

Abstract
Purpose
Excessive and continuously produced free radicals in the outer retina are implicated in retinal aging and the pathogenesis of sight-threatening retinopathies, yet measuring outer retinal oxidative stress in vivo remains a challenge. Here, we test the hypothesis that continuously produced paramagnetic free radicals from the outer retina can be measured in vivo using high-resolution (22-μm axial resolution) 1/T1magnetic resonance imaging (MRI) without and with a confirmatory quench (quench-assisted MRI).
Methods
Low-dose sodium iodate–treated and diabetic C57Bl6/J mice (and their controls), and rod-dominated (129S6) or cone-only R91W;Nrl−/− mice were studied. In dark-adapted groups, 1/T1 was mapped transretinally in vivo without or with (1) the antioxidant combination of methylene blue (MB) and α-lipoic acid (LPA), or (2) light exposure; in subgroups, retinal superoxide production was measured ex vivo (lucigenin).
Results
In the sodium iodate model, retinal superoxide production and outer retina-specific 1/T1 values were both significantly greater than normal and corrected to baseline with MB+LPA therapy. Nondiabetic mice at two ages and 1.2-month diabetic mice (before the appearance of oxidative stress) had similar transretinal 1/T1 profiles. By 2.3 months of diabetes, only outer retinal 1/T1 values were significantly greater than normal and were corrected to baseline with MB+LPA therapy. In mice with healthy photoreceptors, a light quench caused 1/T1 of rods, but not cones, to significantly decrease from their values in the dark.
Conclusions
Quench-assisted MRI is a feasible method for noninvasively measuring normal and pathologic production of free radicals in photoreceptors/RPE in vivo.
Keywords: diabetic retinopathy, MRI, oxidative stress, retinitis pigmentosa, sodium iodate
Free radical production that continually exceeds the quenching capacity of a cell defines oxidative stress. Many studies report evidence to support the hypothesis that oxidative stress is a major contributor to the damaging effects of aging, and to the pathogenesis of many neurodegenerative diseases, including photoreceptor-based retinopathies.1,2 For example, excessive and continuous free radical production in rod photoreceptor cells has been implicated as an important early event in the course of at least two major sight-threatening retinal diseases: retinal degeneration (RD) and diabetic retinopathy (DR).2–12 However, most retinal and brain studies have so far been performed ex vivo or are invasive, and so it has not been possible to confirm the presence of oxidative stress during senescence or disease in vivo.13 This has also limited optimization of antioxidant (AO) treatment efficacy in individuals over time.
Free radicals, such as superoxide and nitric oxide, have an unpaired electron, making them intrinsically paramagnetic and thus detectable by 1/T1 magnetic resonance imaging (MRI).14,15 However, because free radicals are famously short-lived, and have a relatively small relaxivity, sensitizing standard MRI machines to detect free radicals usually involves exogenous injection of a longer-lived contrast agent that traps free radicals and reports with higher relaxivity.14 However, such exogenous agents are currently limited by difficulties in crossing blood-brain/retina barriers and/or concerns about their pharmacokinetics and tissue concentration.14,16
Here, we investigate an alternative approach that takes advantage of the fact that many neurons of interest produce free radicals continuously as either part of normal function or during oxidative stress.1,2,17,18 This steady “geyser” of free radicals likely produces an endogenous, pseudo-constant, and highly localized paramagnetic relaxation mechanism detectable with 1/T1 MRI. To confirm a free radical contribution to the 1/T1 signal, data are collected in the absence and presence of a quenching condition.15 Two strengths of this quench-assisted MRI approach are that many AOs readily cross blood-brain/retina barriers, and are approved by the Food and Drug Administration for safe use in humans.
The present study takes advantage of the 22-μm axial resolution available to MRI that allows measurement of several rod cell compartment-specific functions in vivo with anatomical confirmation provided by coregistration with optical coherence tomography (OCT) images.19–24 Quench-assisted MRI was tested in two established oxidative stress–based retinopathy models using a combination of clinically relevant AOs that operate with different quenching mechanisms: methylene blue (MB, an alternate electron transporter that effectively inhibits superoxide generation by mitochondria2,12,25,26 ) and α-lipoic acid (LPA, a potent free radical scavenger2,12,25,27). To evaluate measurement sensitivity, healthy photoreceptors and RPE were studied as well, because in the dark, rod and cone cells continually produce high but nonpathologic levels of free radicals, a byproduct of the continuous production of ATP needed to maintain open ion channels in their outer segments; this ATP is generated by mitochondria in the inner segment, the location of approximately 75% of the mitochondria in the retina.17,28 The quench condition in this portion of the study is light. In the light, rod cell ion channels close, thus negating the demand for as much ATP, and leading to a substantial reduction in free radical production compared with that in the dark.2 In contrast to rods, cone cells do not saturate with light, and are expected to continue to generate high levels of free radicals in the light.17,28
Materials and Methods
All animals were treated in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and Institutional Animal and Care and Use Committee authorization. Animals were housed and maintained in 12 hour:12 hour light-dark cycle laboratory lighting, unless otherwise noted.
