenic. The c.-7C>T variant was found in a member of a hereditary prostate cancer family who met revised Bethesda guidelines [Fredriksson et al., 2006]. However, the description of a case of CRC that was associated with a c.-28A>T variant in a 29-year-old individual from a family with multiple cancer-affected members [Isidro et al., 2003] suggests that loss of the c.-28A nucleotide may be pathogenic. Whether the c.-28A>T variant in this family was associated with reduced MLH1 expression or cosegregation with disease was not determined. Furthermore, a variant of the adjacent nucleotide (c.-27C>A) also segregates with Lynch syndrome in several unrelated families [Hitchins et al., 2011; Kwok et al., 2014]. Collectively, this suggests that c.-27 and c.-28 sites are located within an important regulatory motif within the MLH1 5′UTR. Further work to identify the DNA-binding protein that binds to this site will help to elucidate the mechanistic basis of how variants within the MLH1 5′UTR affect expression.
The presence of the c.-93G>A, c.-28A>G, c.-7C>T, and c.655A>G variants in both probands allowed us to distinguish between wild-type and variant alleles and transcripts and to investigate the mechanisms of reduced MLH1 expression. We conclusively demonstrate that the variant MLH1 promoter remains unmethylated despite partial loss of expression. This contradicted the pathology report from Proband 32, which reported methylation in tumor DNA. Using several approaches, we demonstrate that the apparent hypermethylation was an artifact caused by the c.-7C>T variant, which lies within a recognition site of the HhaI restriction enzyme utilized as part of the MS-MLPA assay. However, reductions in gene expression can correlate with other chromatin changes at transcription initiation sites, such as increased nucleosome occupancy, which can physically occlude an initiation site from the transcriptional machinery [Jiang and Pugh, 2009]. To determine whether the variant allele was associated with increased nucleosome occupancy, we precisely defined sites of MLH1 transcription initiation and compared nucleosome occupancy between the variant and wild-type alleles using an allele-specific NOMe-Seq assay. Our finding that the variant allele showed no increase in nucleosome occupancy when compared with the wild-type allele, or the MLH1 promoter in other cells expressing high levels of MLH1, confirmed that it remained epigenetically unaltered. The continued recruitment of DNA-binding factors is thought to protect CpG island promoters from nucleosome occupancy [Struhl and Segal, 2013] and de novo methylation [Lienert et al., 2011; Krebs et al., 2014]. This suggests that the variant allele may be protected from epigenetic silencing because it retains the ability to recruit DNA-binding proteins such as transcription factors. Due to the proximity of the c.-28A>G and c.-7C>T variants to transcription initiation sites, we hypothesize that diminished expression may be a result of impaired ability to initiate transcription.
In summary, our study describes further cases of suspected Lynch syndrome associated with the c.-28A>G and c.-7C>T variants. Our analysis shows that these variants may be pathogenic due to partial loss of MLH1 expression. These findings reinforce the potential importance of sequence variants in the MLH1 promoter in Lynch syndrome and the need for future studies to sequence the entire 5′UTR when searching for pathogenic sequence alterations.
Acknowledgments
Disclosure statement: The authors declare no conflicts of interest.
Supporting Information
Disclaimer: Supplementary materials have been peer-reviewed but not copyedited.
Figure S1. Single molecule sequencing identifies a variant haplotype within the MLH1 5’UTR.
Figure S2. Variants in the MLH1 and EPM2AIP1 genes identified by targeted high-throughput sequencing.
Figure S3. Confirmation of a VILL c.1164del1 variant within Proband 32.
Figure S4. Co-segregation of the c.-93G>A, c.-28A>G, c.-7C>T and VILL c.1164del1 variants in a first degree relative of Proband 32 confirms they are located on the same chromosome.
Figure S5. The VILL c.1164del1 variant is absent in Proband N.
Figure S6. Transcription initiation sites within exon 1a of the MLH1 gene.
