

Abstract
Purpose
Type 2 diabetes is a risk factor for meibomian gland dysfunction (MGD). We hypothesize that this diabetic impact is due, at least in part, to the effects of insulin resistance/deficiency and hyperglycemia on human meibomian gland epithelial cells (HMGECs). To begin to test this hypothesis, we examined whether insulin and high glucose influence immortalized (I) HMGECs.
Methods
Immortalized HMGECs were cultured in serum-containing or -free media and treated with insulin, insulin-like growth factor–1 (IGF-1), IGF-1 receptor (R) blocking antibody, and glucose or mannitol for varying time periods. Specific proteins were detected by Western blots, cell proliferation was evaluated by manual cell counting and lipids were assessed with LipidTOX and high performance thin layer chromatography.
Results
We found that insulin induces a dose-dependent increase in phosphatidylinositide 3-kinase/Akt (AKT) signaling in IHMGECs. This effect involves the IGF-1R, but not the insulin receptor (IR), and is associated with a stimulation of cell proliferation and neutral lipid accumulation. In contrast, high glucose exposure alters cell morphology, causes a progressive cell loss, and significantly reduces the levels of IGF-1R, phospho (p)-AKT, Foxhead box protein O1 (FOXO1), and sterol-regulatory element binding protein (SREBP-1) in IHMGECs.
Conclusions
Our data show that insulin stimulates, and that high glucose is toxic for, IHMGECs. These results support our hypothesis that insulin resistance/deficiency and hyperglycemia are deleterious for HMGECs and may help explain why type II diabetes is a risk factor for MGD.
Keywords: insulin, glucose, meibomian gland, epithelial cell, diabetes, dry eye disease
Meibomian glands play an extremely important role in the health and well-being of the ocular surface. The acinar epithelial cells of these glands secrete a proteinaceous lipid mixture (i.e., meibum) that promotes the stability and prevents the evaporation of the tear film.1 Conversely, meibomian gland dysfunction (MGD), and the resulting meibum insufficiency, destabilizes the tear film, increases its evaporation and osmolarity, and is the most common cause of dry eye disease (DED). Dry eye disease afflicts tens of millions of people in the United States alone and is one of the most frequent causes of patient visits to eye care practitioners.2
Recently, investigators have discovered that diabetes is a significant risk factor for the development of MGD.1,3 Such an association might have been predicted, given that the meibomian gland is a large sebaceous gland,4 and that sebaceous gland structure and function are compromised in diabetic patients.5–7 However, the molecular mechanisms that underlie the impact of diabetes on the human meibomian gland are unknown.
We hypothesize that insulin resistance/deficiency and hyperglycemia, which are pathognomonic features of diabetes, are critical factors in the pathogenesis of MGD in diabetic patients. Our rationale is 2-fold. First, insulin is essential for optimal sebaceous gland activity, and is known to induce glandular cell proliferation and lipid accumulation.8 Lack of insulin, in turn, would promote dysfunction. Second, hyperglycemia contributes to lipolysis in adipocytes,9,10 and this response, if occurring in the meibomian gland, could dramatically reduce the quality of meibum.
The purpose of this study was to begin to test our hypotheses by examining the influence of insulin and high glucose on immortalized human meibomian gland epithelial cells (IHMGECs).11 We evaluated whether insulin, as it does in other cells,12 promotes activation of the AKT pathway by binding to the insulin receptor (IR) or insulin-like growth factor–1 (IGF-1) receptor (IGF-1R). We also determined whether insulin stimulates cell proliferation and lipid accumulation in, and whether high glucose is toxic to, IHMGECs. For the toxicity studies, we focused on high glucose's impact on the levels of IR, IGF-1R, AKT, FOXO1, as well as the lipogenesis regulator SREBP-1.
Methods
Cell Culture and Treatment
Immortalized HMGECs (passages 20–26) were maintained in keratinocyte serum free medium (KSFM) supplemented with 5 ng/mL epidermal growth factor (EGF) and 50 μg/mL bovine pituitary extract (BPE; Life Technologies, Grand Island, NY, USA), as previously described.13 When indicated, cells were cultured in basal KSFM alone, or DMEM/F12 (50:50) (Mediatech, Inc. A Corning Subsidiary, Manassas, VA, USA), supplemented with 1 or 10% fetal bovine serum (FBS; Life Technologies).
