

Abstract
Purpose
The carcinogenic capacity of B[a]P/B[a]PDE is supported by epidemiologic studies. However, the molecular mechanisms responsible for B[a]P/B[a]PDE-caused lung cancer have not been well investigated. We evaluated here the role of novel target PHLPP2 in lung inflammation and carcinogenesis upon B[a]P/B[a]PDE exposure.
Experimental Design
We used the Western blotting, RT-PCR, [35S]methionine pulse and immunohistochemistry staining to determine PHLPP2 downregulation following B[a]P/B[a]PDE exposure. Both B[a]PDE-induced Beas-2B cell transformation model and B[a]P-caused mouse lung cancer model were used to elucidate the mechanisms leading to PHLPP2 downregulation and lung carcinogenesis. The important findings were also extended to in vivo human studies.
Results
We found that B[a]P/B[a]PDE exposure downregulated PHLPP2 expression in human lung epithelial cells in vitro and in mouse lung tissues in vivo. The ectopic expression of PHLPP2 dramatically inhibited cell transformation upon B[a]PDE exposure. Mechanistic studies showed that miR-205 induction was crucial for inhibition of PHLPP2 protein translation by targeting PHLPP2-3′-UTR. Interestingly, PHLPP2 expression was inversely associated with tumor necrosis factor alpha (TNFα) expression, with low PHLPP2 and high TNFα expression in lung cancer tissues compared with the paired adjacent normal lung tissues. Additional studies revealed that PHLPP2 exhibited its antitumorigenic effect of B[a]P/B[a]PDE through the repression of inflammatory TNFα transcription.
Conclusions
Our studies not only first time identify PHLPP2 downregulation by lung carcinogen B[a]P/B[a]PDE, but also elucidate a novel molecular mechanisms underlying lung inflammation and carcinogenesis upon B[a]P/B[a]PDE exposure.
Introduction
Lung cancer, as a leading cause of cancer-related mortality, is responsible for approximately 1.4 million deaths annually throughout the world (1). The major etiological agent for lung cancer is tobacco smoke and air pollution (2–6). Although more than 60 carcinogenic compounds have been identified so far in each single cigarette, B[a]P and its metabolite B[a]PDE are the most important and the strongest complete lung carcinogens (7). The carcinogenic capacity of B[a]P is supported by epidemiologic studies and has been proven in studies both in vitro and in vivo (8, 9). The sustained existence of chronic lung inflammation is a major driving force for the development of lung cancers (10). Although the link between chronic airway inflammation and lung carcinogenesis is well established, the molecular mechanisms responsible for the generation of steady increases in chronic lung inflammation have not been investigated.
Growing evidence indicates that a chronic inflammatory micro-environment in the lung is a major driving force for the development of lung cancer (11). PAHs and their derivatives, such as B[a]P/B[a]PDE, are strong carcinogens that actively promote inflammation and cancers of the lung (12, 13). Our previous studies demonstrate that TNFα is an essential inflammatory mediator for cell neoplastic transformation upon B[a]PDE exposure (14). Although our published studies reveal that transactivation of transcription factor NFAT plays an important role in mediating TNFα induction upon B[a]PDE exposure, NFAT transactivation is not consistent with the sustained TNFα transcriptional induction (14). Thus, current studies aim to explore the molecular mechanisms underlying the sustained TNFα transcriptional induction due to B[a]P/B[a]PDE exposure in vitro and in vivo. PHLPP (PH domain leucine-rich repeat protein phosphatase) proteins act as tumor suppressors due to their phosphatase activities, and can inhibit AKT (15, 16), PKC (17), and p70S6 kinase phosphorylation and activation by specifically dephosphorylating these kinases at their key regulatory domains (18). Because these PHLPP substrates positively regulate growth factor-induced signaling, PHLPP exhibits its specific characterization as a suppressor of cancer cell proliferation (19). Although PHLPP2 expression has been reported to be downregulated in cancer tissues (20–22), its potential contribution to environmental carcinogenesis has not yet been studied. Moreover, association of PHLPP2 with lung inflammation and tumorigenesis has never been explored to the best of our knowledge. Thus, our studies address these important questions in both in vitro cell culture model and in vivo animal model. The majority of the important findings were also extended to in vivo human studies.
Materials and Methods
Chemical reagents and plasmids
Benzo[a]pyrene (B[a]P) was purchased from Sigma–Aldrich. Benzo[alpha]pyrene-7,8-diol-9,10- epoxide (B[a]PDE) was a generous gift from Dr. Shantu Amin, Department of Pharmacology, School of Medicine, the Pennsylvania State University (Hershey, PA) as described in our previous studies (23). Protein synthesis inhibitor cyclohexamide (CHX) was bought from Calbiochem. The dual luciferase assay kit was purchased from Promega. TRIzol reagent and SuperScript First-Strand Synthesis system were bought from Invitrogen. PolyJet DNA In Vitro Transfection Reagent was purchased from SignaGen Laboratories. Both miR-Neasy Mini Kit and the miScript PCR system for miRNA detection were bought from Qiagen. The shRNA plasmids specifically targeting PHLPP2 and CREB were purchased from Open Biosystems (Thermo Fisher Scientific). Another set of shRNA plasmids that target human TNFα was bought from Sigma–Aldrich. HA-PHLPP2 expression construct was obtained from Addgene (16). The PHLPP2-3′ -UTR luciferase reporter has been cloned into pGL3 vector to detect the 3′-UTR activity as described in our previous study (24). TNFα promoter-driven luciferase reporter was described in our previous study (14). miR-494 and its control vector were a kindly gift from Dr. K. Yoshida (Meiji University, Kawasaki, Kanagawa, Japan; ref. 25). miR-205 expression construct was obtained from Addgene (26).
Cell culture and transfection
Beas-2B and A549 cell lines were used for in vitro experiment (Cell lines were originally obtained from ATCC; both cell lines are regularly authenticated on the basis of viability, recovery, growth, morphology, and chemical response as well, most recently confirmed 3–4 months before use). Cells were cultured as previously described (27). Cell transfections were performed by using PolyJet DNA In Vitro Transfection Reagent, according to the manufacturer's instruction.
RT-PCR
Cells were treated with B[a]PDE for the indicated time points, and then 5.0 μg total RNA was used for first-strand cDNA synthesis with oligodT (20) primer by SuperScript First-Strand Synthesis system (Invitrogen). The PHLPP2 and TNFα were determined by PCR. The results were imaged with α Innotech SP image system (α Innotech Corporation).
Western blot analysis
The antibodies specific against p-c-JUN(Ser63), p-c-JUN(Ser73), c-JUN, p-CREB(Ser133), CREB, p-ELK(Ser383), PTEN, p-AKT (Ser473), TNFα, and PARP were purchased from Cell Signaling Technology. Antibodies specific to PHLPP1 and PHLPP2 were bought from Bethyl Laboratories, Inc.. HA antibody was purchased from Covance Inc. Antibodies against NFAT3, SP1, and GAPDH were bought from Santa Cruz Biotechnology. Antibodies against β-ACTIN or α-TUBULIN were purchased from Sigma. Western blotting was performed as described in our previous publication (27).
[35S]methionine pulse assay
The pulse assay was performed as described in our previous publication (28).
Animal experiments and lung tissue
The C57BL/6J female mice at age of 6 to 8 weeks were randomly divided into four groups as indicated in Supplementary Table S2. B[a]P exposure groups (Tnfα+/+; Tnfα−/−), each of mice was exposed to B[a]P (1 mg/mouse, dissolved in 50 μL tricaprylin solvent) by intratracheal instillation. The intratracheal instillation was repeated weekly for 4 weeks and control groups (Tnfα+/+; Tnfα−/−) received the same amount of tricaprylin solvent only. The mice from the B[a]P and tricaprylin control groups were then sacrificed at different periods after last exposure to evaluate the expression of PHLPP2 and TNFα and/or lung pathologic analysis.
