

Abstract
The complement system is an essential element of the innate immune response that becomes activated upon recognition of molecular patterns associated with microorganisms, abnormal host cells, and modified molecules in the extracellular environment. The resulting proteolytic cascade tags the complement activator for elimination and elicits a pro‐inflammatory response leading to recruitment and activation of immune cells from both the innate and adaptive branches of the immune system. Through these activities, complement functions in the first line of defense against pathogens but also contributes significantly to the maintenance of homeostasis and prevention of autoimmunity. Activation of complement and the subsequent biological responses occur primarily in the extracellular environment. However, recent studies have demonstrated autocrine signaling by complement activation in intracellular vesicles, while the presence of a cytoplasmic receptor serves to detect complement‐opsonized intracellular pathogens. Furthermore, breakthroughs in both functional and structural studies now make it possible to describe many of the intricate molecular mechanisms underlying complement activation and the subsequent downstream events, as well as its cross talk with, for example, signaling pathways, the coagulation system, and adaptive immunity. We present an integrated and updated view of complement based on structural and functional data and describe the new roles attributed to complement. Finally, we discuss how the structural and mechanistic understanding of the complement system rationalizes the genetic defects conferring uncontrolled activation or other undesirable effects of complement.
Keywords: complement, inflammation, innate immunity, proteolytic regulation, structural biology
Subject Categories: Immunology
Glosssary
- 3MC
Malpuech, Michels, and Mingarelli‐Carnevale
- 7TM
Seven transmembrane
- aHUS
Atypical hemolytic uremic syndrome
- AMD
Age‐related macular degeneration
- AP
Alternative pathway of complement
- Bb
Activated factor B
- C3aR
C3a anaphylatoxin chemotactic receptor
- C5aR1
C5a anaphylatoxin chemotactic receptor 1
- C5aR2
C5a anaphylatoxin chemotactic receptor 2
- CCP
Complement control protein
- CFHR
Complement factor H‐related protein
- CL
Collectin
- CP
Classical pathway of complement
- CR
Complement receptor
- CRD
Carbohydrate‐recognition domain
- CUB
Complement C1r/C1s, Uegf, Bmp1
- CVF
Cobra venom factor
- DAF
Decay‐accelerating factor
- DAMP
Danger‐associated molecular pattern
- EGF
Epidermal growth factor
- EM
Electron microscopy
- FB
Factor B
- FBG
Fibrinogen
- FD
Factor D
- FDC
Follicular dendritic cell
- FH
Factor H
- FHL‐1
Factor H‐like protein 1
- FI
Factor I
- FP
Properdin
- GAG
Glycosaminoglycan
- GPCR
G protein‐coupled receptor
- IR
Ischemia–reperfusion
- LP
Lectin pathway of complement
- MAC
Membrane attack complex
- MAp19
MBL‐associated protein of 19 kDa
- MAp44
MBL‐associated protein of 44 kDa
- MASP
MBL‐associated serine protease
- MBL
Mannan‐binding lectin
- MCP
Membrane cofactor protein
- MDA
Malonedialdehyde
- MG
Macroglobulin
- MPGNII
Membranoproliferative glomerulonephritis type II
- PAMP
Pathogen‐associated molecular pattern
- PNH
Paroxysmal nocturnal hemoglobinuria
- PRM
Pattern recognition molecule
- SAXS
Small‐angle X‐ray scattering
- SCR
Short consensus repeat
- SLE
Systemic lupus erythematosus
- SP
Serine protease
- SSM
Subcapsular sinus macrophages
- TP
The terminal pathway of complement
- vWA
Von Willebrand factor A
Introduction
The complement system is canonically regarded as a major effector within innate immunity. As a universally distributed defense mechanism in blood and interstitial fluids, complement is one of the first lines of defense against pathogenic microorganisms that breach the mechanical and chemical barriers of the body. It is a germ line‐encoded system of more than 50 circulating and membrane‐bound proteins. The majority of the circulating proteins are produced in the liver, although extrahepatic complement biosynthesis does occur in many other cell types including fibroblasts, T and B cells, adipocytes and endothelial cells (Morgan & Gasque, 1997). The local production of complement proteins appears to be sufficient for the generation of humoral immune responses and is the main source of complement in immune‐privileged sites such as the brain and the eye (Barnum, 1995; Gadjeva et al, 2002).
Complement was first identified as the heat‐sensitive fraction of human plasma that “complemented” antibodies in their ability to kill bacteria. Although complement is usually considered pro‐inflammatory, it has also proven important in the homeostatic processes leading to the removal of dying cells presenting danger‐associated molecular patterns (DAMPs) where complement triggers a sterile inflammatory response leading to essentially the same vascular and cellular inflammatory state (Rock et al, 2010). More recent research additionally associates complement with transport of immune complexes, and regulation of humoral immunity (Carroll & Isenman, 2012). Furthermore, recent findings that implicate complement in angiogenesis and synaptic pruning highlight the role of complement during development in mice (Schafer et al, 2012; Stephan et al, 2012; Haynes et al, 2013). Complement receptors, effectors, and regulators intertwine into a complex network interacting with other crucial pathways such as the coagulation pathway and Toll‐like receptor sensing and signaling (Hawlisch & Kohl, 2006; Amara et al, 2008). Clearly, this intricate network and its interplay with other systems need to be carefully controlled. If this fails, complement can target host tissues and cause organ damage leading to autoimmune and chronic inflammatory diseases.
A long‐standing observation in the clinic that complement deficiencies lead to autoimmune diseases is now being explained by the genetic, functional, and structural studies of complement regulators. One of the complement pattern recognition molecules (C1q) has been identified as an important player in the clearance of autoantigens offering an explanation as to why deficiency of C1q presents the strongest known genetic predisposition for development of the autoimmune disease systemic lupus erythematosus (SLE) with near complete penetrance (Pickering et al, 2000). Acquired complement deficiencies are also frequently observed in SLE and thought to contribute to pathogenesis. Examples of such acquired deficiencies are lowered C1q levels caused by autoantibodies against C1q, or a lowered concentration of two other pivotal complement proteins (C3 and C4). Thus, a well‐established tool to monitor SLE activity used worldwide is measurements of C3 and C4 levels (also as part of disease scoring systems, e.g., SLEDAI) (Bombardier et al, 1992; Gladman et al, 2002).
After the emergence of genome sequencing, it likewise became clear that polymorphisms of complement genes were quite frequent (> 5%). Recent elaborate reviews on the genetics underlying complement deficiencies are found in references (de Cordoba et al, 2012; Rodriguez et al, 2014; de Cordoba, 2015; Liszewski & Atkinson, 2015). The individual‐specific ensemble of polymorphisms in genes encoding complement proteins and regulators can significantly influence the balance between complement activation and regulation, and the set of polymorphisms which determines the intrinsic complement activity is referred to as the complotype of an individual [for review, see Harris et al (2012)]. In the following, we provide an overview of the molecular mechanisms of complement activation and regulation and couple this to the rapidly growing information concerning the structure of complement proteins and their complexes with particular emphasis on understanding the role of complement proteins in health and disease.
Complement activation
Upon complement activation, structural rearrangements, proteolytic cleavages, and the assembly of proteolytic and lytic complexes occur. In this way, complement can be ubiquitously present in an inactive form but become activated locally. Many of the molecules and processes we describe in this Review are illustrated in Fig 1. Complement is activated through the classical pathway (CP), the lectin pathway (LP), and the alternative pathway (AP). The recognition of invading microorganisms by the complement system can occur directly via recognition of pathogen‐associated molecular patterns (PAMPs) by soluble pattern recognition molecules (PRMs). In humans, these are complement protein C1q, mannan‐binding lectin (MBL), collectin‐LK (CL‐LK), or the three ficolins L/M/H (also denoted ficolin‐1, ficolin‐2, and ficolin‐3) (Degn & Thiel, 2013). In the classical and lectin pathways, binding of PRMs to a PAMP or a DAMP (the activator) confers activation of zymogen proteases in complex with the PRMs. Within the CP, the C1 complex consists of the PRM C1q associated with the serine proteases C1r and C1s organized as a calcium‐dependent C1r2s2 tetramer (Arlaud et al, 2001). In antibody‐dependent CP activation, the globular heads of C1q bind to the Fc moieties of multivalent IgG–antigen complexes or to antigen‐bound IgM (Nayak et al, 2012) (Fig 1A). In addition to antibody–antigen complexes, a variety of other ligands have been suggested for C1q (Fig 1A). This includes molecular patterns on certain bacteria, viruses, parasites, and mycoplasma, indicating a role as an antibody‐independent PRM. C1q has also been reported to bind to C‐reactive protein (CRP) in complex with exposed phosphocholine residues on bacteria (Szalai et al, 1999), providing a further means of host defense (Fig 1A). Other C1q ligands are pentraxin‐3 (PTX‐3), serum amyloid P component, β‐amyloid fibrils, as well as tissue damage elements such as DNA and mitochondrial membranes (Kang et al, 2009) (Fig 1A). C1q likewise recognizes a variety of DAMPs exposed by apoptotic cells explaining its linkage to SLE (see below). C1q has been reported to directly bind phosphatidylserine exposed on apoptotic cells (Paidassi et al, 2008), although more recent data suggest that the binding targets are rather DNA, histones, and Annexins A2 and A5 on the apoptotic cell surface (Martin et al, 2012). Recently, the proteins SCARF1 and LAIR‐1 were invoked as immunomodulatory receptors for C1q‐opsonized apoptotic cells, potentially explaining the role of C1q in SLE (Son et al, 2012; Ramirez‐Ortiz et al, 2013).
Figure 1. Molecular view of complement activation, amplification, and regulation.