Groups
The following nondiabetic groups, a mix of male and female mice, were studied: (1) untreated (rod-dominant) C57Bl/6J mice (wild-type [wt]; Jackson Laboratories, Bar Harbor, ME, USA), (2) wt mice treated with sodium iodate with and without MB+LPA AO therapy, (3) diabetic mice with and without MB+LPA antioxidant therapy, and (4) 6-week-old 129S6 rod-dominant control mice (Jackson Laboratories), and (5) R91W;Nrl−/− mice that have a rosette-free and functional all-cone photoreceptor retina.29
For the diabetic study, at 2 months of age, 20-g male wt mice were randomly divided into the following groups and studied after 1.2 or 2.3 months of diabetes (or age-matched): (1) nondiabetic control group (wt), (2) diabetic group (D), and (3) diabetic + antioxidant group (D+AO, details on the AO treatment are below). In all cases, diabetes was similarly induced and maintained in mice. Mice with starting weights of 16 to 20 g were injected with streptozotocin (STZ; 60 mg/kg), 10 mM citrate buffer (pH 4.5) intraperitoneally (IP), within 10 minutes of preparation, once a day for 5 consecutive days. Body weight and blood glucose levels were monitored weekly. Insulin (neutral protamine Hagedorn, Lilly Humulin N; Eli Lilly, Indianapolis, IN, USA), administered to mice as needed based on body weight and blood glucose levels but not more than twice weekly, allowed slow weight gain while maintaining hyperglycemia (blood glucose levels higher than 400 mg/dL). Mice that lost weight and/or had blood glucose levels greater than 600 mg/dL were given 0.1 to 0.2 units of insulin. Normal rodent chow (Purina TestDiet 5001; Purina, Richmond, IN, USA, which contains 11.2% fat, 26% protein, and 62.7% carbohydrate) and water were provided ad libitum. Glycated hemoglobin (A1c) was measured from blood collected before each experiment. A small sample of blood was obtained from a tail puncture and hemoglobin A1c (HbA1c) measured using kits (either Helena Laboratories, Beaumont, TX, USA, or Crystal Chem, Downers Grove, IL, USA). We did not mix results from each kit but did confirm that only mice that exhibited a greater than 2.5-fold difference between control and diabetic A1c values were studied.
Treatments
Sodium Iodate.
Sodium iodate (Sigma-Aldrich Corp., St. Louis, MO, USA) was constituted in saline, and administered as a bolus IP (20 mg/kg) approximately 24 hours before the MRI examination.
Methylene Blue + LPA Antioxidant Therapy.
A combination of two antioxidants was used: MB (an alternate electron transporter that effectively inhibits superoxide generation by mitochondria2,12,25,26) and LPA, a potent free radical scavenger. Thirty minutes after sodium iodate treatment, or approximately 24 hours before the MRI examination of the diabetic group, mice were treated with 1 mg/kg MB (dissolved in saline) IP. The next day, 1 hour before the MRI examination, each MB-treated mouse was also treated with 50 mg/kg LPA (dissolved in saline and pH adjusted to ∼7.4) IP.
Magnetic Resonance Imaging
The general mouse preparation for high-resolution MRI is well established in our laboratory.19 All animals were maintained in darkness for at least 16 hours before and during the dark phase of the MRI examination. In all groups, immediately before the MRI experiment, animals were anesthetized with urethane (36% solution IP; 0.083 mL/20-g animal weight, prepared fresh daily; Sigma-Aldrich Corp.) and treated topically with 1% atropine to ensure dilation of the iris during light exposure followed by 3.5% lidocaine gel to reduce sensation that might trigger eye motion, and to keep the ocular surface moist. High-resolution 1/T1 data (details below) were acquired on a 7-T system (Bruker ClinScan, Bruker Corporation, Billerica, MA, USA) using a receive-only surface coil (1.0-cm diameter) centered on the left eye; 1/T1 MRI data sets were collected in dark-adapted mice because free radical production by rod cells is greater in the dark than in the light.17,30,31 Each 1/T1 data set takes 15 minutes to collect. In some experiments, 1/T1 data also were collected at 29 minutes (midpoint of acquisition) after turning on the light. The end of a fiberoptic bundle was attached to a light source (Mark II Light Source; Prescott's, Inc., Monument, CO, USA) placed caudal to the eye, projecting at a white screen approximately 1 cm from the eye, similar to that previously described.32 We exposed the eye to 0 (i.e., dark) or approximately 500 lux (confirmed outside the magnet using a Traceable Dual-Range Light Meter [Control Company, Friendswood, TX, USA] placed against a 1-cm-diameter aperture; measured this way, room lighting is approximately 300 lux). A combination of two factors prevented us from examining repeated or different dark/light cycles: (1) maintenance of anesthetized mouse physiology for extended periods inside the MRI machine is difficult and urethane provided a reliable but limited hour-long stable preparation; and (2) the slow response time of rods limits adaptation to 30 minutes. In all cases, animals were humanely euthanized as detailed in our DLAR-approved protocol.
The 1/T1 MRI procedure has been described in detail previously.19 Retinal partial saturation T1 data were acquired using a dual-coil mode on a 7-T Bruker ClinScan system: several single spin-echo (time to echo [TE] 13 ms, 7 × 7 mm2 , matrix size 160 × 320, slice thickness 600 μm, in-plane resolution 21.875 μm) images were acquired at different repetition times (TRs) in the following order (number per time between repetitions in parentheses): TR 0.15 second (6), 3.50 s (1), 1.00 second (2), 1.90 seconds (1), 0.35 second (4), 2.70 seconds (1), 0.25 second (5), and 0.50 second (3). To compensate for reduced signal-noise ratios at shorter TRs, progressively more images were collected as the TR decreased. Animals were studied in pseudo-random order between controls and experimental mice.
Magnetic Resonance Imaging Data Analysis
In each animal, we confirmed ocular dilation based on the iris position on the MRI data.33 The 1/T1 MRI data from the central retinal (±1 mm from the center of the optic nerve) were analyzed as previously described.19 Single images acquired with the same TR were first registered (rigid body) and then averaged. These averaged images were then registered across TRs. The same regions of interest as above were analyzed by calculating 1/T1 maps by first fitting to a three-parameter T1 equation (y = a + b*[exp(−c*TR)], where a, b, and c are fitted parameters) on a pixel-by-pixel basis using R (v.2.9.0, R Development Core Team [2009], R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria) scripts developed in-house, and the minpack.lm package (v.1.1.1, Timur V; Elzhov and Katharine M. Mullen minpack.lm: R interface to the Levenberg-Marquardt nonlinear least-squares algorithm found in MINPACK. R package version 1.1–1). The reciprocal (1/T1) values directly reflect paramagnetic free radical levels.34 Central intraretinal 1/T1 profiles were obtained as detailed elsewhere.33 Values from the superior and inferior retina were averaged.