Immunohistochemistry paraffin of human and mouse lung specimens
Lung tissues obtained from the sacrificed mice or human lung cancer specimens were formalin-fixed and paraffin-embedded. For immunohistochemical staining (IHC), we used antibodies specific against PHLPP2 (LifeSpan BioSciences) or TNFα (Abcam). The resultant immunostaining images were captured using the AxioVision Rel.4.6 computerized image analysis system (Carl Zeiss). Protein expression levels were analyzed by calculating the integrated optical density per stained area (IOD/area) using Image-Pro Plus version 6.0 (Media Cybernetics). More detailed procedure was described as Supplementary information.
Luciferase reporter assay
Luciferase activity determination was performed as described in our studies previously (29).
Cell transformation
Cell transformation was performed as described in our studies previously (27). Cells were exposed to 1 μmol/L B[a]PDE for 12 to 24 hours and then B[a]PDE-containing medium was removed and B[a]PDE-treated cells were recultured in fresh 10% FBS DMEM for 2 days. The cultures were split and subjected to another round of treatment. Such B[a]PDE exposure was repeated twice a week for 3 to 4 months. For the TNFα-induced cell transformation, the cells were treated with 10% FBS DMEM containing 5 U/mL TNFα for 2 days and such treatment was applied to cells twice per week for total 3 to 4 months, then the anchorage-independent growth capability of the treated cells was subjected to soft agar assay (14, 30).
Soft agar assay
Anchorage-independent growth ability of B[a]PDE- or TNFα-treated cells was evaluated in soft agar as described in our previous study (31).
Statistical methods
Statistical analysis was performed by Prism 5.0 Software (GraphPad software). ANOVA was used to determine the significance of differences between various groups. The differences will be considered significant at P < 0.05.
Results
Downregulation of PHLPP2 expression occurs by B[a]P/B[a]PDE exposure in both in vitro and in vivo, and is observed in human lung cancer tissues as well
Benzo[a]pyrene (B[a]P) and its ultimate carcinogenic metabolite, (±)-anti-benzo[a]pyrene-7,8-diol-9,10-epoxide (B[a]PDE), are among the complete and the strongest lung carcinogens in tobacco-smoke and environmental polycyclic aromatic hydrocarbon (PAH) pollution (32). The doses of B[a]P and B[a]PDE used for previous studies vary depending on the different types of cells tested in vitro and animals models tested in vivo (33, 34). For example, administration of B[a]P at 40 to 100 mg/kg body weight has been used successfully in many in vivo mouse studies (33, 34). In the current study, we used 1 μmol/LB[a]PDEfor cell treatment, and 1 mg/mouse (about 50 to 75 mg/kg body weight) of B[a]P for animal experiments, which are based on the levels of environmental exposure and previously published studies (14, 33, 34). Because the metabolic activation of B[a]P to B[a]PDE is dependent on p450 activities of its treated cells in vitro (35, 36), B[a]PDE was used to evaluate its effect on an in vitro cell culture model, whereas B[a]P exposure was used for in vivo animal studies. Although lung inflammation involves events from inflammatory cells, such as macrophages, the lung epithelial cells are the cells that are first exposed and response to B[a]P. The current studies, therefore, focus on the effects of B[a]P/B[a]PDE exposure in human lung epithelial cells. Although PHLPP2 has been reported to inhibit cancer cell proliferation in previous studies (19), its potential involvement in lung carcinogenesis due to environmental carcinogen exposure has never been explored. To evaluate this notion, human bronchial epithelial Beas-2B cells were exposed to B[a]PDE, and PHLPP2 protein expression was assessed by Western blotting. As shown in Fig. 1A and B, B[a]PDE exposure resulted in a reduction of PHLPP2 protein expression in a time- and dose-dependent manner, while there is only a slight effect observed in PHLPP1 expression and marked induction of PTEN expression under same experimental conditions. PHLPP2 downregulation exhibited a unique specificity to B[a]PDE, whereas B[a]P did not show effect on PHLPP2 protein expression under same experimental conditions (Fig. 1C), revealing that B[a]P metabolic activation was crucial for this inhibition. Moreover, another human lung carcinogen arsenite failed to suppress PHLPP2 expression (Fig. 1C), suggesting that PHLPP2 downregulation by B[a]PDE is carcinogen specific. We also extended the observation to human lung cancer A549 cells (Fig. 1D and E).
Figure 1.
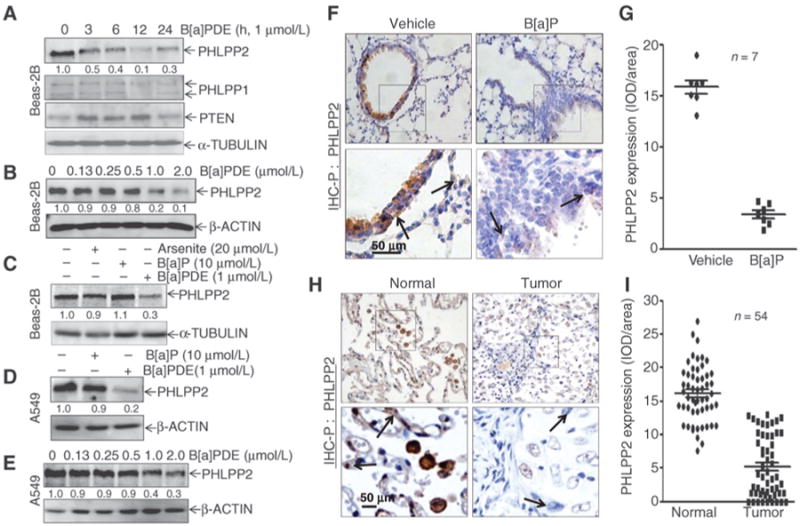
B[a]P/B[a]PDE downregulates PHLPP2 protein expression in vitro and in vivo. A and B, Beas-2B cells were exposed to 1 μmol/L of B[a]PDE for the times indicated (A), or various concentrations of B[a]PDE for 12 h (B); C, Beas-2B cells were treated with either arsenite, B[a]P, or B[a]PDE for 12 hours; D, A549 cells were treated with either B[a]P or B[a]PDE for 12 hours; E, A549 cells were exposed to the various doses of B[a]PDE for 12 hours; whole-cell extracts from each of above experiments were subjected to Western blotting for the determination of protein expression as indicated. Fand G, IHC-P was carried out to evaluate PHLPP2 expression in lung tissues from mice exposed to B[a]P for 180 days and the optical density was calculated as described in Materials and Methods section. Normal lung epithelial cells and pulmonary malignant epithelial cells were indicated in arrows. H and I, PHLPP2 expression in lung tissues from 54 pairs of human primary lung cancer was evaluated by IHC-P and the optical density was calculated as described in Materials and Methods. Arrows, normal lung epithelial cells and pulmonary malignant epithelial cells.
To evaluate the in vivo effect of B[a]P on PHLPP2 expression, we exposed mice to B[a]P by instillation as described in Materials and Methods. IHC showed that PHLPP2 expression was impaired in the mouse lung tissues following B[a]P exposure in comparison with the lung tissues obtained from vehicle control–treated mice (Fig. 1F and G; n = 7; P < 0.05). To determine whether PHLPP2 downregulation was relevant to human lung cancer development, we extended our investigation to human lung cancer tissues. As shown in Fig. 1H and I, PHLPP2 expression in lung cancer tissues obtained from the human lung cancer tissues was profoundly downregulated when compared with that of the corresponding adjacent normal lung tissues (n = 54; P < 0.05). Collectively, our results demonstrated that PHLPP2 downregulation was not only observed in human lung epithelial Beas-2B cells and in mouse lung tissues following B[a]P/B[a]PDE exposure, but was also exhibited in human lung cancer tissues.
