

Abstract
MUTYH-associated polyposis (MAP) is an adenomatous polyposis transmitted in an autosomal-recessive pattern, involving biallelic inactivation of the MUTYH gene. Loss of a functional MUTYH protein will result in the accumulation of G:T mismatched DNA caused by oxidative damage. Although p.Y179C and p.G396D are the two most prevalent MUTYH variants, more than 200 missense variants have been detected. It is difficult to determine whether these variants are disease-causing mutations or single-nucleotide polymorphisms. To understand the functional consequences of these variants, we generated 47 MUTYH gene variants via site-directed mutagenesis, expressed the encoded proteins in MutY-disrupted Escherichia coli, and assessed their abilities to complement the functional deficiency in the E. coli by monitoring spontaneous mutation rates. Although the majority of variants exhibited intermediate complementation relative to the wild type, some variants severely interfered with this complementation. However, some variants retained functioning similar to the wild type. In silico predictions of functional effects demonstrated a good correlation. Structural prediction of MUTYH based on the MutY protein structure allowed us to interpret effects on the protein stability or catalytic activity. These data will be useful for evaluating the functional consequences of missense MUTYH variants detected in patients with suspected MAP.
Keywords: MUTYH, MAP, functional assay, polyposis, prediction
Introduction
MUTYH-associated polyposis (MAP; MIM #608456) is an autosomal-recessive inherited familial colorectal cancer (CRC) and polyposis syndrome [Nielsen et al., 2010]. Biallelic mutations in MUTYH genes (MIM #604933) predispose patients to the development of polyps, generally between 10 and a few hundred polyps. The MUTYH gene encodes a base excision repair (BER) glycosylase, which is involved in the repair of the major base lesion 8-oxo-guanine (8-oxoG), caused by oxidative damage and prevents G:C to T:A transversion [David et al., 2007]. The MUTYH protein recognizes oxoG:A mismatches and excises the undamaged adenine. MUTYH consists of different functional domains (Fig. 1A). The N-terminal domain includes a catalytic region with a helix–hairpin–helix (HhH) motif, as well as a pseudo-HhH region and iron–sulfur cluster loop (FCL) motif, which function in the recognition and excision of adenine moieties opposite 8-oxoG [Guan et al., 1998; Lukianova and David, 2005]. The C-terminal domain is known as a MutT-like domain that shares the homology with the nudix-type motif 1 and functions in the recognition of the 8-oxoG lesion [Noll et al., 1999].
Figure 1.

Functional assay for MUTYH variants. A: Germline mutations observed in MUTYH related to MAP. Alignment of E. coli MutY and human MUTYH is shown; 47 mutations observed in MUTYH related to MAP are indicated. B: Suppression of spontaneous mutations by expression of wild-type MUTYH and 47 variants in E. coli CC104mutY. The control shows transfectants with empty vectors. Green line shows the cut-off value between functionally retained and partially defective. Red line shows the cut-off value between functionally defective and partially defective.
A biallelic variation p.Y179C and/or p.G396D has been reported in up to 70% of patients with MAP in Caucasian populations [Nielsen et al., 2010]. However, these mutations have not been identified in Asian populations (e.g., Japanese and Korean), suggesting the existence of founder mutations and ethnic differentiation. Indeed, the p.A359V is a common and likely founder variant in Japanese and Korean patients [Yanaru-Fujisawa et al., 2008]. Other locally common variants have been identified (e.g., p.P405L in Dutch and p.E480del in Italian populations) [Gismondi et al., 2004; Nielsen et al., 2005].
The Leiden Open Variation Database (LOVD) (http://www.lovd.nl/3.0/home) is a well-designed, open-access database of MUTYH variants [Out et al., 2010]. The majority of MUTYH alterations are missense variants. Currently, more than 200 unique missense variants have been registered in LOVD. Because the significance of these missense variants with respect to protein function is difficult to evaluate, functional assays are required to understand the pathogenesis of MUTYH variants.
One of the most common assays is the in vitro glycosylase assay, which examines the enzymatic activity of recombinant MUTYH proteins to excises adenines opposite 8-oxoG on the oligonucleotide substrates [Ali et al., 2008; D’Agostino et al., 2010]. Another common assay is the in vivo E. coli complementation assay, which examines the abilities of exogenously expressed MUTYH proteins to suppress MutY-deficient E. coli mutation [Kundu et al., 2009].
Functional analyses of two major variants, p.Y179C and p.G396D, have been performed using cells from several species with the equivalent variants. Furthermore, the structural resolution of Bacillus stearothermophilus MutY has provided a structural basis for these variants [Fromme et al., 2004]. Y179 is located on the DNA-binding interface, where it recognizes 8-oxoG:A mispairings and stabilizes protein–DNA complexes. G396 also contributes to 8-oxoG:A mispair recognition and imparts flexibility to the MUTYH conformation. In addition to the two major variants, functional assays have been performed for a number of MUTYH variants. However, the results have been inconsistent [Bai et al., 2005; Ali et al., 2008; Goto et al., 2010]. Moreover, almost all studies have assayed relatively small numbers of variants.
To evaluate the functional significance of many variants comprehensively, we assayed 47 variants differentiated by only subtle variations such as missense mutations or one amino acid deletion. The present study will provide useful information to understand the pathogeneses of these 47 MUTYH variants.
Materials and Methods
E. Coli Strain and Plasmids
The E. coli strain CC104mutY::Tn10 was used [Takao et al., 1998]. Human cDNA encoding MUTYH (type 2, isoform 4) cDNA was subcloned into pMAL-c2 (NEB, Ipswich, MA) to generate pMAL-cY2. CC104mutY was transformed with pMAL-cY2 or the empty pMAL-c2 vector [Takao et al., 1999]. The reference sequence for the MUTYH gene encoding type 2 protein is accession number NM_001048171.1.