Table S1. Primers used throughout this study
Table S2. Microsatellite and single nucleotide markers used to assess zygosity
Table S3. Sequence variants identified using targeted sequencing across the MLH1 locus
References
- Curia MC, Palmirotta R, Aceto G, Messerini L, Veri MC, Crognale S, Valanzano R, Ficari F, Fracasso P, Stigliano V, Tonelli F, Casale V, et al. Unbalanced germ-line expression of hMLH1 and hMSH2 alleles in hereditary nonpolyposis colorectal cancer. Cancer Res. 1999;59:3570–3575. [PubMed] [Google Scholar]
- Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999;59:2029–2033. [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- FANTOM Consortium and the RIKEN PMI and CLST (DGT) Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, Anderson R, et al. A promoter-level mammalian expression atlas. Nature. 2014;507:462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–563. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- Fredriksson H, Ikonen T, Autio V, Matikainen MP, Helin HJ, Tammela TL, Koivisto PA, Schleutker J. Identification of germline MLH1 alterations in familial prostate cancer. Eur J Cancer. 2006;42:2802–2806. doi: 10.1016/j.ejca.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, Nickerson DA, Bamshad MJ, et al. Analysis of 6515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Nguyen TP, Leung HC, Nagasaka T, Rhees J, Hotchkiss E, Arnold M, Banerji P, Koi M, Kwok CT, Packham D, Lipton L, et al. De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer. 2011;128:869–878. doi: 10.1002/ijc.25422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Green AG, Simms M, Pater A, Robb JD, Green JS. Germline hMLH1 promoter mutation in a Newfoundland HNPCC kindred. Clin Genet. 2003;64:220–227. doi: 10.1034/j.1399-0004.2003.t01-1-00110.x. [DOI] [PubMed] [Google Scholar]
- Hesson LB, Dunwell TL, Cooper WN, Catchpoole D, Brini AT, Chiaramonte R, Griffiths M, Chalmers AD, Maher ER, Latif F. The novel RASSF6 and RASSF10 candidate tumour suppressor genes are frequently epigenetically inactivated in childhood leukaemias. Mol Cancer. 2009;8:42. doi: 10.1186/1476-4598-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesson LB, Patil V, Sloane MA, Nunez AC, Liu J, Pimanda JE, Ward RL. Reassembly of nucleosomes at the MLH1 promoter initiates resilencing following decitabine exposure. PLoS Genet. 2013;9:e1003636. doi: 10.1371/journal.pgen.1003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesson LB, Sloane MA, Wong JW, Nunez AC, Srivastava S, Ng B, Hawkins NJ, Bourke MJ, Ward RL. Altered promoter nucleosome positioning is an early event in gene silencing. Epigenetics. 2014;9:1422–1430. doi: 10.4161/15592294.2014.970077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesson LB, Ward RL. Discrimination of pseudogene and parental gene DNA methylation using allelic bisulfite sequencing. Methods Mol Biol. 2014;1167:265–274. doi: 10.1007/978-1-4939-0835-6_18. [DOI] [PubMed] [Google Scholar]
- Hinrichsen I, Brieger A, Trojan J, Zeuzem S, Nilbert M, Plotz G. Expression defect size among unclassified MLH1 variants determines pathogenicity in Lynch syndrome diagnosis. Clin Cancer Res. 2013;19:2432–2441. doi: 10.1158/1078-0432.CCR-12-3299. [DOI] [PubMed] [Google Scholar]
- Hitchins MP, Rapkins RW, Kwok CT, Srivastava S, Wong JJ, Khachigian LM, Polly P, Goldblatt J, Ward RL. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5’UTR. Cancer Cell. 2011;20:200–213. doi: 10.1016/j.ccr.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Isidro G, Matos S, Goncalves V, Cavaleiro C, Antunes O, Marinho C, Soares J, Boavida MG. Novel MLH1 mutations and a novel MSH2 polymorphism identified by SSCP and DHPLC in Portuguese HNPCC families. Hum Mutat. 2003;22:419–420. doi: 10.1002/humu.9192. [DOI] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TK, Miranda TB, Liang G, Berman BP, Lin JC, Tanay A, Jones PA. H2A.Z maintenance during mitosis reveals nucleosome shifting on mitotically silenced genes. Mol Cell. 2010;39:901–911. doi: 10.1016/j.molcel.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs AR, Dessus-Babus S, Burger L, Schubeler D. High-throughput engineering of a mammalian genome reveals building principles of methylation states at CG rich regions. Elife. 2014;3:e04094. doi: 10.7554/eLife.04094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok CT, Vogelaar IP, van Zelst-Stams WA, Mensenkamp AR, Ligtenberg MJ, Rapkins RW, Ward RL, Chun N, Ford JM, Ladabaum U, McKinnon WC, Greenblatt MS, et al. The MLH1 c.