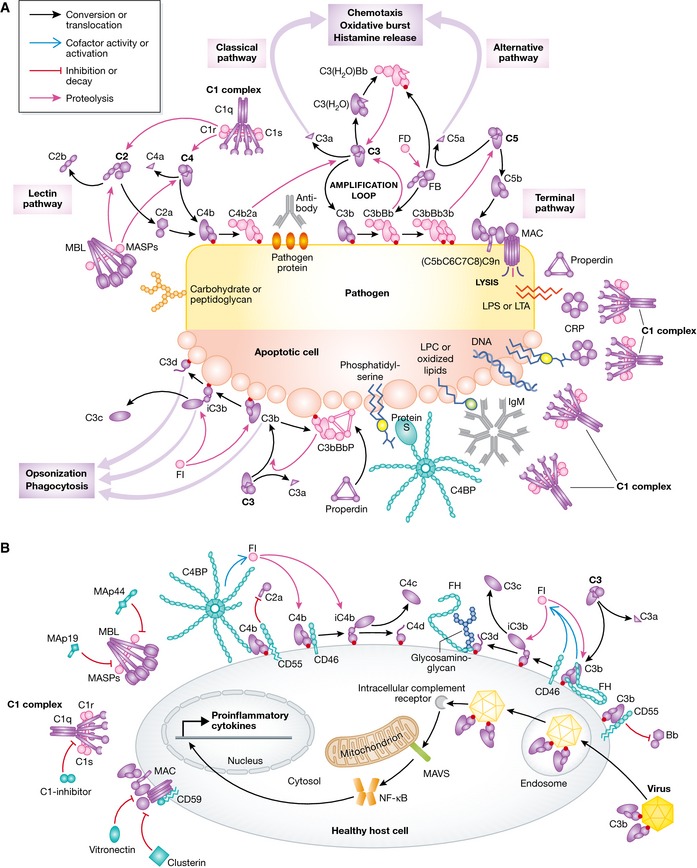
(A) Pattern recognition molecules sense the presence of pathogens and altered self. In the classical pathway, C1q (within the C1 complex) recognizes PAMPs (pathogen‐specific proteins, lipopolysaccharide (LPS), lipoteichoic acid (LTA), peptidoglycan) or DAMPs (DNA, phosphatidylserine, oxidized lipids, lysophosphatidylcholine (LPC)) either directly or antibody‐bound. This recognition induces autoactivation of C1r, which subsequently activates C1s. This is followed by cleavage of C4 and C2 by C1s and the subsequent formation of the CP C3 convertase C4b2a. Cleavage of C4 exposes an internal thioester, which causes C4b to become covalently attached to the activator surface, in turn tethering the convertase activity to the activator. In the lectin pathway, patterns of glycans are detected via MBL, CL‐LK, or ficolins leading to activation of MASPs and formation of the same C3 convertase, C4b2a. C3 convertases cleave C3 into C3b, which also becomes covalently attached to the activator surface. Surface‐associated C3b recruits FB, which leads to FB activation and the formation of C3bBb, the AP C3 convertase, which cleaves more C3 and amplifies complement activation. In addition to the surface‐bound C3 convertase, a fluid‐phase convertase can be formed by association of water‐reacted C3, termed C3(H20), to FB thus constantly maintaining a low level of complement activation in solution (tick‐over). Both of the surface‐bound C3 convertases can bind a C3b molecule whereby the C5 convertases are formed. These cleave C5 into C5a and C5b, thus initiating the terminal pathway and leading to formation of the membrane attack complex (MAC). Complement opsonins and PRMs are shown in purple, whereas the proteolytically active complexes are shown in light pink. (B) Complement activation and amplification are attenuated on host surfaces. The healthy cells express membrane‐bound or attract soluble regulators that irreversibly dissociate convertases (DAF, CR1, FH, C4BP) and act as cofactors for FI‐mediated degradation of C3b and C4b (MCP, FH, CR1, C4BP) or prevent MAC assembly (CD59). Soluble regulators also prevent formation of the MAC (clusterin, vitronectin). Recently, it was discovered that complement mediates a potent intracellular immune response to non‐enveloped viruses. Deposition and covalent attachment of C3 onto pathogens in the extracellular environment serve as a marker of cellular invasion because C3 products in the cytosol are detected by an as yet unidentified receptor. This receptor signals through MAVS and induces an antiviral state by triggering the transcription of pro‐inflammatory cytokines. Intracellular complement immunity is independent of professional immune cells and is conserved in mammals.
Following C1q‐ligand binding, C1r autoactivates and subsequently cleaves C1s, which may then cleave C4 into the fragments C4a and C4b (Fig 1A). The nascent C4b can be covalently bound to the activator via an exposed internal thioester leading to irreversible tagging of the activator. C2 binds activator‐bound C4b and is cleaved by C1s to generate the active serine protease C2a bound to C4b resulting in the CP C3 convertase C4b2a (Muller‐Eberhard et al, 1967). The C3 convertase cleaves C3 into the anaphylatoxin C3a and the major opsonin of the complement system, C3b, which like C4b, becomes covalently coupled to the activator through its exposed thioester (Law & Dodds, 1997).
Activation of the lectin pathway (LP) is initiated by the collectins MBL and CL‐LK or one of the three ficolins (Fig 1A). MBL and CL‐LK harbor Ca2+‐dependent carbohydrate‐recognition domains (CRDs) and collagen‐like regions through which they trimerize. Such trimers oligomerize in larger complexes (Figs 1A and 2A), allowing high‐avidity binding (K D ≈ 10−9 M) based on multiple low‐affinity interactions of their CRDs (K D ≈ 10−3 M) (Kawasaki et al, 1983; Degn & Thiel, 2013). Ficolins are structurally similar to collectins, but instead of C‐type lectin domains they possess fibrinogen (FBG)‐like domains for PAMP recognition (Matsushita, 2013). Ficolins recognize motifs containing acetylated groups, including non‐sugars such as N‐acetyl‐glycine, N‐acetyl‐cysteine, and acetylcholine. Besides conferring avidity, the oligomerization of collectins and ficolins allows these PRMs to discriminate not only specific monosaccharides or acetylated groups but more importantly, in an immunological sense, also specific patterns of sugars and acetyl groups characteristic to pathogens.
Figure 2. Large macromolecular complexes of complement proteins assembled upon complement activation.
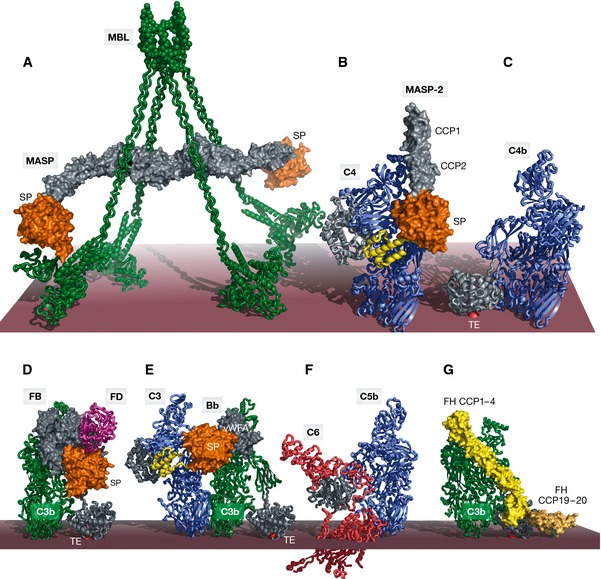
The order of panels (A–F) reflects the order of appearance starting from activation in the LP and ending with MAC assembly in the TP. (A) SAXS model of the MBL:MASP‐1 complex with MBL (green) associated with a MASP‐1 homodimer with its serine protease domains (orange) protruding away from the MBL collagen stems in agreement with an intercomplex activation mechanism. (B) Crystal structure of the C4:MASP‐2 complex (RCSB ID 4FXG) with the substrate (C4, blue with the anaphylatoxin domain in yellow) making contacts at two distinct sites; the CCP domains (gray) and the SP domain (orange). (C) Crystal structure of C4b (RSCB ID 4XAM) with the TE domain colored in gray and the reactive thioester covalently bound to the membrane shown as a red sphere. (D) Crystal structure of the ternary C3bB:D complex (RSCB ID 2XWB). FB binds C3b (green, with the TE domain in gray) via its vWA and 3 CCP domains (gray). The SP domain (orange) is in the closed state. FD (magenta) is recruited to FB. (E) Structural model of the AP C3 convertase in complex with a C3 substrate (blue) generated by superimposing C3bBb stabilized with SCIN (RCSB ID 2WIN) and the C5:CVF complex (RCSB ID 3PVM). The anaphylatoxin moiety (yellow) is released upon cleavage. (F) Crystal structure of the C5b6 complex (RCSB ID 4A5W) revealing conformational rearrangements occurring upon C5 cleavage to C5b (blue), reminiscent of those observed in the C3/C4 to C3b/C4b conversion. (G) Structural model of FH binding to C3b (green) generated by superimposing C3b bound to FH CCP1–4 (light yellow, RCSB ID 2WII) and TE domain bound to FH CCP19–20 (dark yellow, RCSB ID 4ONT), CCP5–18 are not illustrated. FH also interacts with host glycans. The binding of FH prepares C3b for FI binding and cleavage. In all panels, the red surface approximates the activator such as the surface of an LPS layer on a pathogenic bacterium. Importantly, this is separated from the cell membrane; thus, panel (F) does not imply that C6 extends into the membrane.