Site-Directed Mutagenesis
The 47 MUTYH variants comprised 46 missense variants and one 3-bp in-frame deletion (p.E480del) and were constructed via site-directed mutagenesis as described previously [Shimodaira et al., 1998] (Table1). LOVD provided information for the majority of MUTYH variants. DNA sequencing confirmed the specific mutation of each MUTYH gene. These MUTYH variants included 47 germ-line variants found in patients with MAP (or suspected MAP) and/or normal populations. DNA variant numbering was based on the cDNA reference sequence by assigning nucleotide +1 to A of the ATG translation initiation codon.
Table 1.
Summary of Functional Assay in Present Study and In Silico Prediction, Functional Assay in Previous Studies, Clinical Information
| Variations | Nucleotide changea | Functional assay (fold to wild type)b | Protein levelc | SIFT (score)d | Polyphen-2 (score)e | Glycosylase assay | Previous rifampicin assay | Detected population (frequency of report) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| p.P18L | c.53C>T | 26.0 | (2.7) | 0.96 | T | (0.50) | B | (0.028) | NE | NE | FAP (1), MP (5), sporadic CRC(6), LC (1), HC (3) |
| p.V22M | c.64G>A | 12.0 | (1.2) | 1.00 | A | (0.00) | B | (0.185) | Proficient | NE | FAP, sporadic CRC, HC (allele frequency = 13%) |
| p.G25D | c.74G>A | 5.00 | (0.5) | 0.82 | A | (0.43) | B | (0.335) | NE | NE | FAP (1), MP (5), sporadic CRC(6), LC (1), HC (3) |
| p.W103R | c.265A>G | 47.0 | (4.9) | 0.89 | A | (0.00) | D (++) | (1.000) | NE | NE | Sporadic HCC (4), HC |
| p.D105N | c.271C>T | 8.67 | (0.9) | 0.84 | A | (0.00) | D (++) | (0.802) | NE | NE | MP (1), sporadic CRC (1) |
| p.Y128H | c.340T>C | 34.7 | (3.6) | 1.17 | A | (0.00) | D (++) | (1.000) | NE | NE | AFAP (1) |
| p.W131R | c.349T>A | 75.3 | (7.8) | 1.22 | A | (0.00) | D (++) | (1.000) | NE | NE | FAP (1) |
| p.P157L | c.428G>A | 31.3 | (3.2) | 0.83 | A | (0.00) | D (++) | (1.000) | NE | NE | AFAP (1) |
| p.Y179C | c.494A>G | 46.7 | (4.8) | 1.17 | A | (0.00) | D (++) | (1.000) | Deficient | Deficient | Common variant in FAP, AFAP |
| p.R182C | c.502G>A | 85.3 | (8.8) | 0.67 | A | (0.00) | D (++) | (1.000) | Deficient | NE | MP (1), sporadic CRC (1) |
| p.R182H | c.503G>A | 63.0 | (6.5) | 1.31 | A | (0.00) | D (++) | (1.000) | NE | NE | AFAP (4) |
| p.R185Q | c.512C>T | 50.7 | (5.2) | 1.10 | T | (0.13) | D (++) | (0.977) | NE | NE | AFAP (1) |
| p.G189E | c.524C>T | 24.5 | (2.5) | 0.77 | A | (0.00) | D (++) | (1.000) | NE | NE | AFAP (1) |
| p.I223V | c.625A>G | 11.0 | (1.1) | 0.71 | T | (0.06) | D (++) | (0.958) | Partially active | NE | FAP (1), MP (1) |
| p.A227V | c.638C>T | 17.5 | (1.8) | 1.08 | A | (0.01) | D (++) | (0.999) | NE | NE | HC (1) |
| p.V234M | c.658G>A | 22.3 | (2.3) | 0.73 | A | (0.01) | D (+) | (0.815) | NE | NE | Familial CRC (1) |
| p.R241W | c.679C>T | 40.7 | (4.2) | 0.76 | A | (0.00) | D (++) | (1.000) | Deficient | Deficient | Sporadic CRC or HC (1) |
| p.R245C | c.691C>T | 40.7 | (4.2) | 0.91 | A | (0.00) | D (++) | (1.000) | NE | NE | MP (2) |
| p.R245H | c.692G>A | 69.5 | (7.0) | 0.58 | A | (0.00) | D (++) | (1.000) | Deficient | NE | FAP (5), AFAP (1) |
| p.V246F | c.694G>T | 11.7 | (1.2) | 1.19 | A | (0.02) | B | (0.143) | Deficient | Slightly deficient | FAP (3), sporadic CRC or HC (1) |
| p.R274W | c.778C>T | 55.0 | (5.7) | 0.55 | A | (0.00) | D (++) | (0.999) | NE | NE | FAP (1), AFAP (1) |
| p.G286E | c.815G>A | 5.00 | (0.5) | 1.24 | A | (0.00) | D (++) | (1.000) | Deficient | NE | FAP (1), MP (1) |
| p.C290W | c.828T>G | 51.0 | (5.3) | 0.87 | A | (0.00) | D (++) | (1.000) | NE | NE | Sporadic LC (1), HC (1) |
| p.P295L | c.842C>T | 55.5 | (5.7) | 0.42 | A | (0.00) | D (++) | (1.000) | Deficient | NE | FAP or AFAP (7), MP (3) |
| p.R309C | c.883C>T | 27.7 | (2.9) | 0.52 | T | (0.11) | B | (0.012) | Proficient | NE | AFAP (2), MP (2) |
| p.Q338H | c.972G>C | 6.67 | (0.7) | 0.83 | T | (0.14) | B | (0.343) | Proficient | Proficient | FAP, sporadic CRC, HC (allele frequency = 30%) |
| p.Q338R | c.971T>C | 12.5 | (1.3) | 0.93 | T | (0.52) | B | (0.039) | NE | NE | HNPCC (-like) family (1) |