To determine the effect of insulin on the AKT pathway in IHMGECs, we treated cells in both differentiating and proliferating culture conditions.14 For differentiation, cells were placed in DMEM/F12 with 10% FBS for 6 days, then starved overnight in media containing 1% FBS. For proliferation, cells were cultured in KSFM containing EGF and BPE for 4 days, then in supplement-free KSFM overnight. Following these time periods, IHMGECs were exposed to various concentrations of insulin for 15 minutes.
To examine whether insulin activity in IHMGECs involves the IGF-1R or IR, cells were cultured under differentiating conditions and treated alone, or in combination, with insulin (200 nM), IGF-1 (10 nM), an IGF-1R blocking antibody (10 nM), or an isotype (i.e., IgG1) control (10 nM). Insulin-like growth factor–1 served as a positive control in this study, given that we have previously shown that it upregulates the AKT pathway by binding to the IGF-1R.13
To assess whether insulin stimulates the proliferation and/or lipid accumulation in IHMGECs, cells were cultured for up to 9 days in the presence or absence of insulin (200 nM) in serum-containing media.
To determine the impact of high glucose on IHMGEC morphology, survival, signaling pathways, and protein expression, we cultured IHMGECs in KSFM containing 5.8 mM glucose (low) as a base medium. We added 20 mM glucose or mannitol (mannitol is not metabolized by cells and serves as an osmotic control) to test a high glucose effect. For comparison, we also cultured cells in standard DMEM/F12 medium, which contains relatively high glucose (17.5 mM). All media were supplemented with 10% FBS. To confirm these high glucose effects, we cultured IHMGECs in either the standard DMEM/F12 with 10% FBS as a high glucose condition (17.5 mM), or a custom-made DMEM/F12 with 10% FBS containing only 5 mM glucose.
Human IGF-1R blocking monoclonal antibody and the corresponding mouse IgG1 isotype control were purchased from R&D Systems (Minneapolis, MN, USA). Recombinant human (rh) insulin and IGF-1 were purchased from the National Hormone and Peptide Program (Torrance, CA, USA), dissolved in PBS (Mediatech, Inc.) and filter-sterilized. For cellular treatment, IGF-1 was used at a final concentration of 10 nM.
Cell Proliferation Assay
Immortalized HMGECs were cultured in 12-well plates in designated media, exposed to insulin for defined time periods and counted manually with a hemocytometer.
Lipidtox Staining of Neutral Lipids
Cells were then exposed to LipidTOX green neutral lipid stain (Life Technologies) and imaged with a Nikon Eclipse E800 (Nikon Instruments, Melville, NY, USA) according to reported procedures.13 For each sample, 10 images were taken at random locations and fluorescent intensities were quantified using ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA).
Lipid Composition Analysis Using High Performance Thin-Layer Chromatography (HPTLC)
Total cellular lipids were extracted from samples containing equivalent amount of cells and isolated on HPTLC plates (Silica Gel 60; Merck, Darmstadt, Germany), as previously described.15
SDS-PAGE and Immunoblots
Cells were lysed in SDS Laemmli buffer (Bio-Rad, Hercules, CA, USA) supplemented with 1% protease inhibitor cocktail, 200 uM sodium orthovanadate and 5% β-mercaptoethanol (all from Sigma-Aldrich Corp., St. Louis, MO, USA). Lysates were heated at 95°C for 10 minutes, separated by SDS-PAGE on 4% to 20% gradient or 10% Tris-glycine precast gels (Life Technologies), and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Waltham, MA, USA). Rabbit or mouse antibodies specific for phospho (p)-AKT (Ser473), AKT, p-FOXO1 (Ser256), FOXO1, β-actin (all from Cell Signaling Technology, Danvers, MA, USA), and the precursor form of SREBP-1 (Santa Cruz Biotechnology, Dallas, TX, USA) were used. Membranes were blocked with 5% bovine serum albumin (for antibodies of p-AKT and p-FOXO1; Sigma-Aldrich Corp.) in Tris-buffered saline containing 0.01% Tween-20 (TBS/T) or with 5% skim milk in TBS/T (for antibodies AKT, FOXO1, β-actin and SREBP-1). All primary antibodies were diluted 1:1000 in blocking buffer except for p-AKT (1:4000), SREBP-1 (1:200), and β-actin (1:10,000). HRP-conjugated secondary antibodies were goat anti-rabbit IgG and Fc-specific goat anti-mouse IgG diluted 1:5000 (Sigma-Aldrich Corp.). Proteins were visualized with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Rockford, IL, USA) using a G-Box gel documentation station (Syngene, Frederick, MD, USA). Image analysis and densitometry were performed using GeneSys software (Syngene).