PHLPP2 downregulation by B[a]PDE-mediated Beas-2B cell transformation
To determine the potential contribution of PHLPP2 downregulation to the lung tumorigenic effect of B[a]PDE, we repeatedly exposed Beas-2B cells to B[a]PDE and the anchorage-independent growth capabilities of B[a]PDE-treated cells were evaluated in a soft agar assay. As shown in Fig. 2A and B, B[a]PDE-induced cell transformation could be observed as early as 60 days after exposure and reached the peak at 90 days of exposure. To test the role of PHLPP2 downregulation in B[a]PDE-induced cell transformation, stable cell transfectant Beas-2B HA-PHLPP2 was established (Fig. 2C) and ectopic expression of HA-PHLPP2 resulted in a dramatic inhibition of Beas-2B cell transformation as compared with that of vector control transfectants due to B[a]PDE exposure (Fig. 2D and E), suggesting that PHLPP2 downregulation played an important role in cell transformation induced by B[a]PDE. This notion was further supported by the results obtained in PHLPP2-knockdown cells (Fig. 2F). Stable introduction of PHLPP2 shRNAs into Beas-2B cells (sh-PHLPP2-1 and sh-PHLPP2-2) alone could lead to an increase in spontaneous cell transformation and could significantly promote B[a]PDE-induced cell transformation incomparison with Beas-2B nonsense control transfectants (Fig. 2G and H). Taken together, we provided solid data to demonstrate that PHLPP2 downregulation contributed to human bronchial epithelial cell malignant transformation due to B[a]PDE exposure.
Figure 2.
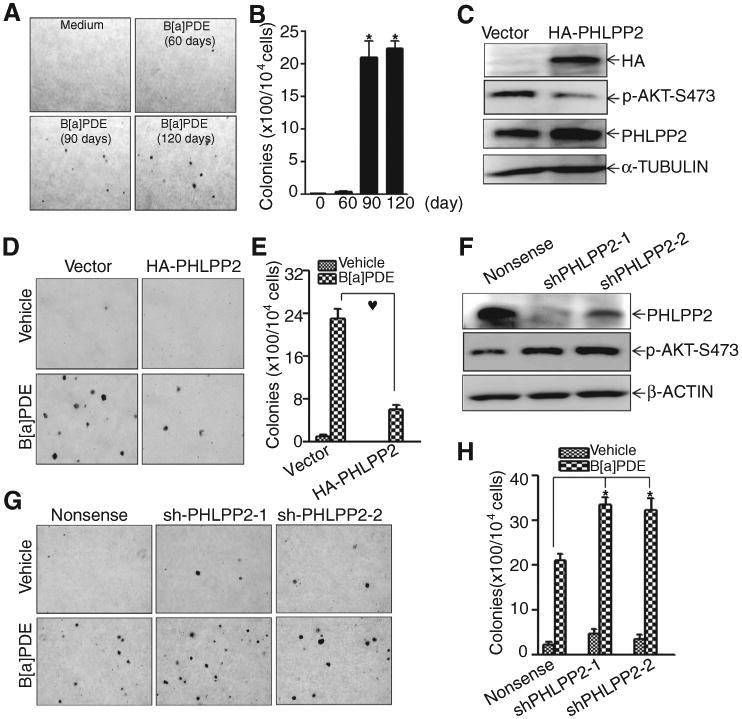
PHLPP2 downregulation is crucial for B[a]PDE-induced Beas-2B cell transformation. A and B, Beas-2B cells were repeatedly exposed to 1 μmol/L of B[a]PDE for induction of cell transformation by the determination of their anchorage-independent growth in soft agar (A) and the number of colonies was scored and presented as colonies per 104 seeded cells (B). C to E, the effect of HA-PHLPP2 overexpression on B[a]PDE-induced cell transformation was determined by soft agar assay as described in Materials and Methods. F to H, the effect of PHLPP2 shRNA on B[a]PDE-induced cell transformation was determined by soft agar assay as described in Materials and Methods.
PHLPP2 protein downregulation is mediated by miR-205 induction following B[a]PDE exposure
To elucidate the molecular mechanisms underlying PHLPP2 downregulation due to B[a]PDE exposure, the effect of B[a]PDE on PHLPP2 mRNA expression and protein degradation were first evaluated. The results revealed that B[a]PDE exposure did not affect PHLPP2 mRNA expression (Fig. 3A) and exogenous HA-PHLPP2 protein expression (Fig. 3B). These results, together with our observation of similar protein degradation rates between Beas-2B cells treated with CHX alone and CHX plus B[a]PDE (Fig. 3C), excluded the possible effect of B[a]PDE on PHLPP2 mRNA expression and protein degradation. Therefore, we sought to determine whether B[a]PDE exposure affected PHLPP2 protein translation. Short-term pulse-labeling assay was performed to examine PHLPP2 protein translational activity between B[a]PDE-exposed and unexposed Beas-2B cells. As shown in Fig. 3D, the incorporation of 35S-methionine/cysteine into newly synthesized PHLPP2 protein was observed in unexposed Beas-2B cells, but was markedly reduced in B[a]PDE-exposed cells, indicating that B[a]PDE exposure caused an inhibition of PHLPP2 protein translation.
Figure 3.
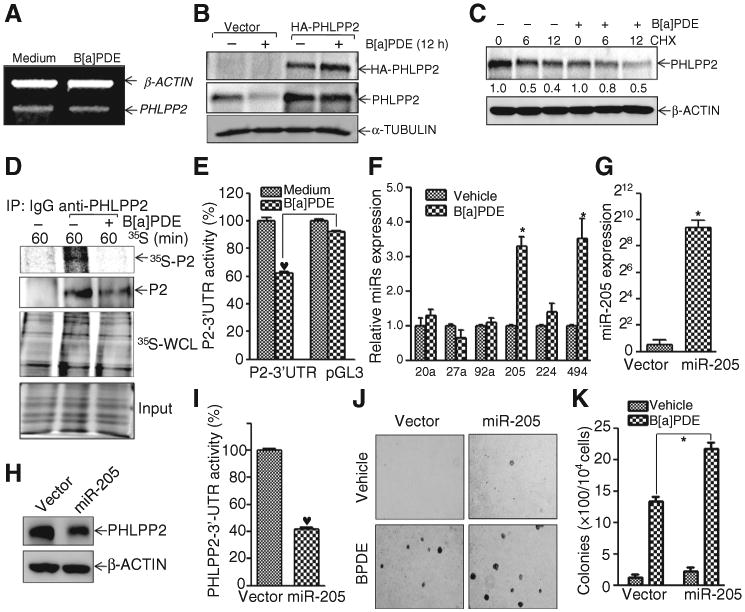
B[a]PDE downregulates PHLPP2 expression by upregulating miR-205. A, Beas-2B cells were treated with 1 μmol/L of B[a]PDE for 12 hours, RT-PCR was carried out for the determination of PHLPP2 mRNA levels. B, indicated cells were exposed to 1 μmol/L of B[a]PDE, exogenous HA-PHLPP2 expression and PHLPP2 expression were analyzed by Western blotting.C, Beas-2Bcellswere treated with CHX (50 μg/mL) together with or without B[a]PDE for indicated times, and the cell extracts were subjected to analysis of PHLPP2 degradation rate by Western blotting. D, newly synthesized PHLPP2 protein was monitored as described in Materials and Methods, and PHLPP2(P2) in IP complex was evaluated by Western blotting. 35S-whole cell extracts (WCL) and total protein Coomassie blue staining were used as controls. E, the transfectants were exposed to B[a]PDE for 12 hours. The results were presented as PHLPP2-3′-UTR luciferase activity relative to medium control. F, quantitative real-time PCR was used to detect the effect of miRNAs induction upon B[a]PDE exposure for 12 hours. miR-205 was stably transfected into Beas-2B cells, and identified by real-time PCR (G). The effect of miR-205 on PHLPP2 expression was evaluated by Western blotting (H). I, the effect of miR-205 on PHLPP2-3′ -UTR activity was determined by the transient cotransfection of PHLPP2-3′-UTR-luciferase reporter, miR-205 and TK. The results were normalized by TK activity. J and K, soft agar assay was performed to evaluate the effect of miR-205 on B[a]PDE-induced cell transformation in soft agar assay.