| p.P380T | c.1096G>T | 18.0 | (1.9) | 0.64 | T | (0.59) | B | (0.004) | NE | NE | FAP (1) |
| p.L388P | c.1121A>G | 69.5 | (7.2) | 0.40 | A | (0.00) | D (++) | (1.000) | Deficient | NE | AFAP (1), MP (2) |
| p.G396D | c.1145G>A | 16.3 | (1.7) | 1.30 | A | (0.00) | D (++) | (1.000) | Partially active | Deficient | Common variant in FAP, AFAP |
| p.P405L | c.1172G>A | 101 | (10.4) | 0.96 | A | (0.00) | D (++) | (1.000) | Deficient | Deficient | FAP (9), AFAP (10), MP (5), CRC (4) |
| p.P405S | c.1172G>A | 32.3 | (3.3) | 0.71 | A | (0.00) | D (++) | (0.999) | NE | NE | GC (1) |
| p.Q414R | c.1199A>G | 12.0 | (1.2) | 1.31 | T | (0.27) | B | (0.000) | NE | NE | GC (1) |
| p.L420M | c.1216G>T | 7.00 | (0.7) | 0.91 | T | (0.14) | B | (0.180) | NE | NE | FAP (1), MP (1), HNPCC (-like) family (1) |
| p.R426C | c.1234C>T | 10.7 | (1.1) | 0.72 | T | (0.05) | B | (0.000) | NE | NE | FAP (1), FAP or AFAP (1), HC (1) |
| p.R437P | c.1268C>G | 21.5 | (2.2) | 0.57 | T | (0.08) | B | (0.445) | NE | NE | Sporadic CRC (1), sporadic CRC or HC (1) |
| p.R437Q | c.1268C>T | 10.0 | (1.0) | 1.51 | T | (1.00) | B | (0.001) | NE | NE | Sporadic CRC (1), sporadic CRC or HC (1) |
| p.A473D | c.1378C>A | 54.0 | (5.6) | 0.93 | T | (0.20) | D (++) | (0.863) | Deficient | NE | FAP (2) |
| p.A473T | c.1375G>A | 12.0 | (1.2) | 0.56 | T | (0.44) | B | (0.139) | NE | NE | MP (1) |
| p.A489T | c.1423G>A | 32.0 | (3.3) | 1.07 | A | (0.01) | D (++) | (0.973) | NE | NE | AFAP (1) |
| p.V493F | c.1435G>T | 37.0 | (3.8) | 1.26 | A | (0.00) | D (+) | (0.73) | NE | NE | FAP or AFAP (1), sporadic CRC (2), HC (1) |
| p.G503E | c.1466G>A | 18.5 | (1.9) | 0.67 | T | (0.08) | B | (0.404) | NE | NE | HC (dbSNP) |
| p.S515F | c.1502C>T | 9.67 | (1.0) | 1.32 | A | (0.05) | B | (0.003) | Proficient | NE | FAP, sporadic CRC, HC (allele frequency = 3%) |
| p.P516L | c.1505C>T | 10.0 | (1.0) | 1.20 | T | (0.47) | B | (0.017) | NE | NE | HC (1) |
| p.L529M | c.1543C>A | 5.50 | (0.6) | 1.10 | T | (0.11) | D (++) | (0.915) | NE | NE | HC (dbSNP) |
| p.R534Q | c.1559G>A | 6.00 | (0.6) | 1.15 | T | (0.33) | B | (0.032) | NE | NE | Sporadic CRC (1), LC or HC (1) |
| p.E480del | c.1395_1397delGGA | 40.5 | (4.2) | 0.84 | NA | NA | Deficient | NE | FAP (6), AFAP, sporadic CRC | ||
The reference sequence for the MUTYH gene-encoding type 2 protein is accession number NM_001048171.1. For cDNA numbering: nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1.
Indicated numbers are rifampicin-resistant rate (/108) in MutY-deficient E. coli.
Indicated numbers are band intensity of western blotting normalized by actin control.
Prediction by SIFT algorism.
Prediction by Polyphen-2 algorism.
T, tolerated; A, affect protein function; NA, not analyzed; NE, not examined; FAP, familial adenomatous polyposis; B, benign, D(+), possibly damaging, D(++), probably damaging; AFAP, attenuated FAP; MP, multiple polyps; CRC, colorectal cancer; GC, gastric cancer; LC, lung cancer; HCC, hepatocellular carcinoma; HC, healthy control; dbSNP, data base of single-nucleotide polymorphism.
Fluctuation Test
Nine independent overnight cultures were analyzed for rifampicin-resistant (RifR) mutations as previously described [Takao et al., 1999]. Aliquots of the cultures were spread on LB agar containing ampicillin (50 μg/ml) and rifampicin (100 μg/ml). The plates were then incubated at 37°C for 24 hr to determine the frequency of RifR mutation.
Western Blot Analysis
HCT 116 human colon cancer cells were cultured in RPMI-1640 with 10% fetal bovine serum at 37°C in a 5% CO2 atmosphere. HCT116 cells were transfected with each Flag-tagged MUTYH expression plasmid by using X-treme GENE HP DNA Transfection Reagent (Roche, Mannheim, Germany). Cell lysates were loaded onto 10% sodium dodecyl sulfate polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA). Western blot analyses were performed using an anti-FLAG antibody (F3165; Sigma–Aldrich, St. Louis, MO), anti-MUTYH antibody (H00004595-M01; Abnova, Taipei City, Taiwan), and anti-β-actin antibody (A5316, Sigma–Aldrich), and signals were detected on an Odyssey Infrared Imaging system (Li-COR, Lincoln, NE). Bands of MUTYH and ACTB were quantified by densitometry.