The LP PRMs recognize the sugar moieties and acetyl groups of a variety of foreign glycoproteins and glycolipids. A plethora of physiological microbial targets of MBL have been defined, encompassing Gram‐positive and Gram‐negative bacteria, viruses, fungi, and protozoa (Kjaer et al, 2013). The second collectin CL‐LK is a heterocomplex of two polypeptide chains, CL‐L1 and CL‐K1 (Henriksen et al, 2013a). CL‐LK has been reported to bind mannan slightly less efficiently than MBL but DNA more efficiently (Henriksen et al, 2013b). However, rather few studies on the specificity of plasma CL‐LK have been published since plasma components seem to inhibit most binding (Henriksen et al, 2013a), but recombinant CL‐K1 shows binding to mannan (i.e., carbohydrate structures on yeast surfaces), to strains of Escherichia coli, Candida albicans, and Pseudomonas aeruginosa (Selman & Hansen, 2012), and also to various oligonucleotides (Henriksen et al, 2013b). Binding of CL‐L1 to mannose‐coated beads has been observed (Axelgaard et al, 2013). H‐ficolin is reported to bind to strains of Mycobacteriae, Aerococcus viridans (Tsujimura et al, 2001), and Hafnia alfvei (Swierzko et al, 2012), Salmonella typhimurium, Salmonella minnesota, and E. coli (Sugimoto et al, 1998). L‐ficolin displays a very promiscuous binding to several bacteria (Krarup et al, 2005) including strains of S. typhimurium, E. coli, Staphylococcus aureus, and Streptococcus pneumoniae. M‐ficolin binds to specific strains of Streptococcus agalactiae and S. pneumoniae (Kjaer et al, 2011). In addition to recognizing foreign glycoproteins and glycolipids, the LP has been shown to recognize host organelles, mitochondria, and cause their opsonization by C4 and C3 activation products. However, the immune handling of opsonized mitochondria was not accompanied by inflammation (Brinkmann et al, 2013). These observations further corroborate complement as a mediator of non‐inflammatory, homeostatic clearance of DAMPs as mentioned above. Conversely, MBL binding to natural IgM recognizing autoantigenic neoepitopes exposed on apoptotic or necrotic cells following, for example, ischemia–reperfusion (IR) injury, can lead to untoward activation and tissue damage (Zhang et al, 2006). A long‐standing observation regarding the LP is that a small subset of lupus patients also harbor anti‐H‐ficolin antibodies (Yoshizawa et al, 1997). Indeed, H‐ficolin was first identified as an autoantigen in SLE and dubbed the Hakata antigen, and based on our recent elucidation of the clustering‐based activation mechanism of the lectin pathway (see later), it is tempting to speculate that antibody‐driven cross‐linking of H‐ficolin:MASP complexes may drive aberrant complement activation (Degn et al, 2014b). An observation of the cell surface association of M‐ficolin by tethering through recognition of membrane‐associated sialic acids by the fibrinogen‐like (FBG) domain of M‐ficolin implies its potential role in cell clearance (Honore et al, 2010).
The LP PRMs form complexes with MBL‐associated serine proteases (MASPs), which are always present as dimers (Fig 2A). MASP‐1 and MASP‐2 are structural and functional homologs of C1r and C1s from the CP, but there are important differences between PRM–protease complexes from the two pathways. Whereas the C1 complex has a defined stoichiometry (a hexamer of the heterotrimeric C1q subunit in complex with a C1r2s2 tetramer), the LP PRMs are polydisperse oligomers of trimers. For MBL, a tetramer is the most abundant oligomer and this carries only a single MASP‐1 or MASP‐2 dimer (Fig 2A), but the more rare, larger oligomers may simultaneously carry both dimers (Dahl et al, 2001; Teillet et al, 2005; Degn et al, 2013a). MASP‐1 in complex with an activator‐bound PRM autoactivates and cleaves MASP‐2 as well as C2, whereas activated MASP‐2 cleaves C4 (Fig 2B) and C2 resulting in the same C3 convertase as in the CP, that is, C4b2a (Matsushita et al, 2000; Rossi et al, 2001; Chen & Wallis, 2004). An efficient catalytic activity of the third MASP, MASP‐3, is yet to be discovered. The substrates suggested so far, insulin‐like growth factor‐binding protein 5 (Cortesio & Jiang, 2006) and pro‐factor D and factor B (Iwaki et al, 2011), are all cleaved slowly at high enzyme to substrate ratio. For example, zymogen MASP‐3 cleavage of pro‐factor D occurred at 20:1 molar ratio over 1 h at 37°C (Iwaki et al, 2011).
Activation through the CP and LP results in deposition of C3b (Fig 1A) on the activator, which recruits factor B (FB) in the first step of the AP. The resulting proconvertase C3bB is subsequently cleaved by factor D (FD), generating the AP C3 convertase C3bBb (Fearon et al, 1973), which is functionally homologous to the CP C3 convertase C4b2a. A positive feedback amplification loop is now initiated as multiple copies of C3b are deposited on the activator leading to further assembly of the AP C3 convertase. Regardless of the initiating pathway, up to 90% of the deposited C3b molecules are generated through the AP (Harboe et al, 2004, 2009). This amplification is rapidly terminated on host cells by various regulators (Fig 1B) (discussed later), but proceeds vividly on pathogens and altered host tissues lacking such regulators (Fig 1A). Importantly, the AP may initiate independently of the CP and LP and without direct pattern recognition. This occurs through constitutive generation of C3(H2O), a fluid‐phase form of C3 in which the thioester has reacted with a water molecule (Pangburn et al, 1981). C3(H2O) resembles C3b and is capable of binding FB and forming a fluid‐phase C3 convertase generating more C3b, and further amplification may now occur on nearby surfaces as described above (Fig 1A). The spontaneous initiation mechanism of the AP is potentially dangerous but is controlled through “reverse recognition” as the AP is specifically inhibited on host cells. C3(H2O) is not long‐lived as it is rapidly inactivated by the FI protease (see below).
Properdin (FP) has been shown to be a positive complement regulator, stabilizing the AP C3 convertase up to 60 min and thus increasing its half‐life 10‐fold (Fearon & Austen, 1975) (Fig 1A). Plasma FP oligomerizes into di‐, tri‐, and tetramers, or even higher order oligomers and its properties vary as a function of its oligomerization state (Agarwal et al, 2010). In addition to stabilizing already formed C3 convertase, FP may direct fluid‐phase C3b or C3(H2O) to bind certain surfaces including activated platelets (Saggu et al, 2013), apoptotic/necrotic cells, and Chlamydia pneumoniae and in this way function as a PRM (Cortes et al, 2012). FP was likewise reported to bind apoptotic T cells and promote complement activation and phagocytosis, exemplifying another complement‐mediated clearance mechanism (Kemper et al, 2008). Although AP activation may occur in their absence (Degn et al, 2014a; Ruseva et al, 2014), it has been demonstrated that MASP‐1 and/or MASP‐3 cleavage of pro‐FD to FD is required for the full activity of the AP in mice (Takahashi et al, 2010), providing an interesting link between the lectin and the alternative pathways.
The terminal pathway (TP) of complement (Fig 1A) is initiated when a threshold density of C3b molecules on an activator has been reached. The C3 convertases can recruit another C3b molecule to form C3bBb3b (Medicus et al, 1976) and C4b2a3b (Takata et al, 1987), the AP and CP C5 convertases, respectively. Through cleavage of C5, they generate the potent chemoattractant C5a (see below) and C5b. The latter forms the lytic membrane attack complex (MAC, also called C5b‐9) together with C6, C7, C8, and multiple C9 molecules in membranes of pathogens lacking a protective cell wall like Gram‐negative bacteria (Fig 1A) (Laursen et al, 2012; Berends et al, 2014). Recent crystal structures of the MAC initiating C5b6 complex and C8 in combination with a cryo‐EM reconstruction of a soluble form of MAC have led to atomic models for the assembly and structure of the entire MAC complex (Hadders et al, 2007, 2012; Aleshin et al, 2012). Exactly how the MAC elicits killing of Gram‐negative pathogens remains unsettled as it is unlikely to span both the outer and the inner membranes simultaneously (Berends et al, 2014). The MAC may not represent a major defense mechanism in adults as evidenced by the successful systemic blockade of C5 cleavage as a therapeutic strategy without significant increase in infectious episodes (see below). Another aspect of the terminal pathway is the ability of sublytic numbers of C5b‐9 complexes, assembled on host cells, to induce signal transduction resulting in cell cycle progression instead of cell death (Tegla et al, 2011). In recent studies, the sublytic MAC was shown to activate the NLRP3 inflammasome and trigger the secretion of pro‐inflammatory cytokines IL‐1β and IL‐18 subsequent to caspase‐1 activation (Laudisi et al, 2013; Triantafilou et al, 2013). In an in vivo model, inflammation was substantially reduced in C6‐deficient mice, strongly implicating the sublytic MAC in inflammatory processes. It will be interesting to test whether therapeutics targeting the inflammasome or caspases would be beneficial in MAC‐associated pathologies.
It has been known for decades that complement fragments can be generated by other means besides the three canonical activation routes, and especially, the cross talk with the coagulation system has regained attention due to studies indicating that thrombin, coagulation factors XIa, Xa, and IXa, and plasmin effectively cleave C3 and C5 and generate C3a and C5a (Huber‐Lang et al, 2006; Amara et al, 2010; Berends et al, 2014). C3 can also be produced intracellularly by the CD4+ T cells. This C3 is processed by the T‐cell lysosomal protease cathepsin L, yielding biologically active C3a and C3b (Liszewski et al, 2013). Tonic intracellular C3a generation is required for homeostatic T‐cell survival, whereas shuttling of this intracellular C3 activation system to the cell surface upon T‐cell stimulation additionally induces autocrine proinflammatory cytokine production. Thus, C3aR activation via intrinsic generation of C3a appears to be an integral part of human Th1 immunity (Ghannam et al, 2014). Thrombin slowly cleaves C5 and generates C5a, but under conditions with normal convertase activity, this is possibly not a physiologically significant reaction. Clotting‐induced production of thrombin instead leads to cleavage of C5 or C5b in the CUB domain. C5a can be released from such CUB‐digested C5 by the conventional C5 convertases, and the combined action of thrombin and convertases appears to enhance the efficiency of the lytic pathway (Krisinger et al, 2012). Conversely, MASP‐1 has been reported to activate coagulation (Takahashi et al, 2011; La Bonte et al, 2012) and to initiate endothelial cell signaling via cleavage of protease‐activated receptor 4 (Megyeri et al, 2009).