For the past decade, miRNA has been a well-documented and pivotal factor for regulating physiologic and pathologic processes through its ability to regulate mRNA stability and protein translation (37, 38). To further uncover the mechanism of B[a]PDE in the modulation of PHLPP2 translation, the PHLPP2-3′-UTR–dependent luciferase reporter was stably transfected into Beas-2B cells and the effect of B[a]PDE exposure on PHLPP2-3′-UTR– dependent luciferase activity was determined. As shown in Fig. 3E, PHLPP2-3′-UTR activity was significantly inhibited upon B[a]PDE exposure in comparison with that of the pGL3 control vector transfectants, indicating that the suppression of PHLPP2-3′-UTR activity was responsible for the decrease in the rate of PHLPP2 protein translation by B[a]PDE. To find which miRNA was involved in the downregulation of PHLPP2 by B[a]PDE, bioinformatics software was used to analyze the potential miRNA binding sites in the PHLPP2 3′-UTR region. The results showed that there were multiple potential miRNA binding sites in the PHLPP2 3′-UTR region, including miR-20a, miR-27a, miR-92a, miR-205, miR-224, and miR-494 (Supplementary Fig. S1A). We then determined the effects of B[a]PDE on the expression of these potential miRNAs and the results indicated that B[a]PDE exposure led to the induction of miR-494 and miR-205, whereas it did not show any observable induction of the other miRNAs in Beas-2B cells (Fig. 3F). To identify the role of miR-494 or miR-205 in the B[a]PDE-mediated downregulation of PHLPP2 expression, either miR-494 or miR-205 plasmids were stably transfected into Beas-2B cells. As shown in Fig. 3G and H and Supplementary Fig. S1, the inhibition of PHLPP2 protein expression was only observed in Beas-2B cells stably transfected with miR-205, whereas the introduction of miR-494 slightly increased PHLPP2 protein expression (Supplementary Fig. S1B and S1C). To provide direct evidence that miR-205 is targeting the PHLPP2-3′-UTR region, we cotransfected miR-205 and PHLPP2-3′-UTR luciferase reporter constructs into Beas-2B cells. As shown in Fig. 3I, miR-205 expression profoundly inhibited PHLPP2-3′-UTR activity, demonstrating that miR-205 inhibited PHLPP2 protein translation by targeting PHLPP2-3′-UTR following B[a]PDE exposure. Consistently, miR-205 overexpression also exhibited a promotion of both basal level and B[a]PDE-induced level of cell transformation in Beas-2B cells (Fig. 3J and K). Collectively, our results conclusively demonstrated that miR-205 upregulation mediated the downregulation of PHLPP2 protein translation following B[a]PDE exposure.
PHLPP2 downregulation contributes to inflammatory TNFα induction by B[a]PDE
The sustained existence of chronic lung inflammation is a major driving force for the development of lung cancers (11, 39). TNFα plays an important role in promoting tumor growth and progression by regulating chronic inflammation (40). Our previous studies also demonstrate that TNFα mediates B[a]PDE-induced mouse epidermal cell transformation (14). To determine the potential correlation of PHLPP2 downregulation with lung inflammatory responses upon B[a]P/B[a]PDE exposure, we investigated the PHLPP2 and TNFα protein expression in human lung cancer tissues in comparison with the normal lung tissues that are adjacent to tumor tissues using both Western blotting and IHC. The results showed an inverse relationship between PHLPP2 and TNFα protein expression (Fig. 4A). The human lung cancer tissues showed a downregulation of PHLPP2 protein expression but high TNFα protein expression, and in striking contrast, the normal lung tissues had high PHLPP2 expression but low TNFα protein expression as demonstrated by both Western blotting (Fig. 4A) and IHC assay (Fig. 4B and C; n = 54; P < 0.05). Consistently, similar inverse relationship between PHLPP2 protein expression and TNFα mRNA induction in B[a]PDE-transformed Beas-2B cells was observed (Fig.4D). The inverse relationship betweenPHLPP2 and TNFα expression both in vitro and in vivo revealed the possible regulatory effect of PHLPP2 in TNFα expression following B[a]P/B[a]PDE exposure. To test this, we evaluated the TNFα mRNA levels and TNFα promoter-driven luciferase reporter activity between Beas-2B (HA-PHLPP2) and Beas-2B (Vector) cells following B[a]PDE exposure, and the results showed that B[a]PDE treatment profoundly led to TNFα mRNA and TNFα promoter-driven luciferase induction in Beas-2B (Vector) cells, whereas such induction was almost impaired by ectopic expression of HA-PHLPP2 under same experimental conditions (Fig. 4E and F). Moreover, knockdown of PHLPP2 by its shRNA significantly elevated TNFα mRNA expression and its promoter transactivation (Fig. 4G and H). Consistent with PHLPP2 regulation of TNFα expression, stable expression of miR-205 in Beas-2B cells markedly promoted TNFα mRNA induction by B[a]PDE exposure (Fig. 4I). These results clearly demonstrated that miR-205–mediated PHLPP2 downregulation played an essential role in B[a]PDE-induced TNFα expression.
Figure 4.
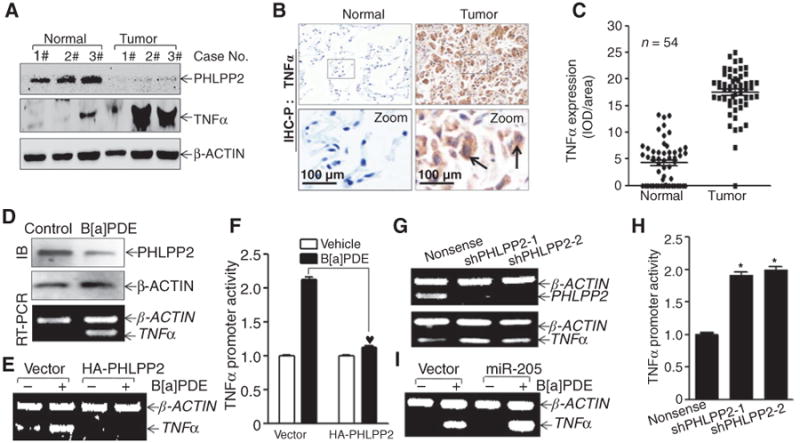
Inverse correlation of the expression of PHLPP2 and TNFα, and PHLPP2 downregulation plays an important role in TNFα induction by B[a]PDE. A, three pairs of human lung cancer tissue extracts were subjected to Western blotting for determination of PHLPP2 and TNFα expression and β-actin was used as protein loading control. B and C, TNFα expression was determined by IHC-P in the paired human lung cancer specimens (n = 54). TNFα expression between normal tissues and tumor tissues was determined by an Image-Pro Plus version 6.0 system and then analyzed by prism 5.0 software using the scatter plot style. Arrows, typical cancer cells.D, the cell extracts or total RNA from normal and B[a]PDE-transformed Beas-2B cells were subjected to Western blotting for the determination of PHLPP2 protein expression or RT-PCR for the determination of TNFα mRNA expression. E and F, Beas-2B cells and its transfectants as indicated were exposed to 1 μmol/L of B[a]PDE for 12 h and TNFα mRNA expression was evaluated by PT-PCR. G and H, Beas-2B TNFα-luciferase reporter stable transfectants as indicated were exposed to 1 μmol/L of B[a]PDE for the determination of TNFα transcription at the time points indicated. The results were presented as luciferase activity relative to vehicle control. I, the effects of overexpressed miR-205 on B[a]PDE-induced TNFα in Beas-2B cells.