Subcellular Localization
HCT116 cells were cultured in the same condition as above. The cells were transfected with each EGFP-fusion MUTYH expression plasmid by the same method as above. An enhanced green fluorescent protein (EGFP)-encoding sequence was added to the N-terminus of the MUTYH gene, and pEGFP-N3 (Clontech Laboratories, Mountain View, CA) was used as a negative control. HCT116 cells transfected with EGFP-fusion plasmids were mounted and then examined under a fluorescent microscope with the appropriate filters. DAPI was used for nuclear staining.
Predictions of Protein Activity and Structure
The Sorting Intolerant From Tolerant (SIFT) algorithm (SIFT Human Protein; http://sift.jcvi.org) and PolyPhen-2 (version2.2.2; http://genetics.bwh.harvard.edu/pph2) were incorporated to predict the effects of amino acid substitutions on protein functions [Ng and Henikoff, 2006; Hicks et al., 2011]. Based on the crystal structure of the B. stearothermophilus MutY [Fromme et al., 2004; Manuel et al., 2004], the MUTYH structure was predicted via a homology modeling method [Cardozo et al., 1995] using ICM-Molsoft software (Molsoft L.L.C., La Jolla, CA).
Results
BER Detection with a MutY-Deficient E. coli Complementation Assay
Because human MUTYH shares close homology with E. coli MutY, human MUTYH is known to complement the deficiency of MutY-deficient E. coli. To analyze the functional significance of MUTYH variants, we monitored this complementation ability according to the frequency of rifampicin-resistant mutation. Exogenous expression of human MUTYH protein in E. coli CC104mutY suppressed the mutation rate (9.67/108) relative to the expression of CC104mutY with an empty vector (137/108) (Fig. 1B). This result confirmed that human MUTYH complemented the BER activity of MutY-deficient E. coli.
The BER activities of the 47 MUTYH variants expressed in MutY-deficient E. coli CC104mutY were then evaluated (Table1). The mutation rates of p.Y179C and p.G396D, two common variants found in Caucasian patients with MAP, were 4.8- and 1.7-fold, respectively. Mutation rates of p.V22M, p.Q338H, and p.S515F, three common variants found in normal populations, were 1.2-, 0.7-, and 1.0-fold, respectively. Based on the mutation rates of these five variants with relatively clear clinical information, we set two cut-off lines at 1.7- and 4.8-fold to determine the function of each variant. Relative to the wild type, we categorized variant that had less than 1.7-fold higher mutation rate as functionally retained, more than or equal to 1.7-fold, and less than 4.8-fold as partially defective, and more than or equal to 4.8-fold as defective. According to this categorization, 17 variants (p.V22M, p.G25D, p.D105N, p.I223V, p.V246F, p.G286E, p.Q338H, p.Q338R, p.Q414R, p.L420M, p.R426C, p.R437Q, p.A473T, p.S515F, p.P516L, p.L529M, and p.R534Q) were functionally retained, 17 variants (p.P18L, p.Y128H, p.P157L, p.G189E, p.A227V, p.V234M, p.R241W, p.R245C, p.R309C, p.P380T, p.G396D, p.P405S, p.R437P, p.A489T, p.V493F, p.G503E, and p.E480del) were partially defective, and 13 variants (p.W103R, p.W131R, p.Y179C, p.R182C, p.R182H, p.R185Q, p.R245H, p.R274W, p.C290W, p.P295L, p.L388P, p.P405L, and p.A473D) were functionally defective (Fig. 1B; Table1).
Protein Expression Levels Among the MUTYH Variants
To evaluate the stability of exogenously expressed MUTYH proteins, FLAG-tagged versions of the wild-type and 47 variants were transiently expressed in HCT116 colon cancer cells. MUTYH proteins were detected by western blot analysis, and the loaded protein amounts were normalized according to the β-actin level. There were minimal differences among the variants with regard to normalized MUTYH protein levels. Representative results of the 10 variants are shown in Figure 2.
Figure 2.

Western blot analyses of 10 representative MUTYH variants. A FLAG sequence was added at the N-terminus of MUTYH gene. Expressed protein in HCT116 cells detected by anti-Flag antibody. The ratios of the variants to the wild type are shown as the relative MUTYH protein level.
Subcellular Localization of the MUTYH Variants in Colon Cancer Cells
To analyze the effects of variants on the subcellular localization of MUTYH protein, we observed the distribution of EGFP-fusion MUTYH protein transiently expressed in HCT116 cells (Fig. 3). We used the nuclear isoform 4 among MUTYH-splicing variants to detect variants that disturbed the nuclear distribution and the BER ability in the nucleus. Wild-type MUTYH localized in the nuclei, whereas EGFP alone was distributed throughout the cells. With the expression of all 47 MUTYH variants, we detected EGFP signals at a similar intensity as that observed with wild-type MUTYH. EGFP-fused MUTYH variants localized mainly in the nuclei and were not distributed as widely throughout the cell as EGFP alone.
Figure 3.
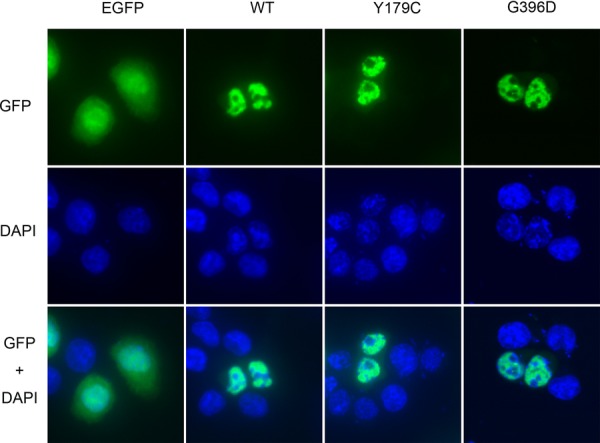
Subcellular localization of wild-type MUTYH and two representative variant MUTYH proteins. An EGFP sequence was added at the N-terminus of MUTYH gene, and pEGFP-N3 was used as a negative control. Localization of the transiently expressed protein in HCT116 cells was determined by fluorescence with GFP. Nuclei stained by DAPI and overlay image of GFP and DAPI are shown.