Statistical Analyses
Student's t-test, nonparametric t-test, one-way or two-way ANOVA were performed when appropriate using Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Insulin Activates the AKT Signaling Pathway in IHMGECs
To examine the effect of insulin on IHMGECs, we treated cells with insulin for 15 minutes in both differentiating and proliferating culture conditions. As shown in Figures 1A and 1B, insulin activated the AKT signaling pathway in a dose-dependent manner in cells cultured in differentiating, but not proliferating, conditions. Stimulation of this pathway became apparent following exposure to 10 nM insulin, and the response increased at higher doses, reaching a plateau at 100 to 333 nM.
Figure 1.

Insulin activates p-AKT in IHMGECs. Immortalized HMGECs were cultured in serum-containing or serum-free media, followed by treatment with various doses of insulin for 15 minutes, as explained in the Methods section. (A, B) Two separate experiments with individual densitometry graphs below.
Insulin Activation of p-AKT in IHMGECs Is Dependent on IGF-1 Receptor
To determine whether insulin activity in IHMGECs involves the IGF-1R or IR, cells were cultured under differentiating conditions and treated alone, or in combination, with insulin, IGF-1, an IGF-1R blocking antibody, or an isotype (i.e., IgG1) control. Insulin-like growth factor–1 served as a positive control in this study. As illustrated in Figure 2B, the ability of insulin or IGF-1 to activate p-AKT in IHMGECs was markedly reduced when cells were pretreated with the IGF-1R blocking antibody, but not the IgG1 isotype control. The IGF-1R blocking antibody specifically, and almost completely, suppressed expression of the IGF-1R (Fig. 2A). In contrast, neither insulin nor IGF-1 had any influence in IGF-1R levels (Fig. 2A). The efficacy of the IGF-1R blocking antibody to decrease insulin action was not dependent upon any interference with the IR (Fig. 2C).
Figure 2.

Insulin activation of p-AKT in IHMGECs is dependent on IGF-1R. (A) Overnight incubation of IHMGECs with IGF-1R blocking antibody, and not IgG1 isotype control or negative control (i.e., nothing added) diminished IGF-1R expression. Treatment with IGF-1 or insulin for 15 minutes did not change IGF-1R expression. (B) Following overnight incubation with IGF-1R blocking antibody, insulin and IGF-1 both showed decreased activation of p-AKT after 15 minutes of stimulation. (C) Treatment with IGF-1R blocking antibody for 7 days diminished IGF-1R expression, but not IR expression. Immortalized HMGECs were cultured in DMEM/F12/10% FBS for 5 to 7 days before protein expressions were evaluated. Insulin, 200 nM; IGF-1, 10 nM; IGF-1R blocking antibody, 10 nM; IgG1 isotype, 10 nM.
Insulin Stimulation of IHMGEC Proliferation and Lipid Accumulation
To evaluate whether insulin stimulates the proliferation and/or lipid accumulation in IHMGECs, cells were cultured for up to 9 days in the presence or absence of insulin in serum-containing media. Our results demonstrate that insulin promotes the proliferation of IHMGECs (Fig. 3), as well as the accumulation of neutral lipids (Fig. 4A). This lipogenic insulin effect was associated with a significant increase in the cellular triglyceride content (Fig. 4B), but not in cholesterol ester, cholesterol, or phospholipid levels (data not shown). The magnitude of the insulin-induced triglyceride enhancement was analogous to that of IGF-1 (Fig. 4B).
Figure 3.