-27C>A and c.85G>T variants are linked to dominantly inherited MLH1 epimutation and are borne on a European ancestral haplotype. Eur J Hum Genet. 2014;22:617–624. doi: 10.1038/ejhg.2013.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok CT, Ward RL, Hawkins NJ, Hitchins MP. Detection of allelic imbalance in MLH1 expression by pyrosequencing serves as a tool for the identification of germline defects in Lynch syndrome. Fam Cancer. 2010;9:345–356. doi: 10.1007/s10689-009-9314-0. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Guo JY, Lim R, Soo R, Koay E, Salto-Tellez M, Leong A, Goh BC. Clinical and molecular characteristics of hereditary non-polyposis colorectal cancer families in Southeast Asia. Clin Genet. 2005;68:137–145. doi: 10.1111/j.1399-0004.2005.00469.x. [DOI] [PubMed] [Google Scholar]
- Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schubeler D. Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet. 2011;43:1091–1097. doi: 10.1038/ng.946. [DOI] [PubMed] [Google Scholar]
- Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–932. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76:1–18. doi: 10.1111/j.1399-0004.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Koch Y, Kopp R, Lohse P, Baretton G, Stoetzer A, Aust D, Daum J, Kerker B, Gross M, Dietmeier W, Holinski-Feder E. Sixteen rare sequence variants of the hMLH1 and hMSH2 genes found in a cohort of 254 suspected HNPCC (hereditary non-polyposis colorectal cancer) patients: mutations or polymorphisms? Eur J Med Res. 2001;6:473–482. [PubMed] [Google Scholar]
- Raevaara TE, Korhonen MK, Lohi H, Hampel H, Lynch E, Lonnqvist KE, Holinski-Feder E, Sutter C, McKinnon W, Duraisamy S, Gerdes AM, Peltomaki P, et al. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–549. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Struhl K, Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol. 2013;20:267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlay PC, Kelly TK, Liu CC, You JS, De Carvalho DD, Miranda TB, Zhou XJ, Liang G, Jones PA. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell. 2011;147:1283–1294. doi: 10.1016/j.cell.2011.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Greenblatt MS, Goldgar DE, Boffetta P Group IUGVW. Assessing pathogenicity: overview of results from the IARC Unclassified Genetic Variants Working Group. Hum Mutat. 2008;29:1261–1264. doi: 10.1002/humu.20903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, Bapat B, Bernstein I, Capella G, den Dunnen JT, du Sart D, Fabre A, et al. Application of a 5-tiered scheme for standardized classification of 2360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014;46:107–115. doi: 10.1038/ng.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A, van der Klift H, Franken P, Wijnen J, Breukel C, Bezrookove V, Smits R, Kinarsky Y, Barrows A, Franklin B, Lynch J, Lynch H, et al. A 10-Mb paracentric inversion of chromosome arm 2p inactivates MSH2 and is responsible for hereditary nonpolyposis colorectal cancer in a North-American kindred. Genes Chromosomes Cancer. 2002;35:49–57. doi: 10.1002/gcc.10094. [DOI] [PubMed] [Google Scholar]
- Ward RL, Dobbins T, Lindor NM, Rapkins RW, Hitchins MP. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med. 2013;15:25–35. doi: 10.1038/gim.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You JS, Kelly TK, De Carvalho DD, Taberlay PC, Liang G, Jones PA. OCT4 establishes and maintains nucleosome-depleted regions that provide additional layers of epigenetic regulation of its target genes. Proc Natl Acad Sci USA. 2011;108:14497–15502. doi: 10.1073/pnas.1111309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Single molecule sequencing identifies a variant haplotype within the MLH1 5’UTR.
Figure S2. Variants in the MLH1 and EPM2AIP1 genes identified by targeted high-throughput sequencing.
Figure S3. Confirmation of a VILL c.1164del1 variant within Proband 32.
Figure S4. Co-segregation of the c.-93G>A, c.-28A>G, c.-7C>T and VILL c.1164del1 variants in a first degree relative of Proband 32 confirms they are located on the same chromosome.
Figure S5. The VILL c.1164del1 variant is absent in Proband N.
Figure S6. Transcription initiation sites within exon 1a of the MLH1 gene.
Table S1. Primers used throughout this study
Table S2. Microsatellite and single nucleotide markers used to assess zygosity
Table S3. Sequence variants identified using targeted sequencing across the MLH1 locus