In each mouse, retinal thicknesses (μm) from the manganese-enhanced MRI images were objectively determined using the “half-height method,” wherein a border is determined via a computer algorithm based on the crossing point at the midpoint between the local minimum and maximum, as detailed elsewhere.33,35 The distance between two neighboring crossing points thus represents an objectively defined thickness. Thickness values were then normalized with 0% depth at the presumptive vitreoretinal border and 100% depth at the presumptive retina-choroid border. As previously discussed, the dark-to-light transition produces a significant increase in choroidal, but not retinal, thickness.36–38 To allow for comparisons between groups and conditions, 1/T1 transretinal profiles in dark and light in each mouse were thus spatially normalized to the anatomical thickness value in the dark.22
Similar to collection and analysis procedures used in the lucigenin superoxide assay below to minimize day-to-day variability, in the MRI experiments, each session consisted of at least three control mice studied alternatively with at least three experimental mice. Correction factors were calculated at each retinal depth to adjust the mean control 1/T1 values of the same-day controls to a reference set of control 1/T1 values. These depth-specific correction factors were then applied to the experimental data from that day. For this reason, the y-axis is labeled as “Adjusted 1/T1.”
Lucigenin Assay of Superoxide
Superoxide levels were measured ex vivo with lucigenin (bis-N-methylacridinium nitrate), as reported previously.2
Statistical Analysis
Superoxide levels were analyzed by two-tailed ANOVA followed by the Fisher post hoc test. Magnetic resonance imaging transretinal profile data between groups was first performed using a one-tailed unpaired t-test at different locations of the adjusted 1/T1 transretinal profiles to objectively identify regions of interest. Then, a generalized estimating equation (GEE) approach was used to compare selected location ranges, identified from the t-tests as significant.39,40 The GEE method is a more powerful two-tailed method that performs a general linear regression analysis using contiguous locations in each subject and accounts for the within-subject correlation between contiguous locations. When the initial t-test identified a location range as likely significant (P ≤ 0.05) at the one-tailed level, GEE was performed on that data in that range. Data are presented as mean ± SEM. Differences were considered statistically significant at P less than 0.05.
Results
Quench-Assisted MRI is Sensitive to Outer Retina Oxidative Stress In Vivo
Model 1: Sodium Iodate.
Sodium iodate is an oxidizing agent that targets RPE cells. At high systemic doses, at least some of the histopathologic features found in the dry form of AMD are observed.41,42 However, at lower doses (e.g., 20 mg/kg), there is good, but indirect, evidence that both RPE and rod cells experience oxidative stress in the absence of histopathology.43,44 To confirm that low-dose iodate increases production of at least one free radical species, we measured superoxide production from the whole retina (Fig. 1A) of low-dose sodium iodate–treated mice. Furthermore, as shown in Figure 1A, sodium iodate superoxide production was supernormal and was corrected by the AO combination of MB and LPA. The laminar retinal architecture was largely normal on visual inspection of OCT data (Fig. 1B) with the RPE layer becoming hyperreflective43; whole retinal thickness MRI measurements also were unremarkable (controls, 219 ± 3 μm [n = 16, mean ± SEM]; iodate-treated mice, 226 ± 6 μm [n = 10]; iodate- and MB+LPA-treated mice, 225 ± 6 μm [n = 9]; P = 0.5; one-way ANOVA). However, these superoxide measurements do not indicate which retina region(s) are generating the excess free radicals in vivo.
Figure 1.

Quench-assisted MRI measurement in vivo of outer retina oxidative stress. (A) Retina superoxide production measured ex vivo from dark-adapted controls (C, black, n = 10); sodium iodate–treated mice (IO3, green, n = 6); IO3 mice treated with AO (IO3+AO, red, n = 6). *Significant difference (P < 0.05). (B) Quench-assisted MRI profiles measured in vivo from dark-adapted controls (black, n = 30); IO3 mice (green, n = 7); IO3+AO mice (red, n = 9). Optical coherence tomography images (control vs. IO3) show mostly unchanged laminar spacing within the retina; dashed vertical lines map outer plexiform layer (48%) and retina/choroid boundary (100%) onto MRI profiles; MRI insert shows regions studied (white boxes); visual inspection of each group's MRI does not allow for easy appreciation of differences in the derived parameter 1/T1 and so only a representative image is presented. **Retinal depth range with significant difference (P < 0.05). Adjusted 1/T1 data at each depth used factors that normalize same-day controls to a control reference data set.
Having established the presence of excessive free radical production in this model, we then examined the outer retina in the low-dose sodium iodate model with quench-assisted MRI (Fig. 1B). The 1/T1 of both RPE and rod cell layers were greater than normal, and MB+LPA treatment effectively corrected these supernormal values to baseline in vivo, confirming that this increased 1/T1 was due to free radicals (Fig. 1B); 1/T1 of the presumptive retinal ganglion cell layer of sodium iodate treatment mice significantly decreased with MB+LPA treatment compared with untreated controls, warranting further study.
Summarizing, in an acutely induced model, excessive free radical production and its suppression with MB+LPA measured ex vivo (Fig. 1A) were also measured in vivo (Fig. 1B) by quench-assisted MRI with localization to the outer retina.
Model 2: Diabetes.