Structure, assembly, and activation of the proteolytic complexes within complement
Much attention has been devoted to understanding, at the atomic level, how giant proteolytic complexes within complement assemble and how they recognize their substrates and generate their products. Crystal structures accumulated over the last 25 years of the involved PRMs, proteases, and their substrates have generated a very rich structural framework for comprehending these complicated proteolytic reactions. In the following, we focus on some of the larger and more recent structures of complement proteins and their complexes.
Despite their quite different ligands, the PRMs in the CP and LP have the same basic architecture. Their subunits form trimers through a collagen‐like region and the C‐terminal ligand‐binding domains. Such trimers oligomerize through their N‐terminal regions, but whereas the C1q trimer contains three distinct chains (A, B and C), CL‐LK contains two polypeptide chains (CL‐K1 and CL‐L1), and only homotrimers are found in MBL and the ficolins. The C1r, C1s, MASP‐1, MASP‐2, and MASP‐3 proteases associated with these PRMs share a modular structure with their N‐terminal CUB‐EGF‐CUB domains (Fig 2A) mediating protease dimerization (Teillet et al, 2008; Venkatraman Girija et al, 2013) as well as association with the PRM collagen stems through a calcium‐dependent interaction with a conserved lysine residue in the collagen region of the PRMs (Lacroix et al, 2009; Gingras et al, 2011; Bally et al, 2013). The MASPs, like C1r and C1s of the C1 complex, encompass the domains CUB1‐EGF‐CUB2‐CCP1‐CCP2‐SP. Upon activation of zymogen MASP (or zymogen C1r and C1s), the polypeptide chain is cleaved between the CCP2 and the SP domains, but the resulting A and B chains remain associated through a disulfide bond. The C‐terminal part of the serine proteases is formed by two CCP domains followed by the catalytic SP domain (Fig 2B).
Regardless of the paramount importance of PRM:serine protease complexes in pattern recognition and complement activation, only low‐resolution structural information is currently available. The prevalent model for the C1 complex, based on a combination of negative stain electron microscopy (EM), biochemical, and biophysical interaction studies (Bally et al, 2009, 2013; Phillips et al, 2009; Brier et al, 2010), states that prior to ligand binding and activation, the SP domains of C1r and C1s are tucked inside a large conical‐shaped void delimited by the collagen stems and the ligand‐binding domains of C1q. Ligand binding is then believed to trigger a conformational change causing intramolecular activation of C1r and C1s followed by exposure of the C1s SP domains in order to make them accessible for the substrates C4 and the proconvertase C4b2 (Wallis et al, 2010). C1q binds antigen‐bound IgM but also single IgGs through their Fc fragment, though with a lower affinity (K D ≈ 10−4 M) (Hughes‐Jones & Gardner, 1979). This low‐affinity interaction is not sufficient to activate the C1 complex, thus binding to several IgG Fc regions by a single C1q is necessary to achieve avidity and activate C1. IgG molecules being monovalent in solution, the only way this can be achieved is through antigen‐driven IgG clustering on an activating surface (Burton, 1985). A recent cryo‐EM tomography study at 66 Å resolution revealed the structure of the full C1 complex bound to IgG arranged in an Fc‐mediated hexamer on hapten‐coated liposomes (Diebolder et al, 2014). The N‐terminal parts of the six collagen stems in C1q assemble in parallel into one compact structure, whereas the C1r2s2 tetramer is located within a flat, elongated structure centrally in C1. The catalytic SP domains of C1r2s2 were not visible, but suggested to protrude from the central axis of the molecule (Diebolder et al, 2014). The globular ligand‐binding domains are seen in contact with the CH2 domains of the Fc hexamer. Presumably this study presents the activated C1 molecule bound to a model IgG immune complex, although the activation status of the C1r2s2 is not described.
Based on the functional and structural homologies between serine proteases and the PRMs from the two pathways, it was suggested that the activation mechanism and conformation of PRM:MASP complexes from the LP resemble those suggested for the C1 complex (Wallis et al, 2010). According to this model, binding of PRM:MASP‐1 to an activator causes a conformational change conferring intramolecular activation of MASP‐1, which may then activate a nearby PRM:MASP‐2 complex. However, a major problem with the intramolecular activation mechanism for the LP is that it requires extensive deviations from known crystal structures of fragments encompassing the CUB‐EGF‐CUB domains of MASPs, in order to place the SP domains of the proteases inside the PRM cone. We recently challenged this concept in two different ways. We first showed that clustering of PRM:MASP‐1 without PRM ligand binding is sufficient to initiate LP activation and that activation of MASP‐1 and subsequently activation of MASP‐2 are intermolecular reactions driven primarily by juxtaposition and orientation of the PRM:MASP complexes (Degn et al, 2014b). In a second study, we used small‐angle X‐ray scattering (SAXS) and EM to demonstrate that non‐activated MBL:MASP‐1 complexes have their MASP‐1 SP domains protruding from the MBL collagen stems (Fig 2A) and separated by more than 200 Å (Kjaer et al, 2015). This is incompatible with intracomplex MASP‐1 cleavage within a PRM:MASP‐1 complex but in agreement with the intercomplex activation mechanism driven by juxtaposition of PRM:MASP complexes (Degn et al, 2014b) and requires no global conformational changes to accompany activation, but is rather dependent on the concentration of PRM:MASP on the activating surface. Future studies will show whether the concept of intermolecular activation can be extended to the classical pathway.
The first common step in the LP and CP is the cleavage of C4 by MASP‐2 or C1s, respectively. Our recent crystal structures of C4 and its complex with a catalytic fragment of MASP‐2 (Fig 2B) provided detailed insight into this crucial step of complement activation (Kidmose et al, 2012). The structure of C4 demonstrated that the protein has the same domain organization and overall structure as C3 and C5 (Fig 2B). In the C4:MASP‐2 complex, the scissile bond region is accommodated in the MASP‐2‐active site as anticipated. Furthermore, the C‐terminal C345c domain of C4 was found to be in contact with a MASP‐2 exosite located between the two CCP domains (Rossi et al, 2005; Duncan et al, 2012) and the corresponding contact between C1s and the C4 domain was also confirmed (Kidmose et al, 2012) (Fig 2B).
A major breakthrough in the structural studies of complement was the structure determination of C3 and C3b (Janssen et al, 2005, 2006; Wiesmann et al, 2006). The most striking observation when comparing these structures is the overwhelming conformational change that C3 undergoes upon release of C3a. In C3, the thioester is concealed and solvent inaccessible, whereas in C3b it is placed 80 Å away from its C3 position and freely accessible to activator nucleophiles. We have recently shown that an almost identical conformational change occurs in nascent C4b resulting from C4 cleavage through the CP and LP (Fig 2C) (Mortensen et al, 2015). Furthermore, nascent C5b undergoes a conformational change resembling that of C3b and C4b but becomes trapped half‐way due to the association of C5b with C6 (Fig 2F) (Aleshin et al, 2012; Hadders et al, 2012). But how are C3 and C5 actually cleaved by the convertases? Establishing the structure of convertases and their complexes with substrates represents a major challenge as the active convertases rapidly dissociate irreversibly. Furthermore, the convertases are surface‐linked enzymes and especially the C5 convertases, which depend on adjacent C3b molecules in order to gain affinity for C5, may not be truly reconstituted as fluid‐phase enzymes. Our recent structure of C4b provides insight into the architecture of the CP C5 convertase C4b2a3b, in which the presence of C3b shifts the specificity from C3 to C5 (Mortensen et al, 2015). By SAXS modeling, we have shown that a conserved loop in the C4b TED (residues 1231–1255) is exposed to the solvent. This loop contains a conserved Ser1236 residue which may play the role of a nucleophile and covalently link C4b to C3b through its thioester, thus forming a covalent C4b‐C3b heterodimer (Kim et al, 1992). If indeed this loop is the main contact of C4b to C3b in the CP C5 convertase, then the longest axes of the two proteins may be roughly parallel, positioning C3b in a way that it can no longer establish interaction with C5. Based on these observations in combination with the structure of a factor H (FH) fragment bound to C3b (Fig 2G) and data suggesting that the FH‐binding site of C3b within the CP C5 convertase is inaccessible (Weiler, 1989; Meri & Pangburn, 1990), we proposed that C3b alters the conformation of the CP C3 convertase rather than offering an additional C5‐specific binding site.