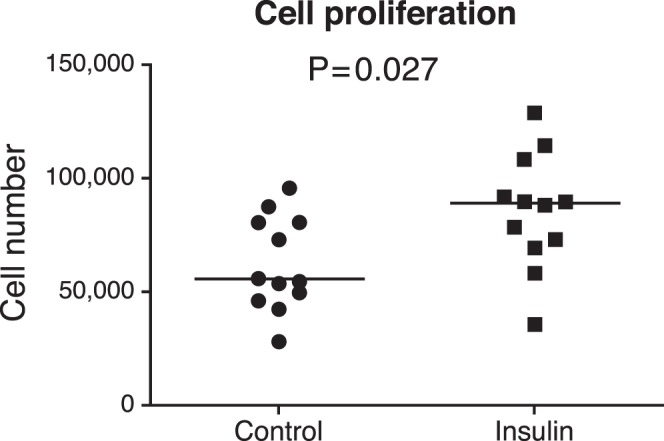
Insulin promotes cell proliferation in IHMGECs.
Figure 4.

Insulin promotes neutral lipid accumulation in IHMGECs. (A) Cells (n = 3 wells/treatment) were cultured with or without 200 nM insulin for 8 days and stained with LipidTOX. (B) Insulin and IGF-1 (10 nM) treatment increased triglyceride content.
High Glucose Impact on IHMGECs
To assess the impact of high glucose on IHMGEC morphology, survival, signaling pathways, and protein expression, we conducted a series of studies.
As shown in Figure 5, exposure to varying glucose levels had no effect on IHMGEC appearance from 1 to 8 days of culture. However, high (17.5 or 25.8 mM), but not low, glucose media caused marked differences in cell morphology by 13 days of treatment, as well as considerable cell loss. To confirm these high glucose effects, we cultured IHMGECs in either high (17.5 mM) or low 5 (mM glucose) for up to 14 days. We found the same cell morphology changes, and a significant progressive cell loss, in the high glucose–treated cells, as compared with those in the low glucose condition (Fig. 6).
Figure 5.

Effect of high glucose levels on IHMGEC morphology. Column (A), DMEM/F12 containing 17.5 mM glucose; column (B), KSFM containing 5.8 mM glucose; column (C), KSFM with 20 mM additional glucose (total glucose 25.8 mM); and (D) KSFM with additional 20 mM mannitol as an osmotic control. All media were supplemented with 10% FBS. The thin arrows indicate newly detached dead cells, and the thick arrows point to edges where cells are retracting from neighboring cells.
Figure 6.

High glucose induces a progressive cell loss in IHMGECs. Cells were cultured in two DMEM/F12/10% FBS media that differed only in glucose content: low glucose = 5 mM and high glucose = 17.5 mM. (A) Cell number at different time points (two-way ANOVA). (B) Cell morphology at 12 days under a phase-contrast microscope.
High glucose exposure also significantly altered the expression of proteins involved in insulin and IGF-1 signaling pathways and lipogenesis in IHMGECs. As demonstrated in Figures 7A and 7B, 1 week of high glucose treatment significantly reduced the levels of IGF-1R, p-AKT, FOXO1, and SREBP-1. In contrast, high glucose exerted no influence on the cellular content of IR, AKT, p-FOXO1, or cyclin D1.
Figure 7.

Influence of high glucose on protein expressions related to IGF-1 signaling, cell cycle, and lipogenesis. Cells were cultured in two types of DMEM/F12/10% FBS media that differed only in glucose content: Low glucose = 5 mM and high glucose = 17.5 mM for 7 days. (A) Western blot results; (B) associated densitometry graphs (Student's t-test). Significantly altered proteins are enclosed in boxes.
Discussion
The present study demonstrates that insulin stimulates, and high glucose is deleterious for, IHMGECs. Insulin treatment exerts a striking, dose-dependent increase in AKT signaling in IHMGECs. This effect involves the IGF-1R, but not the IR, and is associated with a rise in cell proliferation and neutral lipid accumulation. In contrast, high glucose exposure induces morphologic alterations in, and a progressive loss of, IHMGECs. This negative influence also leads to significantly reduced levels of cellular IGF-1R, p-AKT, FOXO1, and SREBP-1. These results support our hypothesis that insulin resistance/deficiency and hyperglycemia are critical factors in the pathogenesis of MGD in diabetic patients.