Retinal-based oxidative stress has been identified as pathogenic for DR.2 Many studies report a diabetes-duration–dependent increase in whole retinal oxidative stress biomarkers (e.g., superoxide and nitric oxide levels) on postmortem examination,11,45 and that systemic AO therapy prevents the appearance of early pathophysiology and later histopathology.2,21,22 Surprisingly, the major contributor to the increased free radical level in the retina from 2-month diabetic mice has been suggested to be cells of the outer retina (i.e., rod and RPE cells) and not endothelial cells.2,46,47
Thus, we asked if rod and RPE cells in 1.2- and 2.3-month diabetic mice with hemoglobin A1c levels greater than 2.5 that of controls would show evidence for progressive increases in oxidative stress on quench-assisted MRI in vivo. The 2.3-month hyperglycemic mice demonstrated a significant increase in 1/T1 signal in the presumptive RPE layer, and MB+LPA treatment effectively corrected this abnormality (Fig. 2C). The duration of diabetes was important because outer retinal 1/T1 profiles of 1.2-month diabetic mice were not different from that in age-matched controls (Fig. 2B). Technique reproducibility was demonstrated by the lack of differences in transretinal 1/T1 profiles between 3- and 5-month nondiabetic control mice (Fig. 2A). Interestingly, 2.3 months of diabetes also produced an increase in 1/T1 in the presumptive inner nuclear layer, but this was not different from the MB+LPA group, suggesting it is not related to free radical production; other small differences were noted with AO therapy that require further study. The 2.3 months of diabetes did not alter the laminar morphology based on visual inspection of single-animal OCT data (Fig. 2); however, whole retinal thickness as measured by MRI was lowered with diabetes. Control retinal thickness values were not different between groups (Kruskal-Wallis test, P > 0.05, 231 ± 3 μm [n = 13]) and not different from 5-month control retinal thickness values (223 ± 4 μm [n = 6]); however, retinal thickness values at 1.2 (203 ± 4 μm [n = 4]) and 2.3 months of diabetes without (209 ± 4 μm [n = 5]) or with MB+LPA AO (202 ± 7 μm [n = 4]) were all lower (P < 0.05) than age-matched control values; no differences (P > 0.05) in thickness values between treated and untreated diabetes were noted. The relatively tight range of A1c values for each group (data not shown) precluded attempts at correlation between level of diabetes and change in outer retinal oxidative stress.
Figure 2.
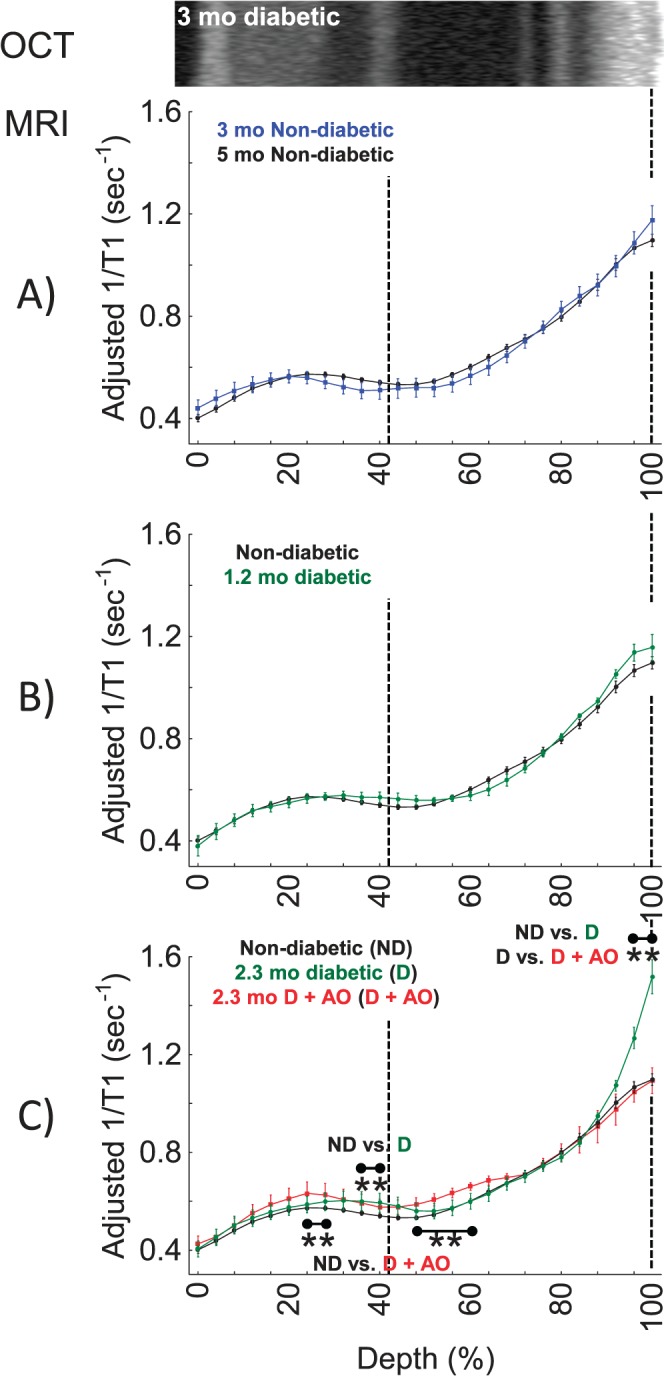
Quench-assisted MRI measurement in vivo of diabetes-induced outer retina oxidative stress. (A) Control mice of 2.3 months (black, n = 30) and 5-month control mice (blue, n = 6). These are the age-matched controls for the 1.2- and 2.3-month diabetic groups, respectively. (B) Nondiabetic mice (black, n = 30) and 1.2-month diabetic mice (green, n = 4), and (C) nondiabetic control mice (black, n = 30), 2.3-month diabetic mice (green, n = 5), and 2.3-month diabetic mice treated with MB+LPA AOs (red, n = 4). Graphs are presented using the conventions of Figure 1B; 2.3-month diabetic and control laminar structure (e.g., Fig. 1) were similar on OCT and only 2.3-month diabetic data are shown. **Retinal depth range with significant difference (P < 0.05).