Several studies have addressed the structure of the AP C3 convertase. Structures of the stable proconvertase C3bB determined by EM and crystallography revealed how FB associates with the C‐terminal C345c domain of C3b through its von Willebrand factor type A (vWA) domain in a Mg2+‐dependent manner (Torreira et al, 2009; Forneris et al, 2010). This proconvertase appears to adopt two conformations, open and closed, differing by a rotation of the FB SP domain. The structure of the ternary complex C3bBD (Fig 2D) revealed that the scissile bond region in FB is only accessible to FD in the open conformation. Furthermore, FD binds primarily through a FB exosite located 25 Å from the scissile bond (Forneris et al, 2010). To determine the crystal structure of the rapidly dissociating C3bBb complex (half‐life of about 90 s at 37°C) formed after C3bB cleavage by FD, the S. aureus protein SCIN was used to stabilize the AP C3 convertase. The only contact between the two convertase subunits is through the C3b C345c domain and the Bb vWA domain, whereas the catalytic SP domain of Bb extends away from C3b (Rooijakkers et al, 2009). Another approach to stabilize the convertase for structural studies is to use FP, and a recent EM study revealed the architecture of FP and the FP‐stabilized AP C3 convertase C3bBbP. The vertices of FP trimers/tetramers were found to contact the C345c domain of C3b and the vWA domain of Bb explaining its stabilizing effect on the AP C3 convertase (Alcorlo et al, 2013).
Structural insight into substrate recognition by the convertases was accomplished with our structure of C5 in complex with the C3b homolog cobra venom factor (CVF) in the absence of FB. C5 and CVF are in contact at two separate interfaces with the largest of these formed between the macroglobulin (MG) 4 and 5 domains from both proteins. The second interface involves the C5 MG7 domain and the CVF MG6 and MG7 domains (Laursen et al, 2011). Binding to CVF requires a conformational change in C5 as compared to the unbound protein (Fredslund et al, 2008) in order to establish the two‐point interaction. By combining the SCIN‐stabilized C3bBb structure and the C5‐CVF structure, it was possible to suggest a general model for convertase–substrate interactions (Fig 2E) applicable to both CP and AP convertases in agreement with existing experimental data (Laursen et al, 2011). This model suggests that C4b/C3b in the CP/AP convertases recognize the substrate MG4, MG5, and MG7 domains in a manner similar to how CVF interacts with C5 and that the substrate undergoes an overall conformational change upon convertase binding. As the catalytic subunits C2a/Bb must function in both the CP/AP C3 and C5 convertases, the orientation of the substrates with respect to the catalytic subunit was also suggested to be similar in C3 and C5 convertases (Laursen et al, 2011).
Regulators of complement activation
Even healthy host cells not recognized by CP or LP PRMs may become tagged by C3b due to the generation of C3(H2O) in the AP or through the “bystander” effect whereby C3b generated in the vicinity becomes attached to a host cell. It is therefore important that the AP is tightly regulated, and a panoply of soluble and membrane‐associated complement regulatory proteins are known (Fig 1B and Table 1). One of the most potent and best‐studied complement regulators is complement factor H (FH) (Ferreira et al, 2010). It possesses decay‐accelerating activity dissociating Bb from the AP C3 convertase, but FH also serves as cofactor for the serine protease factor I (FI) that cleaves C3b into iC3b, unable to form C3 convertase (Fig 1B). FH is a very strong negative regulator of complement on host surfaces, to which FH binds through sialic acid, heparin, and sulfated glycosaminoglycans (GAGs) (Figs 1B and 3A and B). By binding to host‐specific glycans, FH is able to distinguish between self and non‐self, thus preventing complement activation on host surfaces. On microorganisms lacking these protective patterns, FH is absent and complement activation is thus allowed to proceed (Makou et al, 2013). FH consists of 20 repeating units of about 60 residues designated as short consensus repeats (SCRs) or complement control protein repeats (CCPs) (Fig 3A). Owing to its modular, flexible structure, full‐length FH is recalcitrant to high‐resolution structural studies. CCPs 1–4 of FH bind C3b and display decay‐accelerating activity by dissociating Bb from C3 convertases. In addition, FH possess cofactor activity (Figs 1B and 3B) probably by inducing a substrate conformation of C3b susceptible to FI‐mediated degradation (Gordon et al, 1995; Kuhn & Zipfel, 1996; Barlow et al, 2008) and by providing a binding platform for FI (Roversi et al, 2011). The C3b:FH CCP1–4 crystal structure provides insight into the molecular basis of these FH functions (Wu et al, 2009). The four FH CCP domains are in contact with C3b, spanning over 100 Å in a linear fashion with a kink between CCP3 and CCP4 giving it an overall L‐shaped form (Fig 2G). The FH‐binding site in C3b is formed by its α′ N‐terminal region, the MG1, 2, 6, 7, CUB, and thioester (TE) domains. As the positions of the CUB and TE domain are dramatically different in C3 and C3b, the structure explains the selectivity of FH for C3b. A comparison of the structures of the C3b:FH CCP1–4 (Fig 2G) and C3bBb (Fig 2E) complexes identifies the competing interfaces on C3b, thus explaining the FH decay‐accelerating activity.
Table 1.
Complement regulators
| Regulator | Ligand | Function |
|---|---|---|
| Soluble regulators | ||
| Factor H (FH) | C3b, C3d | AP C3 convertase decay and cofactor |
| FHL‐1 | C3b | AP C3 convertase decay and cofactor |
| CFHR1 | C3b, C3d, C5 convertase | Inhibits C5 convertase, competes with FH, acts as decoy for certain pathogens |
| CFHR2 | C3b, C3d, AP C3 convertase | AP C3 convertase decay, competes with FH |
| CFHR3 | C3b, C3d | Competes with FH |
| CFHR4 | C3b | AP C3 convertase decay |
| CFHR5 | C3b, C3/C5 convertase? | Competes with FH |
| Factor I (FI) | Cofactor bound C3b and C4b | Serine protease, degrades C3b, iC3b, C4b, and iC4b |
| Properdin (FP) | C3b, C3bB, C3bBb, GAGs | Stabilizes AP convertases |
| C4BP | C4b, S protein | CP C3 convertase decay and cofactor |
| C1‐INH | C1r, C1s, MASPs | Serine protease inhibitor |
| MAp19 | MBL and ficolins | Competes with MASP‐2 in vitro |
| MAp44 | MBL and ficolins | Blocks MASP transactivation |
| Clusterin | C7, C8, C9, MAC | Inhibits MAC |
| Vitronectin | C5b‐C7, MAC, GAGs | Inhibits MAC |
| Membrane‐associated and transmembrane regulators | ||
| MCP (CD46) | C3b, C4b | Cofactor activity, T‐cell differentiation and activation |
| DAF (CD55) | C3b, C4b, AP and CP C3 convertase | AP and CP C3 convertase decay |
| CD59 | C8, MAC | Inhibits MAC |
| CR1 | C3b, C4b | AP and CP C3 convertase decay and cofactor |
Figure 3. Complement factor H family regulators.
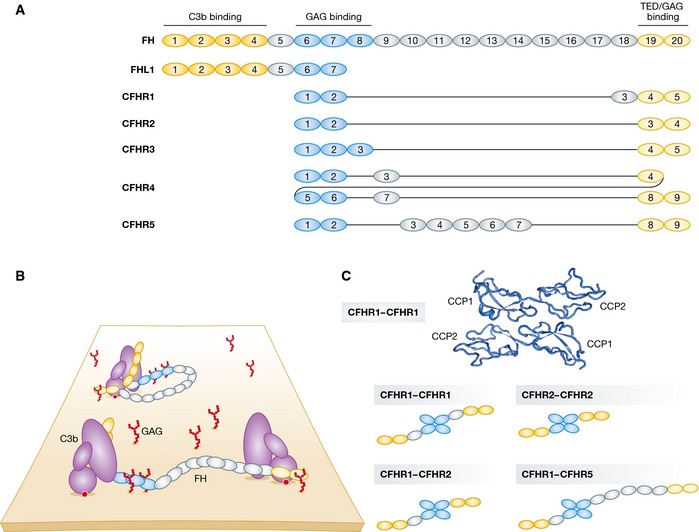
(A) Domain organization of complement factor H (FH). C3‐binding domains are highlighted in yellow and glycosaminoglycan (GAG)‐contacting domains in blue. Complement factor H‐like 1 (FHL1) and complement factor H‐related (CFHR) proteins are represented below according to their sequence similarity to FH. CFHRs share high sequence similarity with each other and with FH. All CFHRs contain domains homologous to the FH GAG‐ and TED‐binding domains but lack the domains homologous to the FH N‐terminal CCP1–4. (B) Models of FH recruited to non‐activating surfaces. FH may be recruited to the self‐surface‐bound C3b and establish bivalent contacts with one C3b molecule. Owing to its flexible structure, FH may bind two surface‐bound C3b molecules (or iC3b and C3d). (C) CFHRs form homo‐ and heterodimers. The N‐terminal CCP1–2 domains of CFHR1 crystallize as head‐to‐tail dimers (RSCB ID 3ZD2). Due to the high sequence identity between the CCP1–2 of CFHR1, 2, and 5, the three proteins are able to form homo‐ and heterodimers in the serum and thus modulate complement activation in a more complex manner.
The FH CCP6–8 are involved in association with self‐surfaces since they bind to heparin, a model for highly sulfated GAGs (Blackmore et al, 1996), whereas FH CCP19–20 interact with both C3b and host surface glycans (Schmidt et al, 2008) (Fig 3A and B). These C‐terminal modules are crucial for distinguishing self from non‐self, and recombinant FH CCP19–20 competitively inhibit full‐length FH deposition on cell surfaces (Ferreira et al, 2006). Furthermore, in the presence of anti‐CCP20 blocking antibody, FH is unable to bind endothelial cells (Oppermann et al, 2006). The recent co‐crystal structure confirmed that discontinuous patches on FH CCP19–20 interact with a contiguous stretch on the C3b TE domain (Morgan et al, 2011). The FH CCP19–20 interaction site is not overlapping with the CCP1–4 site (Fig 2G), consistent with the cooperativity of CCP1–4 and CCP19–20 in C3b binding (Schmidt et al, 2008). A recent study that assayed the binding of FH to a variety of sialosides by NMR spectroscopy (Blaum et al, 2015) showed that FH CCP20 binds specific structures present on host surfaces, and not simply polyanions as previously postulated, as exemplified by the binding to Siaα2‐3Galβ1‐3GalNAc in contrast to lack of binding to Siaα2‐6Galβ1‐4GalNAc. CCP6–8 and CCP19–20 cooperate in binding to host surfaces since separately they bind to heparin with a K D ≈ 5–10 μM, whereas the affinity increases to as high as K D ≈ 30 nM for intact FH (Khan et al, 2012; Zaferani et al, 2012). For recent comprehensive reviews concerning FH binding to host surfaces, see Makou et al (2013) and Perkins et al (2014).