PHLPP2 inhibits TNFα transcription by targeting c-JUN activation following B[a]PDE exposure
To explore the molecular mechanisms underlying PHLPP2 regulation of TNFα transcription following B[a]PDE exposure, the bioinformatics analysis of the TNFα promoter was performed. As shown in Fig. 5A, multiple potential transcription factor binding sites were observed in the TNFα promoter region, including SP1, EGR-1, ELK-1, AP-1, NFAT3, and NFκB. Thus, we then identified which of the transcription factors' nuclear transloca-tionswereinhibitedinBeas-2B (HA-PHLPP2) cells in comparison with the Beas-2B (Vector) cells. The results indicated that ectopic expression of HA-PHLPP2 in Beas-2B cells inhibited the nuclear translocation of NFAT3, c-JUN, and CREB due to B[a]PDE exposure, whereas it did not show any observable effect on other transcription factors as indicated (Fig. 5B). Because miR-205 also regulated TNFα induction via modulation of PHLPP2 expression, the effect of miR-205 on the nuclear translocation of NFAT3, c-JUN, and CREB was further investigated. As shown in Fig. 5C, the expression of miR-205 specifically increased the nuclear translocation of c-JUN and CREB, and not NFAT3, revealing that CREB and c-JUN might be the transcription factors that are responsible for miR-205/PHLPP2 -mediated TNFα induction following B[a]PDE exposure. As shown in Fig. 5D, knockdown of CREB expression by its shRNAs did not show observable effect on TNFα mRNA induction due to B[a]PDE exposure, which excluded the possibility of CREB's contribution to the regulation of this biologic pathway. Interestingly, blockage of c-JUN activation by ectopic expression of TAM67 profoundly impaired TNFα mRNA induction due to B[a]PDE exposure (Fig. 5E). Consistently, the results obtained from using AP-1 binding site point mutated TNFα promoter-driven luciferase reporter together with ectopic expression miR-205 revealed that AP-1 is the miR-205 downstream transcription factor mediating TNFα transcription (Supplementary Fig. S2). Collectively our results demonstrated that c-JUN is the miR-205/PHLPP2 downstream transcription factor that is responsible for inflammatory factor TNFα induction following B[a]PDE exposure.
Figure 5.

c-JUN is a miR-205/PHLPP2 downstream transcription factor responsible for TNFα transcription due to B[a]PDE exposure. A, potential transcriptional factor binding sites in TNFα promoter region that were analyzed using the TRANSFAC 8.3 engine online. B, Beas-2B HA-PHLPP2 or miR-205 (C) transfectants were exposed to 1 μmol/L of B[a]PDE for the indicated time points and cell extracts were used to isolate cytoplasmic and nuclear fractions according to the protocol of the nuclear/cytosol fractionation kit. The isolated protein fractions were subjected to Western blotting to determination the translocation of various transcription factors as indicated. D, specific shRNAs target CREB were stably transfected into Beas-2B cells, and the whole-cell lysates were subjected to Western blotting for the determination of CREB protein level. Beas-2B transfectants as indicated were exposed to1 μmol/L of B[a]PDE for 12 hours and TNFα mRNA levels were determined by RT-PCR (D, bottom and E, bottom), whereas cell protein extracts was subjected to Western blotting (E, top) identification of TAM67 inhibition of c-JUN phosphorylation due to B[a]PDE exposure.
TNFα induction plays an important role in cell transformation in vitro and lung carcinogenesis in vivo following B[a]P/B[a]PDE exposure
To determine the role of TNFα induction in B[a]P/B[a]PDE-induced lung epithelial cell transformation, we tested whether TNFα repeated treatment alone could result in Beas-2B cell transformation. As shown in Fig. 6A and B, TNFα repeated treatment alone did lead to the anchorage-independent growth of Beas-2B cells in a time-dependent manner. Importantly, knockdown of TNFα with stable transfection of TNFα shRNA profoundly inhibited Beas-2B (shTNFα) cell transformation following B[a]PDE-repeated exposure (Fig. 6C–E), whereas PHLPP2 down-regulation observed in TNFα-knockdown cells was similar in Beas-2B (Nonsense) transfectants (Supplementary Fig. S5A). The results suggested that TNFα acts as a PHLPP2 downstream mediator that is essential for B[a]PDE-induced lung epithelial cell transformation. To further investigate the role of TNFα induction in B[a]P/B[a]PDE-induced lung cancer development, the C57BL/6J TNFα−/− mice and its control TNFα+/+ mice were identified (Fig. 6F) and exposed to B[a]P as described in Materials and Methods. Consistent with our above findings in vitro, B[a]P exposure significantly induced TNFα expression in the lung tissues of TNFα+/+ mice in time-dependent manner, as observed in both Western blotting and IHC paraffin (IHC-P) assay (Fig. 6G–I, Supplementary Table S2). Consistent with TNFα induction, various lung lesions, including inflammation, hyperplasia/pre-carcinoma, and carcinoma, were observed in mice after being exposed to B[a]P for 180 days in TNFα+/+ mice (n = 7, Fig. 6J, Supplementary Figs. S3 and S4; Supplementary Tables S2 and S3), whereas there was only one mouse with lung hyperplasia that was observed in total 6 TNFα−/− mice under same experimental conditions (Supplementary Figs. S3 and S4; Supplementary Tables S2 and S3). These data clearly demonstrated that TNFα induction was critical for lung inflammation and various lung lesions, including lung carcinogenesis, due to B[a]P exposure.
Figure 6.