Summarizing, in a chronic disease model, quench-assisted MRI data in vivo are consistent with a diabetes-duration–dependent increase in free radical production over that in controls in the outer retina. It is possible that quench-assisted MRI did not capture the contribution of all types of free radical species, including those in the rod cells. Nonetheless, the present finding clearly highlights cells of the outer retina as a major contributor to retinal oxidative stress in vivo, consistent with previous measurements performed ex vivo2,46,47 and other studies in vivo.21,22,48–50 One important implication of these findings is that treatments that correct retinal oxidative stress or its sequela (e.g., inflammation, retinal microvasculopathy) in diabetes cannot be assumed to act only or primarily on endothelial cells, the focus of clinical evaluation of DR. In other words, based on these considerations, it seems prudent going forward to consider how treatment affects diabetes-induced oxidative stress in both the inner and outer retina.
Quench-Assisted MRI Is Sensitive to Continuous Production of Free Radicals in Healthy Outer Retina In Vivo
To test measurement sensitivity in the absence of injury, we then examined healthy photoreceptors that continuously produce free radicals in the dark.17,30,31,51 Unlike rods, cone cells do not saturate in the light, and previous work predicts that their generation of free radicals will be much higher than in light-adapted rods.17,17,28 In support of this prediction, outer retina–specific 1/T1 values of healthy control mice were higher in the dark and reduced by a light quench (Figs. 3A, 3C); maintaining control mice in the dark for the same time as in the light did not cause outer retinal 1/T1 values to decrease (data not shown) thus ruling out a possible duration-of-anesthesia confounder. Furthermore, in a cone-only mouse model (R91W;Nrl−/−),29 dark-adapted cone-cell–specific 1/T1 values did not decrease with light (Figs. 3B, 3C). Based on Okawa et al.,17 we predicted a higher outer retinal 1/T1 in these cone-only mice. The increased values in the presumptive RPE layer on MRI were unexpected and need additional evaluation. Retinal thickness values were different (P < 0.05) between 129S6 (224 ± 3 μm [n = 7]) and R91W;Nrl−/− mice (205 ± 3 μm [n = 5]). No evidence for RPE hyperreactivity on OCT of the mutant mice was noted (data not shown).
Figure 3.

Quench-assisted MRI measurement of healthy outer retina free radical production in vivo. (A) Magnetic resonance imaging profiles from 129S6 mice exposed in a paired-fashion to dark (n = 7) then light (n = 7) inside the magnet. (B) Magnetic resonance imaging profiles from cone-only R91W;Nrl−/− double transgenic mice exposed in a paired-fashion to dark (n = 5) then light (n = 5) inside the magnet. Adjusted 1/T1 data at each depth used factors that normalize same-day 129S6 controls to a 129S6 reference data set. Graphs are otherwise presented using the conventions of Figure 2B. **Retinal depth range with significant difference (P < 0.05). (C) Bar graph summary of 1/T1 data in dark (D) and light (L) averaged over the regions spanning the outer limiting membrane (OLM) to the RPE in 129S6 and R91W;Nrl−/− mice based on their OCT data; these regions are indicated by the heavy white lines on the respective OCT images. *Significant difference (P < 0.05).
Summarizing, in healthy retina, the results of quench-assisted MRI in dark- and light-exposed rod and cone cells in vivo were in line with the light-evoked redox physiology of the outer retina predicted from studies ex vivo.
Discussion
Collectively, the data in this study strongly suggest that continuous free radical production in the outer retina with injury and in health, as measured on postmortem examination, also can be evaluated with quench-assisted MRI in vivo. Depending on the metrics evaluated, postmortem and in vivo assays may be more or less sensitive to different free radical species, although likely with substantial overlap. Quench-assisted MRI likely measures the continuous production of several species of free radicals, and thus is a potentially useful global readout of free radical burden in retinopathies in which sources and species of free radicals are expected to vary over time.6,8,52 If desired, it might be possible to unravel the contribution of each free radical source (e.g., mitochondria, peroxisome, nicotinamide adenine dinucleotide phosphate oxidase) and/or species (e.g., superoxide, peroxide, nitric oxide) with a combination of targeted AO therapies and genetically modified animal models. The results of this study thus represent an important advance over currently available, noninvasive technologies used in vision research because neither OCT nor electrophysiology (ERG) are sensitive to free radicals. Redox-sensitive methods are available but usually require either highly specialized, nonstandard equipment (e.g., electron paramagnetic resonance imaging), or injection of stable free radical–sensitive reporter contrast agents.13,14,16 Quench-assisted MRI represents a relatively simple approach with high potential to improve experimental and clinical evaluation of devastating photoreceptor-based oxidative stress diseases such as RD and DR, and improve early AO intervention and treatment.
Could alternative factors, such as PO2, pH, or hemodynamics, explain the quench-assisted MRI results? For example, oxygen is a paramagnetic molecule, and can also modify 1/T1.53,54 If PO2 is the dominant relaxation mechanism in the present studies, we would predict an increase in 1/T1 with light because PO2 of rod cells increases in the light (compared with that in the dark).55 Instead, as shown in Figure 3, rod cell 1/T1 decreases in the light. Changes in pH also occur with dark and light, but are unlikely to contribute to present observations because light-dependent fluctuations in rod cell pH are relatively small (∼0.2 pH units) and 1/T1 in the absence of a pH-sensitive contrast agent is not particularly sensitive to pH.56 In addition, dark/light changes in rod 1/T1 could not be due to hemodynamic changes because the photoreceptor layer is avascular. These considerations further support the interpretation that quench-assisted MRI directly detects continuous production of paramagnetic free radicals in photoreceptors and RPE in vivo.