There are six other proteins related to FH: the product of CFH alternative splicing, complement factor H‐like protein (FHL‐1), and complement factor H‐related proteins (CFHRs) 1–5 (Jozsi & Zipfel, 2008). The CFHRs are encoded by separate genes and are composed of different number of CCP domains (Table 1 and Fig 3A). CFHR1 is able to regulate the terminal pathway of complement, but it lacks decay and cofactor activities (Timmann et al, 1991; Heinen et al, 2009). CFHR1 binds to several microbes, and since it can compete with FH for the same binding sites, it is believed that it can act as a decoy preventing complement subversion by certain pathogens (Haupt et al, 2007; Kunert et al, 2007). CFHR2 regulates complement by inhibiting the AP C3 convertase, but it does not compete with FH for C3b binding (Eberhardt et al, 2013). CFHR3 competes with FH for C3b binding, but its role as cofactor remains debated (Hellwage et al, 1999; Fritsche et al, 2010). C3b is also bound by CFHR4, which can apparently stabilize the AP C3 convertase and prevent its decay by FH (Hebecker & Jozsi, 2012). CFHR5 binds C3b, CRP, and heparin, but its possible role as a cofactor is rather poorly supported by the data. Although not a potent complement regulator, CFHR5 associates with C3 activation products and localizes to complement deposits of diseased kidney glomeruli (Murphy et al, 2002). CFHR1, CFHR2, and CFHR5 can form homo‐ and heterodimers due to the conserved dimerization interface (Fig 3C) (Goicoechea de Jorge et al, 2013). Furthermore, because of their conserved C3b/C3d binding interface and the avidity conferred by their dimerization, CFHRs can effectively compete with FH and interfere with complement inhibition even though their relative plasma concentrations are lower than that of FH [FH 0.7–3.6 μM (Esparza‐Gordillo et al, 2004); CFHR1 1.7–2.5 μM (Heinen et al, 2009); CFHR5 0.05–0.09 μM (McRae et al, 2005)]. Considering that FH levels in the blood are not actively regulated, it is reasonable to assume that altering CFHR levels and the composition of the different dimeric species could provide an intricate way of controlling complement activation. However, additional studies are needed to fully understand the role of CFHRs in health and disease. Genetic variations of CFH and some of the CFHR genes have been associated with chronic inflammatory diseases such as age‐related macular degeneration (AMD) and atypical hemolytic uremic syndrome (aHUS) (discussed later).
C4‐binding protein (C4BP) is a fluid‐phase regulator of the CP and LP similar to FH in its regulatory properties, but directed at C4b. It is a large glycoprotein consisting of seven α‐ and one β‐chain, both made of CCP modules, and with a spider‐like appearance (Fig 1) (Blom et al, 2004; Hofmeyer et al, 2013). C4BP is both a decay‐accelerating factor dissociating C2a from the CP C3 convertase and a cofactor, promoting FI‐mediated cleavage into iC4b and further to C4c and C4d (Fig 1B). Another regulator of the CP and LP is C1 inhibitor (C1‐INH), a member of the serine protease inhibitor (serpin) family. It irreversibly blocks C1r and C1s in the C1 complex as well as MASP‐1 and MASP‐2 (Davis, 1988; Degn et al, 2013b) (Fig 1B). Two splice variants of the MASP2 and MASP1 genes, respectively, MAp19 and MAp44 lacking the SP domain regulate the LP (Degn et al, 2009; Skjoedt et al, 2010) (Fig 1B). MAp44 has been shown to inhibit complement by preventing MBL:MASP co‐complex formation, thereby precluding MASP transactivation (Degn et al, 2013a). MAp19 has been reported to compete with MASP‐2 for binding to MBL and prevent cleavage of C4, thus downregulating complement activation (Iwaki et al, 2006). This finding has recently been disputed (Degn et al, 2011) as it was found that MAp19 possessed 10‐fold lower affinity toward MBL compared to MASP‐2. Taking into account the similar physiological abundance of MASP‐2 (at 7 nM) and MAp19 (at 11 nM) and the fact that other molecules associate more strongly with MBL, it is unlikely that MAp19 can serve as a complement inhibitor under physiological conditions, unless unique conditions are present at specific sites of the body. Other soluble complement regulators include clusterin and vitronectin, both regulating complement by inhibiting MAC assembly or insertion into membranes within the terminal pathway (Fig 1B). Clusterin prevents C9 from binding the C5b‐8 complex, whereas vitronectin inhibits the association of C5b‐7 with membranes. Upon binding of C8 and C9 to the soluble C5b‐7 complex, a fluid‐phase MAC complex is formed instead, also known as soluble MAC (sMAC) (Podack et al, 1978; Tschopp & French, 1994; Moskovich & Fishelson, 2007).
Host cells express transmembrane and membrane‐associated factors that protect them from complement activation (Fig 1B and Table 1). Membrane cofactor protein (MCP or CD46) is a ubiquitously expressed CCP module‐based protein (absent only on erythrocytes) that serves as cofactor for FI cleavage of C3b and C4b (Andrews et al, 1985) (Fig 1B). CD59 and decay‐accelerating factor (DAF or CD55) are glycosylphosphatidylinositol (GPI)‐anchored complement regulators. CD59 is a small (20 kDa), widely expressed glycoprotein that blocks C9 from incorporating into the C5b‐8 complex as well as C9 polymerization in a preformed C5b‐9 complex (Meri et al, 1991; Morgan et al, 2005) (Fig 1B). DAF, a 70‐kDa CCP‐based glycoprotein, accelerates the decay of both the C3 and C5 AP and CP convertases by binding to C3b and C4b (Medof et al, 1984) (Fig 1B).
Complement receptors
The anaphylatoxins C3a and C5a, released when the convertases cleave C3 and C5, exert their biological functions upon binding to seven‐transmembrane domain (7TM) receptors in the membranes of host cells. Two of these receptors, C3aR and C5aR1 (CD88), are G protein‐coupled receptors (GPCR), whereas the third, C5aR2 (previously known as C5L2), is structurally similar to C5aR1 but does not couple to heterotrimeric G proteins (Li et al, 2013). C5aR2 was first considered as a decoy receptor, limiting the availability of the C5a and C5adesArg ligands to C5aR1. Decoy receptors do not undergo ligand‐induced internalization but are rather continuously recycled between the cell membrane and the intracellular compartments, thereby removing their extracellular ligand (Weber et al, 2004). Thus, it has been suggested that C5aR2 may reduce the cellular responses to pro‐inflammatory molecules and thereby actively regulate inflammatory processes (Rittirsch et al, 2008). Additionally, some studies report concerted action of C5aR1 and C5aR2 in adipocyte metabolism and immunity as well as formation of C5aR1/C5aR2 heterocomplexes (Bamberg et al, 2010; Poursharifi et al, 2014).
Signaling through C3aR and C5aR1 triggers chemotaxis, oxidative burst, histamine release, and leukotriene and interleukin synthesis (Klos et al, 2009). Through such pro‐inflammatory properties, anaphylatoxins play an important role in chronic inflammatory diseases including rheumatoid arthritis, inflammatory bowel disease, as well as in asthma and allergy (Linton & Morgan, 1999; Woodruff et al, 2003; Hawlisch et al, 2004). However, C3a may instead elicit an anti‐inflammatory response depending especially on the cell type activated through C3a–C3aR signaling and the phase of inflammation (Coulthard & Woodruff, 2015). In addition, anaphylatoxins are implicated in tissue regeneration and development (Mastellos et al, 2001; Strey et al, 2003; Hillebrandt et al, 2005). In the liver, C3a induces STAT3 activation and an increased IL‐6 production (He et al, 2009). In the eye, C3a signaling also activates STAT3 and promotes retinal regeneration (Haynes et al, 2013). In addition, C3a is implicated in neural stem cell migration and regeneration (Shinjyo et al, 2009). An interesting study recently implicated C3aR in alloreactivity (Asgari et al, 2013). By examining renal allograft rejection patients, the authors reported increased C3a generation and thus C3aR stimulation. Upon C3aR activation, phosphorylation of extracellular signal‐regulated kinase 1 and 2 (ERK1/2) and increased efflux of ATP were observed in monocytes. Subsequent caspase‐1 activation led to IL‐1β secretion, thus linking C3aR potentiation to Th17 responses. Lately, C3a has been suggested to act as a secretagogue on islets stimulating insulin secretion by pancreatic β‐cells in mice (Lo et al, 2014). Activation of C3aR on β‐cells leads to an intracellular increase in ATP, mitochondrial respiration, and an elevated Ca2+ flux. Islets in FD knockout mice with a resulting lower AP activity therefore secrete less insulin under high glucose conditions. Furthermore, type 2 diabetes patients with β‐cell failure were observed to express less FD (also known as adipsin) than patients without β‐cell failure (Lo et al, 2014). C5a has been shown to offer neuroprotection against glutamate‐mediated cell death (Mukherjee et al, 2008). It is widely accepted that C5a harbors a higher pro‐inflammatory potency, as compared with C3a (Guo & Ward, 2005), which may correlate with the findings that implicate C5a in pathogenesis of sepsis, rheumatoid arthritis, ischemia–reperfusion injury, allergy, and asthma (Guo & Ward, 2005; Klos et al, 2009). The biological activity and potency of anaphylatoxins are regulated by carboxypeptidases that cleave off the C‐terminal arginine residue yielding C3adesArg and C5adesArg (Bokisch & Muller‐Eberhard, 1970; Matthews et al, 2004). C3adesArg does not signal through C3aR, whereas C5adesArg retains up to 10% of C5a activity (Wilken et al, 1999; Sayah et al, 2003). A role of C3adesArg (acylation‐stimulating protein) in triglyceride synthesis (Cianflone et al, 2003) mediated by its binding to C5aR2 (Cui et al, 2009) has been suggested though strongly disputed (Klos et al, 2009).