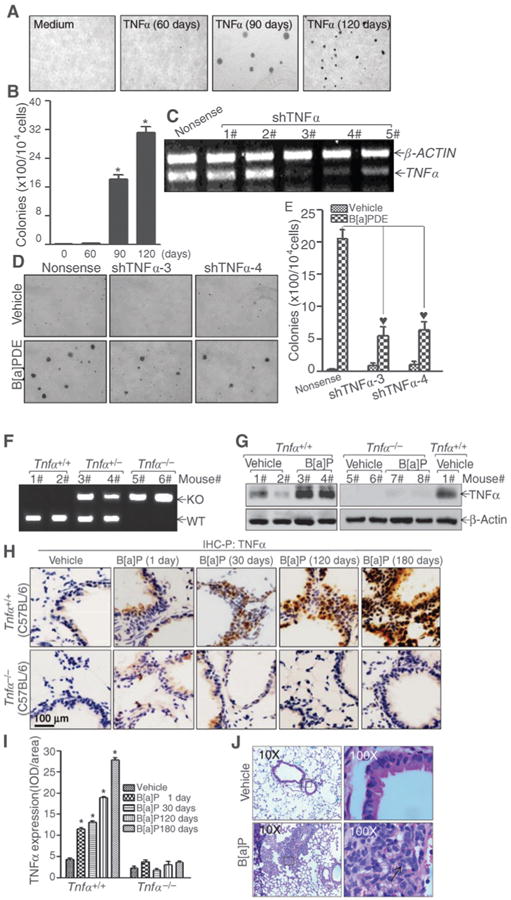
TNFα induction is critical for B[a]P/B[a]PDE-induced carcinogenesis. A and B, Beas-2B cells were repeatedly exposed to TNFα for the indicated times and TNFα-treated cells were used to determine their anchorage-independent growth ability in soft agar. The results were presented as described in Materials and Methods section. C, specific TNFα shRNAs were stably transfected into Beas-2B cells and the knockdown efficiency in Beas-2B cells was evaluated by RT-PCR assay. D and E, TNFα shRNAs stable transfectants were repeatedly exposed to 1 μmol/L of B[a]PDE for 120 days. The effect of TNFα shRNA on B[a]PDE-induced cell transformation was determined in soft agar. F, Tnfα−/− mice were identified by genomic DNA PCR using three primers of oIMR4182-5′-TAG CCA GGA GGG AGA ACA GA-3′, oIMR4183-5′-AGT GCC TCT TCT GCC AGT TC-3′, and oIMR7297-5′-CGT TGG CTA CCC GTG ATA TT-4′. G, mice were exposed to B[a]P for 180 days as described in Materials and Methods, and ung tissue extracts were then subjected to Western blotting. H and I, IHC-P was performed to evaluate TNFα expression in lung tissues from mice exposed to B[a]P at the indicated times. J, the images representing lung carcinoma in mice exposed to B[a]P. Arrow, heteromorphism.
Discussion
B[a]P/B[a]PDE causes lung cancer mainly by either causing DNA mutations (41) or by activating signaling cascades that regulate gene expression (42). Because the DNA binding and damage activity of B[a]PDE has well been characterized in many previous studies (43, 44), current studies focused on the mechanisms underlying signaling alterations in gene regulatory pathways that subsequently result in human lung bronchial epithelial cell transformation and lung carcinogenesis following B[a]P/B[a]PDE exposure. Our studies identified that B[a]P/B[a]PDE exposure could lead to miR-205 induction, which targets PHLPP2-3′-UTR and inhibits PHLPP2 protein translation and expression, subsequently resulting in c-JUN activation and ultimately increasing TNFα transcription and inflammatory responses to further contribute to lung carcinogenesis. These findings not only identifies PHLPP2 as a new target for B[a]P/B[a]PDE initiation of lung carcinogenic effects, but also provides a novel scenario linking PHLPP2 to the generation of steady increases in chronic lung inflammation caused by environmental B[a]P/B[a]PDE exposure (Supplementary Fig. S6).
Although PHLPP2 downregulation has been reported in a variety of cancers, the involvement of PHLPP2 in carcinogenesis has been largely unexplored, except for a study reporting PHLPP2 inhibition of cancer progression through suppression of IKKβ/NFκB activation (45). A PHLPP2 point mutation at Leu 1016 has been found in breast cancer, mediated by a dramatic decrease of its phosphatase activity (46). Our current studies are the first demonstration of PHLPP2 as a novel target for B[a]P/B[a]PDE's promotion of cell transformation in vitro and mouse lung carcinogenesis in vivo. It was noted that PHLPP2 downregulation was only observed in Beas-2B cells treated with B[a]PDE, but not with B[a]P, whereas B[a]P did inhibit PHLPP2 expression in lung tissues of mice exposed to B[a]P. The explanation of this might be due to defect of related p450s in Beas-2B cells, by which leads to low generation of B[a]PDE in the cells exposed to B[a]P. This explanation has also been supported by the findings in our previous studies in comparison of B[a]P and B[a]PDE for their AP-1 transactivation in mouse epidermal Cl41 cells (42, 47).
The exploration of the mechanisms underlying B[a]P/B[a]PDE regulation of its target's gene expression would be helpful to understand its carcinogenetic effects. Previous studies reported that PHLPP2 expression is negatively regulated by hypoxia in colon cancer cells via HIF-1α-dependent cascade by promotion of PHLPP2 protein degradation (48). Hypoxia could downregulate the expression of USP46, a deubiquitinase identified for mediation of deubiquitination and stabilization of the PHLPP2 protein (20). In this study, we found that B[a]PDE exposure inhibited PHLPP2 protein expression in a protein degradation-independent pathway. This notion is also supported by our previous finding that B[a]PDE treatment is not able to induce HIF-1α activation in Cl41 cells (47). The result of that B[a]PDE downregulated 35S-lebeled PHLPP2 protein synthesis, suggesting that B[a]PDE down-regulates PHLPP2 due to the inhibition of protein translation. It is well known that miRNA plays a key role in the regulation of protein translation by binding to 3′-UTR region of its targeted mRNA. The potential miRNAs of miR-17∼92 cluster and miR-224 had been reported to enhance tumor growth in mantle cell lymphoma or colorectal cancer by downregulating PHLPP2 (24, 49), however, B[a]PDE failed to upregulate miR-20a, miR-92a, and miR-224 expression in Beas-2B cells. By determining the effect of B[a]PDE on miRNA expressions, and subsequent miRNA overexpression on PHLPP2 protein expression, we identified miR-205 as a critical mediator for the inhibition of PHLPP2 protein translation following B[a]PDE exposure. This result is consistently supported by previous reports showing the upregulation of miR-205 in human lung cancer (22).
TNFα plays an important role in promoting tumor growth and progression by regulating chronic inflammation responses (40). In current studies, we discovered a novel function of PHLPP2 in the attenuation of TNFα transcriptional induction by B[a]PDE exposure. The inverse relationship between PHLPP2 and TNFα expression was observed in both human lung cancer tissues and normal human lung tissues, B[a]P-exposed mouse lung tissues, as well as B[a]PDE-treated Beas-2B cells. Importantly, the results obtained from the utilization of ectopic PHLPP2 expression or knockdown approaches demonstrated PHLPP2 downregulation was crucial for B[a]PDE-induced TNFα induction. Further investigation identified c-JUN as a key PHLPP2 downstream target responsible for TNFα induction upon B[a]PDE exposure. Moreover, TNFα induction upon B[a]PDE was essential for cell transformation in an in vitro cell model. Considering our previous studies showing that TNFα could cause feedback to activate NFAT3 and NFκB, and in turn promoted TNFα transcription (14, 30), it is possible that there is a positive loop to sustain TNFα production and promote COX-2 overexpression and its products PGs-mediated chronic inflammation to maintain the inflammatory microenvironment as schematically depicted in Supplementary Fig. S6. Moreover, further elucidation of the mechanisms of TNFα in the regulation of B[a]PDE-induced cell transformation, such as regulation of tumor cell cycle/proliferation, autophagy/apoptosis, and angiogenesis, will provide more insight into understanding of TNFα action in lung carcinogenic effect of B[a]P/B[a]PDE. Based on this B[a]P/B[a]PDE-initiated inflammatory feedback loop, TNFα is a central key player for B[a]P-caused lung carcinogenesis. Therefore, we further evaluated this notion in lung carcinogenesis in an in vivo animal model utilizing Tnfα-knockout mice. We found that, chronic lung inflammation and the formation of atypical hyperplasia and carcinoma upon B[a]P exposure observed in wild-type mice were almost impaired in Tnfα-knockout mice, although PHLPP2 expression was still downregulated in Tnfα KO mouse lung tissues (Supplementary Fig. S5B and S5C).