Quench-assisted MRI is likely to be most useful in the presence of an excessive and continuous production of free radicals, a condition of interest that suggests the presence of high levels of oxidative stress. Is measurement of supernormal free radical production necessary and sufficient to identify oxidative stress? Oxidative stress is the continual generation of free radicals exceeding a cell's quenching capacity. However, it is not currently possible to image the endogenous ability of cells to quench free radicals. It is likely that when free radical production exceeds free radical removal, redox-regulated neuronal signaling will be substantially altered and this will induce downstream neuronal pathophysiology.57,58 We expect that a diagnosis of pathologic oxidative stress in vivo might be improved by measuring if (1) there is greater than normal production of free radicals, (2) if such supernormal free radical production is associated with neuronal dysfunction, and (3) if a quench condition corrects (1) and (2). Notably, MRI has matured to the point in which more key functional indices of the health of rod and RPE cells can be measured in mice in vivo than is currently possible by any other imaging approach.19–24 These indices include measurement of rod outer nuclear layer L-type calcium channel (LTCC) function in dark and light, outer nuclear layer arrestin-1 and its light-evoked translocation, and light-dependent expansion of the extracellular space surrounding rod outer segments (a process controlled in part by rod cells and in part by RPE cells).19–24 Future studies that combine quench-assisted MRI with MRI measurements of impaired LTCC function and light-evoked subretinal space expansion in the same set of cells during the same imaging session will uniquely evaluate oxidative stress as it relates to disease pathogenesis in vivo. Because much of the free radical and functional contrast is based on endogenous mechanisms without the need for injection of a contrast agent, clinical translation of this approach into humans is expected.
In summary, quench-assisted MRI is a feasible method for noninvasively evaluating continuous free radical production in photoreceptors and RPE in vivo. In addition, quench-assisted MRI can be readily combined with other MRI assays of photoreceptor function to aid in the identification of pathogenic oxidative stress in vivo. We anticipate that quench-assisted MRI will be translatable to humans because high-resolution images of the retina are possible without sedation using a cued-blinking procedure, and LPA and MB are clinically relevant, as are many other AOs.26,59–62 Future applications of this technology may include the study of continuous free radical production in other persistently active neurons (e.g., dopaminergic pacemaker cells), in neurons engineered to be photosensitive (optogenics), and in cells particularly vulnerable to generating oxidative stress (e.g., CA1 region of the hippocampus).
Acknowledgments
BAB thanks Robia Pautler, PhD, (Baylor College of Medicine, Houston, TX, USA) for originally drawing his attention to the potential of measuring free radical production using a quench assay based on her liver studies.
Supported by National Institutes of Health Grant EY021619 (BAB), an unrestricted grant from Research to Prevent Blindness (BAB), and the Swiss National Science Foundation (SNF 31003A_149311).
Disclosure: B.A. Berkowitz, None; B.X. Bredell, None; C. Davis, None; M. Samardzija, None; C. Grimm, None; R. Roberts, None
References
- 1. Wang X,, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci. 2010; 2: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Du Y,, Veenstra A,, Palczewski K,, Kern TS. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. PNAS. 2013; 110: 16586–16591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Campochiaro PA,, Strauss RW,, Lu L,, et al. Is there excess oxidative stress and damage in eyes of patients with retinitis pigmentosa? Antioxid Redox Signal. 2015; 23: 643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Punzo C,, Xiong W,, Cepko CL. Loss of daylight vision in retinal degeneration: are oxidative stress and metabolic dysregulation to blame? J Biol Chem. 2012; 287: 1642–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rohrer B,, Pinto FR,, Hulse KE,, Lohr HR,, Zhang L,, Almeida JS. Multidestructive pathways triggered in photoreceptor cell death of the RD mouse as determined through gene expression profiling. J Biol Chem. 2004; 279: 41903–41910. [DOI] [PubMed] [Google Scholar]
- 6. Sanz MM,, Johnson LE,, Ahuja S,, Ekstrom PA,, Romero J,, van Veen T. Significant photoreceptor rescue by treatment with a combination of antioxidants in an animal model for retinal degeneration. Neuroscience. 2007; 145: 1120–1129. [DOI] [PubMed] [Google Scholar]
- 7. Usui S,, Komeima K,, Lee SY,, et al. Increased expression of catalase and superoxide dismutase 2 reduces cone cell death in retinitis pigmentosa. Mol Ther. 2009; 17: 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Komeima K,, Rogers BS,, Lu L,, Campochiaro PA. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. PNAS. 2006; 103: 11300–11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Galbinur T,, Obolensky A,, Berenshtein E,, et al. Effect of para-aminobenzoic acid on the course of retinal degeneration in the rd10 mouse. J Ocul Pharmacol Ther. 2009; 25: 475–482. [DOI] [PubMed] [Google Scholar]
- 10. Berkowitz BA,, Gradianu M,, Bissig D,, Kern TS, Roberts R. Retinal ion regulation in a mouse model of diabetic retinopathy: natural history and the effect of Cu/Zn superoxide dismutase overexpression. Invest Ophthalmol Vis Sci. 2009; 50: 2351–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng L,, Du Y,, Miller C,, et al. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia. 2007; 50: 1987–1996. [DOI] [PubMed] [Google Scholar]
- 12. Berkowitz BA,, Roberts R,, Stemmler A,, Luan H,, Gradianu M. Impaired apparent ion demand in experimental diabetic retinopathy: correction by lipoic Acid. Invest Ophthalmol Vis Sci. 2007; 48: 4753–4758. [DOI] [PubMed] [Google Scholar]
- 13. Prunty MC,, Aung MH,, Hanif AM,, et al. In vivo imaging of retinal oxidative stress using a reactive oxygen species-activated fluorescent probe imaging of retinal oxidative stress using H-800CW. Invest Ophthalmol Vis Sci. 2015; 56: 5862–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhelev Z,, Bakalova R,, Aoki I,, Lazarova D,, Saga T. Imaging of superoxide generation in the dopaminergic area of the brain in Parkinson's disease, using mito-TEMPO. ACS Chem Neurosci. 2013; 4: 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stinnett G,, Moore K,, Samuel E,, et al. A novel assay for the in vivo detection of reactive oxygen species using MRI. In: ISMRM 23rd Annual Meeting & Exhibition. Toronto, Ontario: International Society for Magnetic Resonance in Medicine; 2015. Abstract 1917.