Relationship Between the Function and Structure of MUTYH
To elucidate the function–structure relationships associated with MUTYH variants, we first compared the BER activities with the functional predictions generated by the SIFT or Polyphen-2 programs. SIFT predicts the effects of amino acid substitutions on protein function, based on sequence conservation during evolution and the natures of amino acids substituted in a gene of interest [Ng and Henikoff, 2006]. As shown in Table1, among the 17 variants determined to be functionally retained variants, 11 were tolerant, and six were sorted as affected by SIFT. Furthermore, 13 variants were sorted as benign and four were sorted as damaged by Polyphen-2. Among the 13 variants determined to be functionally defective, two were sorted as tolerant and 11 were sorted as affected by SIFT. Polyphen-2 sorted all 13 of these variants as damaged.
We then focused on the relationships between MUTYH functions and the secondary structures. As shown in Figure 1A, mapping of the 47 MUTYH variants on MUTYH cDNA indicated that the majority of functionally defective MUTYH variants were located within the homologous region shared with E. coli MutY, whereas functionally retained variants were distributed throughout the whole gene. Functionally defective variants were located in the two functional domains—the N-terminal catalytic domain, including the FCL motif, and the C-terminal MutT-like domain.
Finally, we constructed a predicted three-dimensional structural model of the MUTYH protein, using a homology modeling method to map variants in the predicted structure (Fig. 4A and B). Most of the variants that were predicted to be situated around the DNA-binding site and the [4Fe-4S] clustering pocket were found to be functionally defective variants (Fig. 4C).
Figure 4.
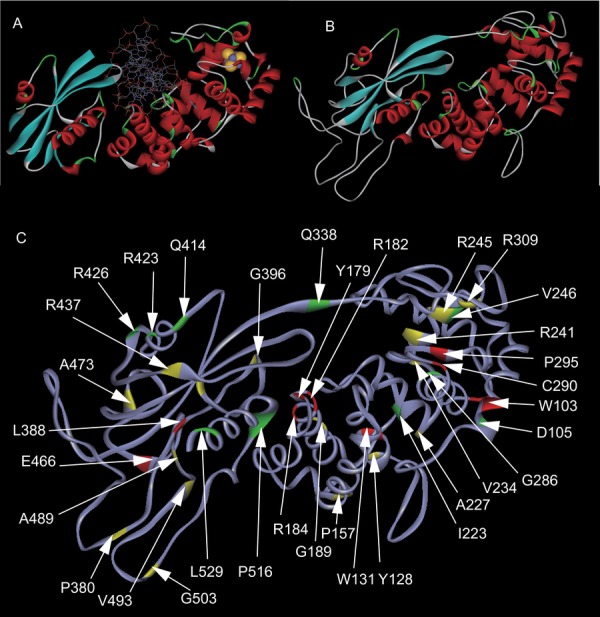
Mapping of MUTYH variants on protein structure. A: Structure of B. stearothermophilus MutY bound to DNA. The iron and sulfur molecules in the [4Fe-4S] cluster are shown as yellow and gray spheres, respectively. B: Structure of MUTYH simulated by homology modeling. C: Mapping of amino acid residues examined in this study and functional information. Red sites show the residues with defective substitution. Yellow sites show the residues with partially defective substitution. Green sites show the residues with retained substitutions. p.R245, p.P405, p.R437, and p.A473 are also highlighted in yellow, as the residues with multiple substitutions that are defective and retained.
Discussion
In this study, we evaluated the functional significance of 47 MUTYH variants by an analysis of E. coli complementation, protein stability, and subcellular localization in mammalian cells to enhance our understanding of the pathogenic effects in MAP.
Several functional assays were performed using a few different methods, of which the most frequently used is the in vitro glycosylase assay. This assay evaluated several variants to be functionally defective (p.Y179C, p.R182H, p.R241W, p.R245H, p.R245L, p.V246F, p.G286E, p.P295L, p.L388P, p.P405L, p.A473D, and p.E480del), partially defective (p.I223V, p.M283V, p.R274Q, and p.G396D), or retained (p.V22M, p.V75E, p.R185W, p.R309C, p.A373V, p.Q338H, and p.S515F) [Wooden et al., 2004; Bai et al., 2005, 2007; Ali et al., 2008; Kundu et al., 2009; Goto et al., 2010]. Our data were consistent with those previously published data for almost all variants, with the exceptions of p.V246F and p.G286E; these were evaluated as retained in our study, but defective by the in vitro study. p.V246F has been shown to be defective in terms of in vitro glycosylase activity, but only slightly defective in the E. coli complementation assay performed in the same study. This suggests that the discrepancy could be attributed to the partial activities assessed by the different methods. The reason for the discrepancy in p.G286E is uncertain, because both an in vitro glycosylase assay and analysis of a knock-in mouse harboring the equivalent variant have shown that this variant is defective; however, it was found to be retained in our study with the clear low mutation rate (0.5-fold) other than border-line
Eight variants (p.Y179C, p.R241W, p.R245L, p.V246F, p.Q338H, p.Q338R, p.G396D, and p.P405L) were examined in previous studies using a different E. coli complementation method than that used in our study [Shinmura et al., 2000; Bai et al., 2005; Bai et al., 2007; Kundu et al., 2009]. We assayed the variants according to complementation ability in MutY-deficient E. coli; in contrast, all the previous studies used MutY and MutM double-deficient E. coli to enhance the mutation rates. Among the eight previously examined variants, seven were assayed in the present study. Both studies found p.Y179C, p.R241W, p.G396D, and p.P405L to be defective and p.Q338H and p.Q338R to be retained. However, p.V246F was evaluated as retained in the present study but defective or slightly defective in the previous studies. These discrepancies are possibly due to the different E. coli backgrounds. Nevertheless, our functional evaluations are consistent with the previous studies for the majority of variants. This corroboration suggests that the functional evaluation for variants newly analyzed in the present study was reliable.