The effects of insulin on IHMGECs required supraphysiological hormone concentrations and appeared to be mediated through the IGF-1R, and not the IR. Typically, insulin acts within the 0.1 to 1.0 nM range, but 10 to 100 nM levels were necessary for insulin activation of p-AKT in IHMGECs. These higher doses of insulin are known to induce IGF-1R signaling.16 Further, IGF-1R and IR form hybrid receptors that are responsive to insulin.16 This insulin-IGF-1R association would account for why the IGF-1 blocking antibody, which did not interfere with IR expression, was able to decrease cellular signaling in response to insulin. As one additional consideration, insulin mimicked the physiological action of IGF-1 on AKT signaling, proliferation and lipid accumulation in IHMGECs.13
Given our data, it may be that IGF-1 is physiologically more important than insulin in the regulation of HMGECs, and that IGF-1 deficiency/resistance may be a contributing factor to the development of MGD. Relevant to this hypothesis are our findings that: (1) systemic IGF-1 deficiency is associated with altered morphology (e.g., keratinization) and reduced size of meibomian glands in growth hormone receptor null and growth hormone antagonist transgenic mice (Liu Y, et al. IOVS. 2015;56:ARVO E-Abstract 3200), and (2) IGF-1R and p-AKT, but not IR, were reduced in IHMGECs in response to high glucose. For comparison, high glucose is known to induce IGF-1 resistance in ventricular myocytes.17 Insulin-like growth factor–1 resistance also plays a role in other pathologies, including renal failure,18 Alzheimer's disease,19 and cardiovascular disease.18–20
Our observation that high glucose exposure causes morphologic alterations in, and a progressive loss of, IHMGECs, suggests that hyperglycemia contributes to the etiology of MGD in diabetic patients. This hypothesis is further supported by our findings that high glucose decreases the levels of IGF-1R, p-AKT, FOXO1, and SREBP-1 in IHMGECs. These detrimental glucose actions appear to be direct, as compared to indirect consequences of hyperosmolarity, because cellular treatment with mannitol did not elicit the same sequelae. Such glucose toxicity has also been demonstrated in a variety of cell types.21,22
In summary, diabetes is a major health problem, afflicting over 25 million people in the United States and costing more than $245 billion annually. A significant complication of diabetes is MGD and its associated evaporative DED. Our study indicates that insulin/IGF-1 deficiency/resistance and hyperglycemia may be key factors in MGD pathophysiology in diabetes. Given these results, it is possible that efforts directed at alleviating this hormonal and glucose imbalance may prove beneficial as a treatment for MGD and DED in diabetic patients. Such a possibility, though, will need to be demonstrated in clinical studies.
Acknowledgments
The authors thank Wendy Kam (Boston, MA, USA) for her constructive critique of the manuscript. Supported by National Institutes of Health Grants 1K99EY023536-01A1 and EY05612 (Bethesda, MD, USA), the Margaret S. Sinon Scholar in Ocular Surface Research Fund (Boston, MA, USA), and the Guoxing Yao Research Fund (Malden, MA, USA).