From a structural perspective, anaphylatoxins are small, cationic peptides (74–79 amino acids) possessing an all‐α‐helical fold stabilized by disulfide bridges. Despite the major differences in biological activity, C3a and C3adesArg present no significant structural differences (Fig 4A) (Bajic et al, 2013a). In contrast, human C5a and C5adesArg are structurally flexible; they seem to oscillate between four‐ and three‐helix bundle conformations, whereas their murine counterparts adopt only the four‐helix bundle conformation (Fig 4A) (Cook et al, 2010; Schatz‐Jakobsen et al, 2014). With respect to receptor binding, numerous mutagenesis studies have been performed for C5a, unlike for C3a. The studies demonstrated that basic residues on C5a are involved in C5aR1 binding (Huber‐Lang et al, 2003) (Fig 4A). This observation is reinforced by the fact that C5aR1 bears N‐terminal sulfotyrosine residues essential for ligand binding (Farzan et al, 2001), thus suggesting that charge–charge interactions drive the binding. The C‐terminally truncated version of the human C5a (residues 1–70) binds C5aR1 with lower affinity than the full C5a but does not activate receptor signaling (Chenoweth et al, 1982). Therefore, it has been suggested that the C5a core is necessary for the receptor binding, but the C‐terminus dictates the signaling [for review, see Klos et al (2013)].
Figure 4. Structural characterization of anaphylatoxins and the role of CR3 and CR2 in antigen trafficking in the lymph node.
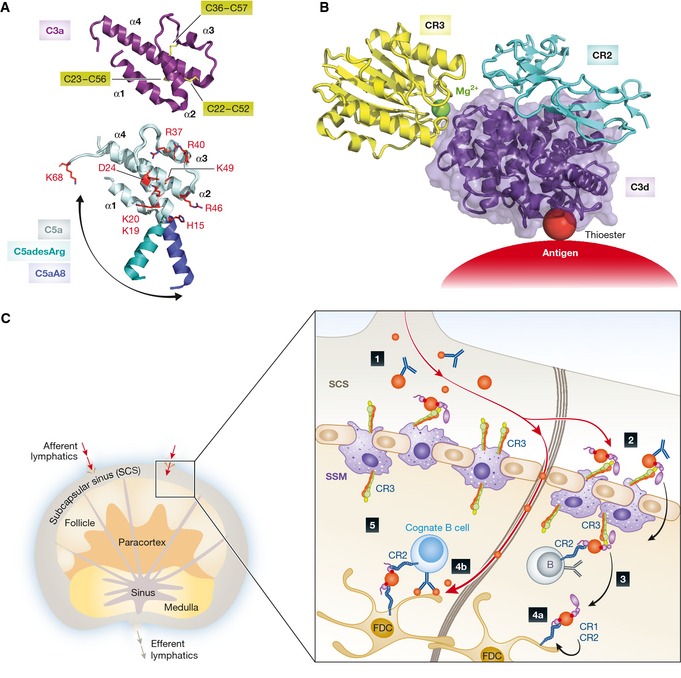
(A) Structure of C3a and C5a anaphylatoxins. The overall structure of human C3a (RCSB ID 4HW5) indicates a four‐helix bundle fold (α‐helices are indicated) stabilized by three disulfide bridges (yellow sticks, cysteine numbering is indicated). Underneath is shown a superimposition of the C5a moiety from the intact C5 (light blue, RSCB ID 3CU7) with C5asdesArg (teal, RSCB ID 3HQB), and the C5aA8 antagonist (dark blue, RSCB ID 4P39). The swing‐out motion of the α1‐helix is indicated with an arrow. C5a residues involved in C5aR1 binding are indicated in red sticks. (B) Atomic model of the CR3 (yellow) and CR2 (light blue) co‐ligation to C3d (purple) generated by superimposing the CR3 I domain:C3d (RCSB ID 4M76) and CR2 CCP1‐2:C3d (RSCB ID 3OED) complexes. A ternary complex between a C3 opsonized antigen, CR2, and CR3 could be of physiological relevance, see the next panel. (C) Complement‐dependent antigen transport, uptake, recycling, and presentation occur in the lymph node. 1) Immune complexes (IC) containing complement‐opsonized antigens drain with the afferent lymphatics into the subcapsular sinus (SCS). 2) Complement‐opsonized antigen is taken up by subcapsular sinus macrophages (SSM) via complement receptor 3 (CR3) and shunted across the subcapsular sinus floor. 3) The antigen is handed off to non‐cognate B cells via complement receptor 2 (CR2), which transport it into the follicle. 4a) Antigen is delivered to follicular dendritic cells (FDCs) via CR1 and CR2. FDCs are subsequently able to retain antigen for long periods of time in a recycling compartment. 4b) Low‐molecular‐weight antigen is delivered directly into the follicle through conduits. 5) Cognate B cells can probe antigen arrayed on the surface of FDCs, and BCR signaling is enhanced by co‐ligation of CR2 by IC‐associated complement fragments. The color coding is consistent with panel (B).
The central effector function of C3b and its degradation products is underscored by the presence of five known receptors on the surface of especially immune cells. Complement receptor 1 (CR1, CD35) is a large CCP module‐based glycoprotein expressed on almost all peripheral blood cells except NK and T cells (Fearon, 1980; Tedder et al, 1983). CR1 binds C3b and C4b with high affinity and iC3b and C3d with a lower affinity (Reynes et al, 1985). CR1 on erythrocytes may bind C3b‐containing immune complexes as part of removal processes, whereas on phagocytic cells it promotes C3b/C4b‐coated particle uptake. CR1 also plays an important role in the germinal centers of lymph nodes where it is found on follicular dendritic cells (FDCs) capturing complement‐opsonized antigens that serve to stimulate B cells (Heesters et al, 2013) (Fig 4B and C). CR2 (CD21), also possessing a CCP architecture, is primarily present on B cells and FDCs. It is important in trapping of C3‐opsonized antigens by FDCs in the germinal centers and stimulating B cells for affinity maturation, isotype switching, and memory (Fang et al, 1998; Carroll, 2000) (Fig 4B and C). CR2 binds C3b, iC3b, and C3d with the same affinity in agreement with the crystal structure of the CR2‐C3d complex revealing recognition of a surface patch on the TE domain accessible in all three ligands but concealed in C3 prior to cleavage (Fig 4B) (van den Elsen & Isenman, 2011).
CR3 and CR4 are integrin‐type heterodimeric receptors (CD11b/CD18 and CD11c/CD18) having distinct α‐chains, αM and αX, respectively, but sharing a common β2‐chain. Both are phagocytic receptors expressed on myeloid leukocytes and NK cells and share iC3b as ligand (Metlay et al, 1990; Ross, 2000). However, structural studies indicate that the receptors bind to different epitopes of iC3b. CR3 was shown to recognize the TE domain of iC3b (Bajic et al, 2013b) (Fig 4B), whereas CR4 binds quite far from this in the C3c moiety of iC3b (Chen et al, 2012). Interestingly, CR3 and CR2 may bind simultaneously to the iC3b TE domain (Bajic et al, 2013b), and since CR3 is expressed on subcapsular sinus macrophages (SSMs), it is plausible that complement‐bearing immune complexes could be conveyed from CR3‐positive SSMs to CR2‐positive naïve B cells within lymph nodes (Phan et al, 2007; Bajic et al, 2013b; Heesters et al, 2014) (Fig 4B and C). Of note, SSM are poorly endocytic, and appear to retain ICs on their surface during the IC shuttling from the sinus‐lining to the follicular side (Phan et al, 2009). The fifth C3b receptor is CRIg (VSIG4), an immunoglobulin‐type receptor expressed on liver‐resident macrophages (Kupffer cells), which plays an important role in the clearance of pathogens from the circulation through interaction with surface‐bound C3b and iC3b opsonins (Helmy et al, 2006). The binding of CRIg to C3b selectively inhibits the interaction of C3 and C5 with the AP, but not with the CP convertases.
An exciting new development is the discovery of a pathway involving an as yet unidentified intracellular complement receptor (Tam et al, 2014). C3b (or its degradation products) is deposited onto viruses or bacterial pathogens such as Salmonella extracellularly and then transported into host cells upon pathogen invasion (Fig 1B). Pathogen‐bound C3 fragments are recognized in the cytosol by an unidentified receptor that signals through the mitochondrial protein MAVS. Subsequently NF‐κB, AP1, and IRF3/5/7 transcription pathways are activated and pro‐inflammatory cytokines such as interferon‐β are produced. This intracellular sensing is conserved in mammals and present in non‐immune cells. Thus, the induction of an antiviral state may be independent of professional immune cells. Interestingly, some pathogens such as picornaviruses, including rhinovirus and poliovirus, are able to subvert this detection system by expression of 3C protease that cleaves C3 and thus prevents interferon and proteasome induction (Tam et al, 2014).