We analyzed a large number of MUTYH variants to comprehensively estimate the pathogenesis of each variant in a simple E. coli assay as a stand-alone method. However, the clinical application of our results might be limited by the difficulty in establishing an appropriate cut-off value, as we could not estimate how intermediate or subtle functional defects would contribute to the pathogenesis of MAP. p.Y179C and p.G396D have the highest incidence among Caucasian patients with MAP. In our assay, p.Y179C exhibited a loss of function, whereas p.G396D demonstrated only a 1.7-fold higher mutation rate relative to the wild type. Several functional assays have shown that p.Y179C induces a more severe functional defect than p.G396D does [Parker et al., 2005; Ali et al., 2008; Kundu et al., 2009]. Recently, the phenotype of patients with MAP harboring a biallelic p.G396D variation was found to be less severe than that of patients harboring a biallelic p.Y179C variation in terms of the hazard ratio for CRC development or the age at the diagnosis [Nielsen et al., 2009]. These genotype–phenotype correlations support our hypothesis that a comprehensive functional assay of a larger number of variants will provide insight into the clinical features, such as the relative risk of CRC or the age of onset. The most common polymorphism is p.Q338H, which has been reported in various countries, although the allelic frequency varies from 4.8% to 64% for a heterozygous allele [Ali et al., 2008]. According to the HapMap project, three common variants have been found in normal populations: p.Q338H (allelic frequency, 30%), p.V22M (13%), and p.S515F (3%). According to our data, p.Q338H, p.V22M, and p.S515F retained similar BER ability to wild type in E. coli. Considering the mutation rates of these five variants (p.Y179C, p.G396D, p.Q338H, p.V22M, and p.S515F), we set the two cut-off lines at 1.7- and 4.8-fold, respectively. These cut-off values were tentatively set for convenience to analyze the data. The appropriate method of categorization should be investigated by further research.
Clinically, the pathogenesis of two common variants (p.Y179C and p.G396D) in MAP is certain, whereas the pathogenesis of three putative polymorphisms (p.V22M, p.Q338H, and p.S515F) is less likely. However, rare variants are difficult to interpret from clinical data. Although biallelic variations predispose patients to the development of polyps and CRC (genotype relative risk: GRR of 117), monoallelic variation carriers have only a slightly increased risk of CRC (GRR of 1.27) [Tenesa et al., 2006]. Accordingly, we could not conclude that monoallelic variations found in a normal population are not pathogenic. Hence, clinical data from monoallelic mutation carriers are not suitable for comparison with functional data. By examining the LOVD database or literature concerning the MUTYH variants, rare variants were categorized into two groups: those reported in patients with only familial adenomatous polyposis, attenuated familial adenomatous polyposis, or multiple polyps; and those reported in healthy controls or patients with sporadic colon cancer (Table1). p.Y128H, p.G189E, p.R245C, p.R245H, p.G286E, p.P295L, p.P405C, p.A473D, and p.V493F were identified in the former group and detected in homozygous states. Our functional assay revealed eight of the nine above-mentioned variants except p.G286E to be defective. Therefore, our functional assays tended to correlate with the clinical features. However, further analysis will be required to understand the significance of these variants. We cannot exclude the possibility that the variant might still be associated with sporadic carcinogenesis, even though a high BER activity will likely not contribute to the pathogenesis of MAP. Even the most common putative polymorphism p.Q338H has been shown to be possibly involved in sporadic colorectal carcinogenesis in a population-based analysis [Picelli et al., 2010]. It is assumed that the reduced ability of this MUTYH variant is caused by a reduced binding capacity for the RAD9–RAD1–HUS1 complex, because the mutation at Q338 residue occurs in the interconnecting domain involved in the interactions with the HUS1 component of that complex [Turco et al., 2013]. Applications of functional information with genetic and clinical features such as the individual incidence of cancer will provide useful information regarding the validity of these functional analyses. Segregation analysis that is usually useful is unsuitable because of the low penetrance of monoallelic MUTYH variations.
Amino acid substitutions in MUTYH are expected to affect both the expression levels and functions of protein. However, our data showed that each variant did not lead to a significant difference in the amount of protein expression in the HCT116 cell lines. Therefore, there was no clear correlation between the MUTYH protein level and BER activity (Table1). A previous study has reported lower levels of endogenous MUTYH in cell lines harboring biallelic p.Y179C/p.G396D variants established from patients with MAP [Parker et al., 2005]. In another study, exogenous MUTYH protein variants in MutY knock-out MEFs were expressed at various levels [Molatore et al., 2010]. It is possible that endogenous protein levels differ among variants and the host cell influences that protein stability. Our results indicate that the variants exert slight effects on protein stability, and the effect of protein stability could almost be excluded from the evaluation of our functional data.
Subcellular localization of MUTYH differs from that of the isoforms generated by alternative splicing [Takao et al., 1998]. Isoforms 2 and 4 are the two major well-described isoforms [Takao et al., 1999]. Isoform 2, which features a mitochondrial targeting signal in the N-terminus, is predominantly located in the mitochondria. Isoform 4 has a nuclear localization signal in the N- and C-termini and is the most abundant nuclear isoform. Our results revealed that variants of MUTYH had no significant effects on the subcellular localization of isoform 4. Although a putative nuclear localization signal was shown to be located in C-terminal 55 residues, five variants (p.G503E, p.S515F, p.P516L, p.L526M, and p.R534Q) that affected this domain did not impair nuclear distribution. Our data suggest that the main reason for the functional defects caused by MUTYH variants is not altered distribution, but rather the loss of protein function itself.