- 16. Ekanger LA,, Allen MJ. Overcoming the concentration-dependence of responsive probes for magnetic resonance imaging. Metallomics. 2015; 7: 405–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Okawa H,, Sampath AP,, Laughlin SB,, Fain GLATP. Consumption by mammalian rod photoreceptors in darkness and in light. Curr Biol. 2008; 18: 1917–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Balaban RS,, Nemoto S,, Finkel T. Mitochondria oxidants, and aging. Cell. 2005; 120: 483–495. [DOI] [PubMed] [Google Scholar]
- 19. Berkowitz BA,, Bissig D,, Patel P,, Bhatia A,, Roberts R. Acute systemic 11-cis-retinal intervention improves abnormal outer retinal ion channel closure in diabetic mice. Mol Vis. 2012; 18: 372–376. [PMC free article] [PubMed] [Google Scholar]
- 20. Berkowitz BA,, Roberts R,, Oleske DA,, et al. Quantitative mapping of ion channel regulation by visual cycle activity in rodent photoreceptors in vivo. Invest Ophthalmol Vis Sci. 2009; 50: 1880–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berkowitz BA,, Bissig D,, Dutczak O,, Corbett S,, North R,, Roberts R. MRI biomarkers for evaluation of treatment efficacy in preclinical diabetic retinopathy. Expert Opin Med Diagn. 2013; 7: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Berkowitz BA,, Grady EM,, Khetarpal N,, Patel A,, Roberts R. Oxidative stress and light-evoked responses of the posterior segment in a mouse model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2015; 56: 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berkowitz BA,, Gorgis J,, Patel A,, et al. Development of an MRI biomarker sensitive to tetrameric visual arrestin 1 and its reduction via light-evoked translocation in vivo. FASEB J. 2014; 29: 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berkowitz BA,, Bissig D,, Roberts R. MRI of rod cell compartment-specific function in disease and treatment in-vivo [published online ahead of print September 4, 2015]. Prog Retin Eye Res. doi:http://dx.doi.org/10.1016/j.preteyeres.2015.09.001. [DOI] [PMC free article] [PubMed]
- 25. Roberts R,, Luan H,, Berkowitz BA. Alpha-lipoic acid corrects late-phase supernormal retinal oxygenation response in experimental diabetic retinopathy. Invest Ophthalmol Vis Sci. 2006; 47: 4077–4082. [DOI] [PubMed] [Google Scholar]
- 26. Wen Y,, Li W,, Poteet EC,, et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J Biol Chem. 2011; 286: 16504–16515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang X,, Rojas JC,, Gonzalez-Lima F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox Res. 2006; 9: 47–57. [DOI] [PubMed] [Google Scholar]
- 28. Perkins GA,, Ellisman MH,, Fox DA. Three-dimensional analysis of mouse rod and cone mitochondrial cristae architecture: bioenergetic and functional implications. Mol Vis. 2003; 9: 60–73. [PubMed] [Google Scholar]
- 29. Samardzija M,, Caprara C,, Heynen SR,, et al. A mouse model for studying cone photoreceptor pathologies R91W;Nrl-/- mouse model. Invest Ophthalmol Vis Sci. 2014; 55: 5304–5313. [DOI] [PubMed] [Google Scholar]
- 30. Wangsa-Wirawan ND,, Linsenmeier RA. Retinal oxygen: fundamental and clinical aspects. Arch Ophthalmol. 2003; 121: 547–557. [DOI] [PubMed] [Google Scholar]
- 31. Yu DY,, Cringle SJ. Oxygen distribution in the mouse retina. Invest Ophthalmol Vis Sci. 2006; 47: 1109–1112. [DOI] [PubMed] [Google Scholar]
- 32. Bissig D,, Berkowitz BA. Light-dependent changes in outer retinal water diffusion in rats in vivo. Mol Vis. 2012; 18: 2561. [PMC free article] [PubMed] [Google Scholar]
- 33. Bissig D,, Berkowitz BA. Same-session functional assessment of rat retina and brain with manganese-enhanced MRI. NeuroImage. 2011; 58: 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vallet P,, Van HY,, Bonnet PA,, Subra G,, Chapat JP,, Muller RN. Relaxivity enhancement of low molecular weight nitroxide stable free radicals: importance of structure and medium. Magn Reson Med. 1994; 32: 11–15. [DOI] [PubMed] [Google Scholar]
- 35. Cheng H,, Nair G,, Walker TA,, et al. Structural and functional MRI reveals multiple retinal layers. Proc Natl Acad Sci U S A. 2006; 103: 17525–17530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Longo A,, Geiser M,, Riva CE. Subfoveal choroidal blood flow in response to light-dark exposure. Invest Ophthalmol Vis Sci. 2000; 41: 2678–2683. [PubMed] [Google Scholar]
- 37. Fuchsjäger-Mayrl G,, Polska E,, Malec M,, Schmetterer L. Unilateral light-dark transitions affect choroidal blood flow in both eyes. Vision Res. 2001; 41: 2919–2924. [DOI] [PubMed] [Google Scholar]
- 38. Fitzgerald ME,, Gamlin PD,, Zagvazdin Y,, Reiner A. Central neural circuits for the light-mediated reflexive control of choroidal blood flow in the pigeon eye: a laser Doppler study. Vis Neurosci. 1996; 13: 655–669. [DOI] [PubMed] [Google Scholar]