In summary, our current study for the first time discovers a PHLPP2 downregulation by environmental lung carcinogen B[a]P/B[a]PDE both in vitro and in vivo, and demonstrate essential role of such PHLPP2 downregulation in lung carcinogenesis. We also identify a novel signaling cascade underlying PHLPP2′s negative modulation of TNFα transcription through the suppression of c-JUN phosphorylation. The results from these current studies, together with our previous studies, clearly demonstrates the formation of inflammatory positive feedback loops by NFAT/NFκB, TNFα, and COX-2, and provides evidence for PHLPP2 and TNFα as essential regulators of the whole network. In addition, these positive feedback loops may be the reason for the persistence and prevalence of these signaling molecules in inflammatory lung tissues, which enhances their effects in lung cancer development. Current studies facilitate our understanding of the molecular mechanism(s) that lead to the formation and maintenance of a chronic lung inflammatory microenvironment, and its role in lung cancer development due to B[a]P/B[a]PDE exposure. Such novel information will spur the development of efficacious preventive and therapeutic approaches for controlling inflammation-associated cancers.
Supplementary Material
Translational Relevance.
PAHs and their derivatives are strong carcinogens that actively promote chronic inflammatory microenvironment, a major driving force for lung cancer development. Although PHLPP2 expression has been found to be repressed in certain cancers, its antitumor role in carcinogen-induced carcinogenesis has never been explored. We found here that environmental lung carcinogen B[a]P/B[a]PDE induced PHLPP2 downregulation in vitro and in vivo, and PHLPP2 was also downregulated in lung cancer tissues. Moreover, we demonstrated that PHLPP2 downregulation was crucial for cell transformation in vitro and lung carcinogenesis in vivo, and PHLPP2 exhibited its antitumor effects through inhibition of TNFα transcription. Our findings not only facilitate our understanding of the molecular mechanism that leads to the formation of chronic lung inflammation, and its role in lung cancer development following B[a]P exposure, but also provide the solid basis of utilization of PHLPP2 and/or TNFα as targets for prevention and therapy of the lung cancer.
Acknowledgments
The authors thank Dr. K. Yoshida (Meiji University, Kawasaki, Kanagawa, Japan) for providing miR-494 constructs and Dr. Shengli Pan (Clinical pathologist, Rui An Hospital of Traditional Chinese Medicine, Wenzhou, Zhejiang, China) for his clinic diagnosis of mouse lung lesions.
Grant Support: This work was partially supported by the grants of NIH/NCI CA112557 and CA 177665, and NIH/NIEHS ES000260; the Natural Science Foundation of China (NSFC81229002, NSFC91029706, and NSFC81372946); the Natural Science Foundation of Zhejiang Province (LZ14H260001) and Key Science and Technology Innovation Team of Zhejiang Province (2013TD10).
Footnotes
Authors' Contributions: Conception and design: H. Huang, J. Gao, C. Huang
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): H. Huang, X. Pan, H. Jin, L. Zhang, C. Yang, P. Liu, Y. Liu, L. Chen, J. Zhu, X. Zeng
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): H. Huang, X. Pan, Y. Li, J. Zhu, G. Chen, J. Gao, C. Huang
Writing, review, and/or revision of the manuscript: H. Huang, X. Pan, H. Jin, Y. Li, G. Chen, J. Gao, C. Huang
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): J. Li
Study supervision: J. Gao, C. Huang
Other (provided essential plasmid constructs for the study): K. Fu
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.She J, Yang P, Hong Q, Bai C. Lung cancer in China: challenges and interventions. Chest. 2013;143:1117–26. doi: 10.1378/chest.11-2948. [DOI] [PubMed] [Google Scholar]
- 2.Parker S, Tong T, Bolden S, Wingo P. Cancer statistics. CA Cancer J Clin. 1996;46:5. doi: 10.3322/canjclin.46.1.5. [DOI] [PubMed] [Google Scholar]
- 3.Hammond E, Seidman H. Smoking and cancer in the United States. Prev Med. 1980;9:169–74. doi: 10.1016/0091-7435(80)90071-7. [DOI] [PubMed] [Google Scholar]
- 4.Wynder EL, Goodman MT. Smoking and lung cancer: some unresolved issues. Epidemiol Rev. 1983;5:177–207. doi: 10.1093/oxfordjournals.epirev.a036258. [DOI] [PubMed] [Google Scholar]
- 5.Saracci R. The interactions of tobacco smoking and other agents in cancer etiology. Epidemiol Rev. 1987;9:175–93. doi: 10.1093/oxfordjournals.epirev.a036301. [DOI] [PubMed] [Google Scholar]
- 6.Vial W. Cigarette smoking and lung disease. Am J Med Sci. 1986;291:130–42. doi: 10.1097/00000441-198602000-00011. [DOI] [PubMed] [Google Scholar]
- 7.Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol. 2002;3:461–9. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- 8.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 9.Yan Y, Wang Y, Tan Q, Hara Y, Yun TK, Lubet RA, et al. Efficacy of polyphenon E, red ginseng, and rapamycin on benzo(a)pyrene-induced lung tumorigenesis in A/J mice. Neoplasia. 2006;8:52–8. doi: 10.1593/neo.05652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao H, Rahman I. Current concepts on the role of inflammation in COPD and lung cancer. Curr Opin Pharmacol. 2009;9:375–83. doi: 10.1016/j.coph.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ballaz S, Mulshine JL. The potential contributions of chronic inflammation to lung carcinogenesis. Clin Lung Cancer. 2003;5:46–62. doi: 10.3816/CLC.2003.n.021. [DOI] [PubMed] [Google Scholar]
- 12.Rubin H. Synergistic mechanisms in carcinogenesis by polycyclic aromatic hydrocarbons and by tobacco smoke: a bio-historical perspective with updates. Carcinogenesis. 2001;22:1903–30. doi: 10.1093/carcin/22.12.1903. [DOI] [PubMed] [Google Scholar]
- 13.Tsay JJ, Tchou-Wong KM, Greenberg AK, Pass H, Rom WN. Aryl hydrocarbon receptor and lung cancer. Anticancer Res. 2013;33:1247–56. [PMC free article] [PubMed] [Google Scholar]
- 14.Ouyang W, Hu Y, Li J, Ding M, Lu Y, Zhang D, et al. Direct evidence for the critical role of NFAT3 in benzo[a]pyrene diol-epoxide-induced cell transformation through mediation of inflammatory cytokine TNF induction in mouse epidermal Cl41 cells. Carcinogenesis. 2007;28:2218–26. doi: 10.1093/carcin/bgm115. [DOI] [PubMed] [Google Scholar]