According to the alignment, human MUTYH shares a well-conserved central region with E. coli MutY and features extended segments toward both the N- and C-termini [David et al., 2007]. The exclusive distribution of the functionally defective MUTYH variants in the central region suggests that this homologous region is important for protein function. Mapping of the secondary structure of MUTYH showed that the functionally defective variants were distributed widely in the two functional domains (i.e., the N-terminal catalytic domain and the C-terminal MutT-like domain) without a clear hot spot (Fig. 1A). In a previous study, the homologous structure of the B. stearothermophilus MutY provided a molecular basis for the functional defects of human MUTYH missense variants [Fromme et al., 2004]. p.W131R, p.Y179C, p.R182C, p.R182H, and p.R185Q can disrupt DNA-binding ability because these are near the equivalent site of the oxo-G or adenine-recognition surface of B. stearothermophilus MutY. p.C290W and p.P295L are in the FCL motif, and p.R241W resides in the equivalent site of the interface of [4F-4S] cluster. Our mapping of variants on the predicted MUTYH structure based on homology modeling indicated that functionally deficient variants surrounded the [4F-4S] cluster. This cluster is well conserved among DNA glycosylases such as E. coli endo III and is believed to play a structural role by searching damaged DNA for base removal activity [Boal et al., 2007]. Our homology modeling provides a structural basis for the functional deficiencies with some variants.
These sets of functional data also allow us to compare the properties of SIFT and PolyPhen-2 predictions. By comparing the BER complementation assays with these algorithms, we could evaluate the abilities of these programs to make functional predictions. Both SIFT and PolyPhen-2 were highly predictive of BER activity in MUTYH. Among the 17 variants deemed as retained and 13 deemed defective, consistent predictions using our functional assay were made for 22 variants (73.3%) with SIFT and 26 variants (86.7%) with Polyphen-2. Based on the data from our functional assay, the false-positive and false-negative rates were 15.4% and 35.3% for SIFT, respectively, and 0% and 23.5% for Polyphen-2, respectively. Our data indicate that the accuracy of in silico predictions can exceed 70%. This high level of accuracy was also supported by our previous study, which compared functional assay data with the SIFT predictions of 101 variants of the mismatch repair gene MLH1 [Takahashi et al., 2007].
In summary, we examined 47 MUTYH variants in functional complementation assay with MutY-disrupted E. coli, and characterized the functional alterations of MUTYH variants. We confirmed that the majority of functionally inactive MUTYH variants were located around the DNA-binding domain and [4Fe-4S] cluster. The results described herein can be applied to evaluate the risk of cancer in individuals or families harboring MUTYH variants, and might provide insight into the functions of MUTYH.
References
- Ali M, Kim H, Cleary S, Cupples C, Gallinger S, Bristow R. Characterization of mutant MUTYH proteins associated with familial colorectal cancer. Gastroenterology. 2008;135:499–507. doi: 10.1053/j.gastro.2008.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, Grist S, Gardner J, Suthers G, Wilson TM, Lu AL. Functional characterization of human MutY homolog (hMYH) missense mutation (R231L) that is linked with hMYH-associated polyposis. Cancer Lett. 2007;250:74–81. doi: 10.1016/j.canlet.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, Jones S, Guan X, Wilson TM, Sampson JR, Cheadle JP, Lu AL. Functional characterization of two human MutY homolog (hMYH) missense mutations (R227W and V232F) that lie within the putative hMSH6 binding domain and are associated with hMYH polyposis. Nucleic Acids Res. 2005;33:597–604. doi: 10.1093/nar/gki209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boal AK, Yavin E, Barton JK. DNA repair glycosylases with a [4Fe-4S] cluster: a redox cofactor for DNA-mediated charge transport? J Inorg Biochem. 2007;101:1913–1921. doi: 10.1016/j.jinorgbio.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo T, Totrov M, Abagyan R. Homology modeling by the ICM method. Proteins. 1995;23:403–414. doi: 10.1002/prot.340230314. [DOI] [PubMed] [Google Scholar]
- D’Agostino VG, Minoprio A, Torreri P, Marioni I, Bossa C, Petrucci TC, Albertini AM, Ranzani GN, Bignami M, Mazzei F. Functional analysis of MUTYH mutated proteins associated with famillial adenomatous polyposis. DNA Repair. 2010;9:700–707. doi: 10.1016/j.dnarep.2010.03.008. [DOI] [PubMed] [Google Scholar]
- David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652–656. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- Gismondi V, Meta M, Bonelli L, Radice P, Sala P, Bertario L, Viel A, Fornasarig M, Arrigoni A, Gentile M, Ponz de Leon M, Anselmi L, Mareni C, Bruzzi P, Varesco L. Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer. 2004;109:680–684. doi: 10.1002/ijc.20054. [DOI] [PubMed] [Google Scholar]