- 39. Berkowitz BA,, Roberts R,, Goebel DJ,, Luan H. Noninvasive and simultaneous imaging of layer-specific retinal functional adaptation by manganese-enhanced MRI. Invest Ophthalmol Vis Sci. 2006; 47: 2668–2674. [DOI] [PubMed] [Google Scholar]
- 40. Liang Z. Longitudinal data analysis using generalized linear models. Biometrika. 1986; 73: 13–22. [Google Scholar]
- 41. Enzmann V,, Row BW,, Yamauchi Y,, et al. Behavioral and anatomical abnormalities in a sodium iodate-induced model of retinal pigment epithelium degeneration. Exp Eye Res. 2006; 82: 441–448. [DOI] [PubMed] [Google Scholar]
- 42. Franco LM,, Zulliger R,, Wolf-Schnurrbusch UE,, et al. Decreased visual function after patchy loss of retinal pigment epithelium induced by low-dose sodium iodate. Invest Ophthalmol Vis Sci. 2009; 50: 4004–4010. [DOI] [PubMed] [Google Scholar]
- 43. Wang J,, Iacovelli J,, Spencer C,, Saint-Geniez M. Direct effect of sodium iodate on neurosensory retina. Invest Ophthalmol Vis Sci. 2014; 55: 1941–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cingolani C,, Rogers B,, Lu L,, Kachi S,, Shen J,, Campochiaro PA. Retinal degeneration from oxidative damage. Free Radic Biol Med. 2006; 40: 660–669. [DOI] [PubMed] [Google Scholar]
- 45. Du Y,, Miller CM,, Kern TS. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic Biol Med. 2003; 35: 1491–1499. [DOI] [PubMed] [Google Scholar]
- 46. Du Y,, Cramer M,, Lee CA,, et al. Adrenergic and serotonin receptors affect retinal superoxide generation in diabetic mice: relationship to capillary degeneration and permeability. FASEB J. 2015; 29: 2194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu H,, Tang J,, Du Y,, et al. Retinylamine benefits early diabetic retinopathy in mice. J Biol Chem. 2015; 290: 21568–21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muir ER,, Chandra SB,, De La Garza BH,, Velagapudi C,, Abboud HE,, Duong TQ. Layer-specific manganese-enhanced MRI of the diabetic rat retina in light and dark adaptation at 11.7 Tesla. Invest Ophthalmol Vis Sci. 2015; 56: 4006–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Giordano C,, Roberts R,, Krentz K,, et al. Catalase therapy corrects oxidative stress-induced pathophysiology in incipient diabetic retinopathy. Invest Ophthalmol Vis Sci. 2015; 56: 3095–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Samuels IS,, Bell BA,, Pereira A,, Saxon J,, Peachey NS. Early retinal pigment epithelium dysfunction is concomitant with hyperglycemia in mouse models of type 1 and type 2 diabetes. J Neurophysiol. 2015; 113: 1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuse Y,, Ogawa K,, Tsuruma K,, Shimazawa M,, Hara H. Damage of photoreceptor-derived cells in culture induced by light emitting diode-derived blue light. Sci Rep. 2014; 4: 5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Usui S,, Oveson BC,, Iwase T,, et al. Overexpression of SOD in retina: need for increase in H2O2-detoxifying enzyme in same cellular compartment. Free Radic Biol Med. 2011; 51: 1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Berkowitz BA. Role of dissolved plasma oxygen in hyperoxia-induced contrast. Magn Reson Imaging. 1997; 15: 123–126. [DOI] [PubMed] [Google Scholar]
- 54. Berkowitz BA. Adult and newborn rat inner retinal oxygenation during carbogen and 100% oxygen breathing. Comparison using magnetic resonance imaging delta Po2 mapping. Invest Ophthalmol Vis Sci. 1996; 37: 2089–2098. [PubMed] [Google Scholar]
- 55. Linsenmeier RA,, Braun RD. Oxygen distribution and consumption in the cat retina during normoxia and hypoxemia. J Gen Physiol. 1992; 99: 177–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yamamoto F,, Borgula GA,, Steinberg RH. Effects of light and darkness on pH outside rod photoreceptors in the cat retina. Exp Eye Res. 1992; 54: 685–697. [DOI] [PubMed] [Google Scholar]
- 57. Poljsak B,, Milisav I. The neglected significance of “antioxidative stress.” Oxid Med Cell Longev. 2012; 2012: 480895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kamata H,, Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999; 11: 1–14. [DOI] [PubMed] [Google Scholar]
- 59. Berkowitz BA,, McDonald C,, Ito Y,, Tofts PS,, Latif Z,, Gross J. Measuring the human retinal oxygenation response to a hyperoxic challenge using MRI: eliminating blinking artifacts and demonstrating proof of concept. Magn Reson Med. 2001; 46: 412–416. [DOI] [PubMed] [Google Scholar]
- 60. Gomes M,, Negrato C. Alpha-lipoic acid as a pleiotropic compound with potential therapeutic use in diabetes and other chronic diseases. Diabetol Metab Syndr. 2014; 6: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lo JCY,, Darracq MA,, Clark RFA. Review of methylene blue treatment for cardiovascular collapse. J Emerg Med. 2014; 46: 670–679. [DOI] [PubMed] [Google Scholar]
- 62. Atamna H,, Nguyen A,, Schultz C,, et al. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2008; 22: 703–712. [DOI] [PubMed] [Google Scholar]