Disclosure: J. Ding, None; Y. Liu, None; D.A. Sullivan, None
References
- 1. Knop E,, Knop N,, Millar T,, Obata H,, Sullivan DA. The international workshop on meibomian gland dysfunction: report of the subcommittee on anatomy, physiology, and pathophysiology of the meibomian gland. Invest Ophthalmol Vis Sci. 2011; 52: 1938–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. The epidemiology of dry eye disease: report of the Epidemiology Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf. 2007; 5: 93–107. [DOI] [PubMed] [Google Scholar]
- 3. Viso E,, Rodriguez-Ares MT,, Abelenda D,, Oubina B,, Gude F. Prevalence of asymptomatic and symptomatic meibomian gland dysfunction in the general population of Spain. Invest Ophthalmol Vis Sci. 2012; 53: 2601–2606. [DOI] [PubMed] [Google Scholar]
- 4. Thody AJ,, Shuster S. Control and function of sebaceous glands. Physiol Rev. 1989; 69: 383–416. [DOI] [PubMed] [Google Scholar]
- 5. Cakmak S,, Gul U,, Gonul M,, Demiriz M,, Cakmak A. Statin therapy and diabetic skin. Adv Ther. 2008; 25: 17–22. [DOI] [PubMed] [Google Scholar]
- 6. Sakai S,, Kikuchi K,, Satoh J,, Tagami H,, Inoue S. Functional properties of the stratum corneum in patients with diabetes mellitus: similarities to senile xerosis. Br J Dermatol. 2005; 153: 319–323. [DOI] [PubMed] [Google Scholar]
- 7. Seirafi H,, Farsinejad K,, Firooz A,, et al. Biophysical characteristics of skin in diabetes: a controlled study. J Eur Acad Dermatol and Venereol. 2009; 23: 146–149. [DOI] [PubMed] [Google Scholar]
- 8. Deplewski D,, Rosenfield RL. Growth hormone and insulin-like growth factors have different effects on sebaceous cell growth and differentiation. Endocrinology. 1999; 140: 4089–4094. [DOI] [PubMed] [Google Scholar]
- 9. Botion LM,, Green A. Long-term regulation of lipolysis and hormone-sensitive lipase by insulin and glucose. Diabetes. 1999; 48: 1691–1697. [DOI] [PubMed] [Google Scholar]
- 10. Green A,, Rumberger JM,, Stuart CA,, Ruhoff MS. Stimulation of lipolysis by tumor necrosis factor-alpha in 3T3-L1 adipocytes is glucose dependent: implications for long-term regulation of lipolysis. Diabetes. 2004; 53: 74–81. [DOI] [PubMed] [Google Scholar]
- 11. Liu S,, Hatton MP,, Khandelwal P,, Sullivan DA. Culture, immortalization, and characterization of human meibomian gland epithelial cells. Invest Ophthalmol Vis Sci. 2010; 51: 3993–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bevan P. Insulin signalling. J Cell Sci. 2001; 114: 1429–1430. [DOI] [PubMed] [Google Scholar]
- 13. Ding J,, Sullivan DA. The effects of insulin-like growth factor 1 and growth hormone on human meibomian gland epithelial cells. JAMA Ophthalmol. 2014; 132: 593–599. [DOI] [PubMed] [Google Scholar]
- 14. Liu S,, Kam WR,, Ding J,, Hatton MP,, Sullivan DA. Effect of growth factors on the proliferation and gene expression of human meibomian gland epithelial cells. Invest Ophthalmol Vis Sci. 2013; 54: 2541–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Y,, Ding J. The combined effect of azithromycin and insulin-like growth factor-1 on cultured human meibomian gland epithelial cells. Invest Ophthalmol Vis Sci. 2014; 55: 5596–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Varewijck AJ,, Janssen JA. Insulin and its analogues and their affinities for the IGF1 receptor. Endocr Relat Cancer. 2012; 19: F63–F75. [DOI] [PubMed] [Google Scholar]
- 17. Ren J,, Duan J,, Hintz KK,, Ren BH. High glucose induces cardiac insulin-like growth factor I resistance in ventricular myocytes: role of Akt and ERK activation. Cardiovasc Res. 2003; 57: 738–748. [DOI] [PubMed] [Google Scholar]
- 18. Fouque D. Insulin-like growth factor 1 resistance in chronic renal failure. Miner Electrolyte Metab. 1996; 22: 133–137. [PubMed] [Google Scholar]
- 19. Talbot K,, Wang HY,, Kazi H,, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012; 122: 1316–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gatenby VK,, Kearney MT. The role of IGF-1 resistance in obesity and type 2 diabetes-mellitus-related insulin resistance and vascular disease. Expert Opin Ther Targets. 2010; 14: 1333–1342. [DOI] [PubMed] [Google Scholar]
- 21. Berlanga-Acosta J,, Schultz GS,, Lopez-Mola E,, Guillen-Nieto G,, Garcia-Siverio M,, Herrera-Martinez L. Glucose toxic effects on granulation tissue productive cells: the diabetics' impaired healing. BioMed Res Int. 2013; 2013: 256043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lorenzi M,, Cagliero E,, Toledo S. Glucose toxicity for human endothelial cells in culture. Delayed replication, disturbed cell cycle, and accelerated death. Diabetes. 1985; 34: 621–627. [DOI] [PubMed] [Google Scholar]