- Goto M, Shinmura K, Nakabeppu Y, Tao H, Yamada H, Tsuneyoshi T, Sugimura H. Adenine DNA glycosylase activity of 14 human MutY homolog (MUTYH) variant proteins found in patients with colorectal polyposis and cancer. Hum Mutat. 2010;31:E1861–E1874. doi: 10.1002/humu.21363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat Struct Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mutat. 2011;32:661–668. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Brinkmeyer MK, Livingston AL, David SS. Adenine removal activity and bacterial complementation with the human MutY homologue (MUTYH) and Y165C, G382D, P391L and Q324R variants associated with colorectal cancer. DNA Repair (Amst) 2009;8:1400–1410. doi: 10.1016/j.dnarep.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukianova OA, David SS. A role for iron-sulfur clusters in DNA repair. Curr Opin Chem Biol. 2005;9:145–151. doi: 10.1016/j.cbpa.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Manuel RC, Hitomi K, Arvai AS, House PG, Kurtz AJ, Dodson ML, McCullough AK, Tainer JA, Lloyd RS. Reaction intermediates in the catalytic mechanism of Escherichia coli MutY DNA glycosylase. J Biol Chem. 2004;279:46930–46939. doi: 10.1074/jbc.M403944200. [DOI] [PubMed] [Google Scholar]
- Molatore S, Russo MT, D’Agostino VG, Barone F, Matsumoto Y, Albertini AM, Minoprio A, Degan P, Mazzei F, Bignami M, Ranzani GN. MUTYH mutations associated with familial adenomatous polyposis: functional characterization by a mammalian cell-based assay. Hum Mutat. 2010;31:159–166. doi: 10.1002/humu.21158. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Franken PF, Reinards TH, Weiss MM, Wagner A, van der Klift H, Kloosterman S, Houwing-Duistermaat JJ, Aalfs CM, Ausems MG, Brocker-Vriends AH, Gomez Garcia EB, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP) J Med Genet. 2005;42:e54. doi: 10.1136/jmg.2005.033217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Joerink-van de Beld MC, Jones N, Vogt S, Tops CM, Vasen HF, Sampson JR, Aretz S, Hes FJ. Analysis of MUTYH genotypes and colorectal phenotypes in patients With MUTYH-associated polyposis. Gastroenterology. 2009;136:471–476. doi: 10.1053/j.gastro.2008.10.056. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Morreau H, Vasen HF, Hes FJ. MUTYH-associated polyposis (MAP) Crit Rev Oncol Hematol. 2010;79:1–16. doi: 10.1016/j.critrevonc.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Noll DM, Gogos A, Granek JA, Clarke ND. The C-terminal domain of the adenine-DNA glycosylase MutY confers specificity for 8-oxoguanine.adenine mispairs and may have evolved from MutT, an 8-oxo-dGTPase. Biochemistry. 1999;38:6374–6379. doi: 10.1021/bi990335x. [DOI] [PubMed] [Google Scholar]
- Out AA, Tops CM, Nielsen M, Weiss MM, van Minderhout IJ, Fokkema IF, Buisine MP, Claes K, Colas C, Fodde R, Fostria F, Franken PF, et al. Leiden Open Variation Database of the MUTYH gene. Hum Mutat. 2010;31:1205–1215. doi: 10.1002/humu.21343. [DOI] [PubMed] [Google Scholar]
- Parker AR, Sieber OM, Shi C, Hua L, Takao M, Tomlinson IP, Eshleman JR. Cells with pathogenic biallelic mutations in the human MUTYH gene are defective in DNA damage binding and repair. Carcinogenesis. 2005;26:2010–2018. doi: 10.1093/carcin/bgi166. [DOI] [PubMed] [Google Scholar]
- Picelli S, Zajac P, Zhou XL, Edler D, Lenander C, Dalen J, Hjern F, Lundqvist N, Lindforss U, Pahlman L, Smedh K, Törnqvist A, et al. Common variants in human CRC genes as low-risk alleles. Eur J Cancer. 2010;46:1041–1048. doi: 10.1016/j.ejca.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Filosi N, Shibata H, Suzuki T, Radice P, Kanamaru R, Friend SH, Kolodner RD, Ishioka C. Functional analysis of human MLH1 mutations in Saccharomyces cerevisiae. Nat Genet. 1998;19:384–389. doi: 10.1038/1277. [DOI] [PubMed] [Google Scholar]
- Shinmura K, Yamaguchi S, Saitoh T, Takeuchi-Sasaki M, Kim SR, Nohmi T, Yokota J. Adenine excisional repair function of MYH protein on the adenine:8-hydroxyguanine base pair in double-stranded DNA. Nucleic Acids Res. 2000;28:4912–4918. doi: 10.1093/nar/28.24.4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Shimodaira H, Andreutti-Zaugg C, Iggo R, Kolodner RD, Ishioka C. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer Res. 2007;67:4595–4604. doi: 10.1158/0008-5472.CAN-06-3509. [DOI] [PubMed] [Google Scholar]
- Takao M, Aburatani H, Kobayashi K, Yasui A. Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res. 1998;26:2917–2922. doi: 10.1093/nar/26.12.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao M, Zhang QM, Yonei S, Yasui A. Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res. 1999;27:3638–3644. doi: 10.1093/nar/27.18.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenesa A, Campbell H, Barnetson R, Porteous M, Dunlop M, Farrington SM. Association of MUTYH and colorectal cancer. Br J Cancer. 2006;95:239–242. doi: 10.1038/sj.bjc.6603239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turco E, Ventura I, Minoprio A, Russo MT, Torreri P, Degan P, Molatore S, Ranzani GN, Bignami M, Mazzei F. Understanding the role of the Q338H MUTYH variant in oxidative damage repair. Nucleic Acids Res. 2013;41:4093–4103. doi: 10.1093/nar/gkt130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooden SH, Bassett HM, Wood TG, McCullough AK. Identification of critical residues required for the mutation avoidance function of human MutY (hMYH) and implications in colorectal cancer. Cancer Lett. 2004;205:89–95. doi: 10.1016/j.canlet.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Yanaru-Fujisawa R, Matsumoto T, Ushijima Y, Esaki M, Hirahashi M, Gushima M, Yao T, Nakabeppu Y, Iida M. Genomic and functional analyses of MUTYH in Japanese patients with adenomatous polyposis. Clin Genet. 2008;73:545–553. doi: 10.1111/j.1399-0004.2008.00998.x. [DOI] [PubMed] [Google Scholar]