- 15.Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly depho-sphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 16.Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917–31. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 17.Gao T, Brognard J, Newton AC. The phosphatase PHLPP controls the cellular levels of protein kinase C. J Biol Chem. 2008;283:6300–11. doi: 10.1074/jbc.M707319200. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Stevens PD, Li X, Schmidt MD, Gao T. PHLPP-mediated dephos-phorylation of S6K1 inhibits protein translation and cell growth. Mol Cell Biol. 2011;31:4917–27. doi: 10.1128/MCB.05799-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Weiss HL, Rychahou P, Jackson LN, Evers BM, Gao T. Loss of PHLPP expression in colon cancer: role in proliferation and tumorigenesis. Oncogene. 2009;28:994–1004. doi: 10.1038/onc.2008.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Stevens PD, Yang H, Gulhati P, Wang W, Evers BM, et al. The deubiquitination enzyme USP46 functions as a tumor suppressor by controlling PHLPP-dependent attenuation of Akt signaling in colon cancer. Oncogene. 2012;32:471–8. doi: 10.1038/onc.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Neill AK, Niederst MJ, Newton AC. Suppression of survival signalling pathways by the phosphatase PHLPP. FEBS J. 2012;280:572–83. doi: 10.1111/j.1742-4658.2012.08537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai J, Fang L, Huang Y, Li R, Yuan J, Yang Y, et al. miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer. Cancer Res. 2013;73:5402–15. doi: 10.1158/0008-5472.CAN-13-0297. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Yan Y, Li J, Ma Q, Stoner GD, Ye J, et al. Differential requirement of signal pathways for benzo[a]pyrene (B[a]P)-induced nitric oxide synthase (iNOS) in rat esophageal epithelial cells. Carcinogenesis. 2005;26:1035–43. doi: 10.1093/carcin/bgi052. [DOI] [PubMed] [Google Scholar]
- 24.Rao E, Jiang C, Ji M, Huang X, Iqbal J, Lenz G, et al. The miRNA-17 approximately 92 cluster mediates chemoresistance and enhances tumor growth in mantle cell lymphoma via PI3K/AKT pathway activation. Leukemia. 2012;26:1064–72. doi: 10.1038/leu.2011.305. [DOI] [PubMed] [Google Scholar]
- 25.Ohdaira H, Sekiguchi M, Miyata K, Yoshida K. MicroRNA-494 suppresses cell proliferation and induces senescence in A549 lung cancer cells. Cell Prolif. 2012;45:32–8. doi: 10.1111/j.1365-2184.2011.00798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang D, Li J, Costa M, Gao J, Huang C. JNK1 mediates degradation HIF-1alpha by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer Res. 2010;70:813–23. doi: 10.1158/0008-5472.CAN-09-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu Y, Zhang D, Huang H, Li J, Zhang M, Wan Y, et al. NF-kappaB1 p50 promotes p53 protein translation through miR-190 downregulation of PHLPP1. Oncogene. 2014;33:996–1005. doi: 10.1038/onc.2013.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang H, Ma L, Li J, Yu Y, Zhang D, Wei J, et al. NF-kappaB1 inhibits c-Myc protein degradation through suppression of FBW7 expression. Oncotarget. 2014;5:493–505. doi: 10.18632/oncotarget.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan Y, Li J, Ouyang W, Ma Q, Hu Y, Zhang D, et al. NFAT3 is specifically required for TNF-alpha-induced cyclooxygenase-2 (COX-2) expression and transformation of Cl41 cells. J Cell Sci. 2006;119:2985–94. doi: 10.1242/jcs.03014. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J, Gao G, Chen L, Deng X, Li J, Yu Y, et al. Cheliensisin A inhibits EGF-induced cell transformation with stabilization of p53 protein via a hydrogen peroxide/Chk1-dependent axis. Cancer Prev Res (Phila) 2013;6:949–58. doi: 10.1158/1940-6207.CAPR-13-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435–51. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 33.Triano EA, Simpson JB, Kratky M, Lang WR, Triolo AJ. Protective effects of trifluralin on benzo(a)pyrene-induced tumors in A/J mice. Cancer Res. 1985;45:601–7. [PubMed] [Google Scholar]
- 34.Morse MA, Zu H, Kresty LA, Stoner GD. Failure of dietary oltipraz to inhibit benzo[a]pyrene-induced lung tumorigenesis in strain a mice. Cancer Lett. 1995;91:133–8. doi: 10.1016/0304-3835(95)03730-k. [DOI] [PubMed] [Google Scholar]
- 35.Conney AH, Chang RL, Jerina DM, Wei SJ. Studies on the metabolism of benzo[a]pyrene and dose-dependent differences in the mutagenic profile of its ultimate carcinogenic metabolite. Drug Metab Rev. 1994;26:125–63. doi: 10.3109/03602539409029788. [DOI] [PubMed] [Google Scholar]
- 36.Shimada T, Gillam EM, Oda Y, Tsumura F, Sutter TR, Guengerich FP, et al. Metabolism of benzo[a]pyrene to trans-7, 8-dihydroxy-7, 8-dihydroben-zo[a]pyrene by recombinant human cytochrome P450 1B1 and purified liver epoxide hydrolase. Chem Res Toxicol. 1999;12:623–9. doi: 10.1021/tx990028s. [DOI] [PubMed] [Google Scholar]
- 37.Wu X, Brewer G. The regulation of mRNA stability in mammalian cells: 2.0. Gene. 2012;500:10–21. doi: 10.1016/j.gene.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zinovyev A, Morozova N, Nonne N, Barillot E, Harel-Bellan A, Gorban AN. Dynamical modeling of microRNA action on the protein translation process. BMC Syst Biol. 2010;4:13. doi: 10.1186/1752-0509-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milara J, Cortijo J. Tobacco, inflammation, and respiratory tract cancer. Curr Pharm Des. 2012;18:3901–38. doi: 10.2174/138161212802083743. [DOI] [PubMed] [Google Scholar]
- 40.Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res. 2006;4:221–33. doi: 10.1158/1541-7786.MCR-05-0261. [DOI] [PubMed] [Google Scholar]
- 41.Kohler SW, Provost GS, Fieck A, Kretz PL, Bullock WO, Sorge JA, et al. Spectra of spontaneous and mutagen-induced mutations in the lacI gene in transgenic mice. Proc Natl Acad Sci U S A. 1991;88:7958–62. doi: 10.1073/pnas.88.18.7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Tang MS, Liu B, Shi X, Huang C. A critical role of PI-3K/Akt/JNKs pathway in benzo[a]pyrene diol-epoxide (B[a]PDE)-induced AP-1 transactivation in mouse epidermal Cl41 cells. Oncogene. 2004;23:3932–44. doi: 10.1038/sj.onc.1207501. [DOI] [PubMed] [Google Scholar]
- 43.Wang A, Gu J, Judson-Kremer K, Powell KL, Mistry H, Simhambhatla P, et al. Response of human mammary epithelial cells to DNA damage induced by BPDE: involvement of novel regulatory pathways. Carcinogenesis. 2003;24:225–34. doi: 10.1093/carcin/24.2.225. [DOI] [PubMed] [Google Scholar]
- 44.Santella RM. Immunological methods for detection of carcinogen-DNA damage in humans. Cancer Epidemiol Biomarkers Prev. 1999;8:733–9. [PubMed] [Google Scholar]
- 45.Agarwal NK, Zhu X, Gagea M, White CLd, Cote G, Georgescu MM. PHLPP2 suppresses the NF-kappaB pathway by inactivating IKKbeta kinase. Oncotarget. 2014;5:815–23. doi: 10.18632/oncotarget.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brognard J, Niederst M, Reyes G, Warfel N, Newton AC. Common polymorphism in the phosphatase PHLPP2 results in reduced regulation of Akt and protein kinase C. J Biol Chem. 2009;284:15215–23. doi: 10.1074/jbc.M901468200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding J, Li J, Chen J, Chen H, Ouyang W, Zhang R, et al. Effects of polycyclic aromatic hydrocarbons (PAHs) on vascular endothelial growth factor induction through phosphatidylinositol 3-kinase/AP-1-dependent, HIF-1alpha-independent pathway. J Biol Chem. 2006;281:9093–100. doi: 10.1074/jbc.M510537200. [DOI] [PubMed] [Google Scholar]
- 48.Wen YA, Stevens PD, Gasser ML, Andrei R, Gao T. Downregulation of PHLPP expression contributes to hypoxia-induced resistance to chemotherapy in colon cancer cells. Mol Cell Biol. 2013;33:4594–605. doi: 10.1128/MCB.00695-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao WT, Li TT, Wang ZG, Wang SY, He MR, Ye YP, et al. microRNA-224 promotes cell proliferation and tumor growth in human colorectal cancer by repressing PHLPP1 and PHLPP2. Clin Cancer Res. 2013;19:4662–72. doi: 10.1158/1078-0432.CCR-13-0244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.