Structural basis for complement‐associated disease
Some of the major players of the complement system, such as C3, FH, and FB, have long been known to be polymorphic. The first C3 polymorphism was described in the late 1960s (Wieme & Demeulenaere, 1967), and the most common FB variants were identified in the early 1970s (Alper et al, 1972). More than a decade later, the most prominent disease‐related FH polymorphism Y402H was reported (Rodriguez de Cordoba & Rubinstein, 1984). All these discoveries were made on the protein level. Mutations in complement genes resulting in more potent activation of complement (i.e. C3, FB) or mutations yielding less active regulators (i.e. FH, FI, MCP) increase the activity of the AP and the inflammatory response triggered upon complement activation. Here, we will focus on some of the most prominent polymorphisms giving rise to complement dysregulation and thus resulting in pathological conditions. By mapping them onto structures of complement proteins, we provide a molecular rationale for complement‐associated diseases.
A developmental phenotype arising from a complement defect is the so‐called 3MC syndrome, a conglomerate diagnosis encompassing the Michels, Malpuech, Mingarelli, and Carnevale syndromes. The four syndromes were initially aggregated based on clinical overlap and similarities, and this was subsequently cemented by two independent reports identifying the etiological basis as inherited deficiency of either MASP‐3 or CL‐K1 (Sirmaci et al, 2010; Rooryck et al, 2011). Patients with 3MC syndrome exhibit a spectrum of developmental defects, including developmental delay, growth, and mental retardation, characteristic facial dysmorphism, skeletal anomalies with radioulnar synostosis, and several other defects. It seems plausible that the basis of the observation of causative mutations in both CL‐K1 and MASP‐3 is based in a physiologically relevant complex of the two proteins, but the exact nature and role of such a complex, and the relevant substrate of MASP‐3 still remains unclear. However, a recent structure of the catalytic domain of zymogen MASP‐3 indicates that some of the 3MC associated mutation perturbs the structure of the catalytic site of this protease (Yongqing et al, 2013) (Fig 5A–C), suggesting that active MASP‐3 is a prerequisite for normal development.
Figure 5. The molecular basis of complement‐associated disease.
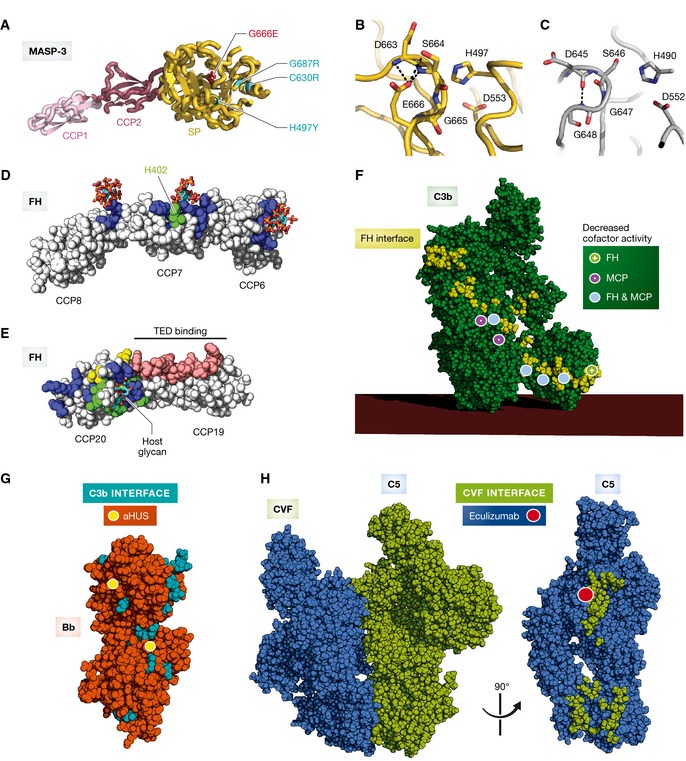
(A) Crystal structure of the zymogen mutant G666E (red sticks) of the MASP‐3 CCP1‐CCP2‐SP fragment (RCSB ID 4KKD). The SP domain polymorphisms associated with the 3MC syndrome (H497Y, C630R, G666E, and G687R) are indicated. (B) Zoom‐in on the active site of the zymogen mutant G666E of MASP‐3. E666 is making multiple polar contacts (dashed black lines) with the α‐turn of the active site restraining the peptide chain in a locked position that keeps the catalytic residues S664 and H497 too far apart. (C) Zoom‐in on the catalytic pocket of the MASP‐1 SP domain (RCSB ID 4IGD). The equivalent of MASP‐3 E666 is G648 in MASP‐1, that makes only a single hydrogen bond, which is insufficient to restrain the catalytic S646 away from the catalytic H490. (D) Crystal structure of FH CCP6–8 in complex with sulfated glycans (RSCB ID 2UWN). All 3 CCP domains interact with host glycans. The carbohydrate‐binding residues are colored in blue. The H402 risk variant is highlighted in green. (E) FH CCP19–20 in complex with host‐specific glycans (RSCB ID 4ONT). The FH residues in contact with the TE domain of C3b, iC3b, or C3d is shown in pink; glycan‐interacting residues are shown in blue; aHUS‐associated mutations are shown in yellow; and aHUS‐associated mutations overlapping with glycan‐binding residues are highlighted in green. (F) Mapping of the aHUS‐associated C3 polymorphisms on C3b responsible for decreased cofactor activity of FH (green dot), MCP (pink dot), and both FH and MCP (light blue dot). Mapped is also the FH interaction area (yellow) explaining why aHUS is triggered when the polymorphisms occur on this C3b interface. (G) The C3b interaction area (cyan) is mapped on the surface of Bb (orange). aHUS‐associated polymorphisms of FB are highlighted with yellow dots. (H) Crystal structure of the CVF:C5 complex (RCSB ID 3PVM). The CVF interaction area (mapped in green) on C5 (blue) overlaps with that of the C5 convertase. Eculizumab prevents C5 recognition by the C5 convertases and is used in treatment of PNH. The red dot indicates the position of C5 Arg885, a polymorphism associated with the lack of response to eculizumab treatment of certain PNH patients. The eculizumab epitope comprises the Arg885 and thus overlaps with the putative convertase binding area.
Age‐related macular degeneration (AMD) is the most frequent cause of blindness in the western civilization affecting elderly persons, with as many as 30 million people affected worldwide. The disease leads to accumulation of immune deposits between the Bruch's membrane and retinal pigment epithelial cells, called drusen, which contain several complement components and regulators suggesting that complement activation is involved in AMD pathogenesis (Zarbin, 2004). In accordance, mutations in FH, FI, and C3, among others, predispose to development of AMD (Zipfel et al, 2010; Seddon et al, 2013; Zhan et al, 2013). Genotyping studies have revealed numerous single nucleotide polymorphisms (SNPs) within the CFH gene and their correlation with susceptibility to AMD (Li et al, 2006). The major SNP variant Y402H increases the risk of developing AMD up to fourfold in heterozygotes and up to sevenfold in homozygotes. A main component of drusen is malondialdehyde (MDA), a common lipid peroxidation product accumulating in many pathophysiological processes. In animal models, FH was found to be able to block MDA‐induced pro‐inflammatory effects. In contrast, the polymorphism Y402H markedly reduced the ability of FH to bind MDA, indicating a causal link to disease etiology (Weismann et al, 2011). The crystal structure of FH CCP6–8 402H variant in complex with a sulfated sugar as a GAG model has suggested how this FH mutation contributes to AMD pathogenesis (Prosser et al, 2007) (Fig 5D). The histidine directly contacts the sulfated sugar whereas a tyrosine, present in FH 402Y, is incompatible with this contact, suggesting that an altered GAG recognition underlies the increased risk for AMD. Nevertheless, the normal 402Y variant is not devoid of GAG binding as such, in fact it binds even more tightly than the 402H to some GAG types (Clark et al, 2006; Herbert et al, 2007). Additionally, the structure also revealed GAG subsites in CCP6 and CCP8, suggesting that the three CCP modules bind GAGs simultaneously (Fig 5D).
Atypical HUS (aHUS) is caused by chronic, uncontrolled activation of the AP, and is characterized by systemic thrombotic microangiopathy and thrombocytopenia that lead to renal failure (Noris & Remuzzi, 2009). It can be caused by rare genetic deficiency or defects within complement components or regulators of the alternative pathway (e.g. FH, CFHR1, CFHR3, MCP, FI, FB, C3) but may also be due to the acquisition of neutralizing autoantibodies against complement proteins, for example, anti‐FH antibodies. Mechanical hemolysis in the microvasculature of the kidney leads to release of free heme, which reacts with C3 leading to AP activation on endothelial cells and in addition induces expression of the C3b binding P‐selectin, thereby further supporting AP activity (Frimat et al, 2013). Mutations in genes encoding FH, FI, FB, and C3 have been found in about 60% of aHUS patients. As already mentioned, FH CCP19–20 are also involved in GAG binding and SNPs within these modules predispose to aHUS (Manuelian et al, 2003; Dragon‐Durey et al, 2004). The recent crystal structure of the ternary complex FH CCP19–20:C3d:3′ sialyl lactose (Fig 5E) offers a molecular rationale for why carriers of FH SNPs are predisposed to aHUS (Blaum et al, 2015). Some mutations directly impair FH binding to the sugar moiety by removing hydrogen bond and hydrophobic stacking interactions, while others are suggested to interfere with the geometry of the GAG‐binding site.
Functional studies of 15 FB mutants identified in aHUS patients showed that only six of these are related to pathogenesis. However, the results of functional studies with these six FB mutants could be rationalized by mapping them to structures of the C3b:FH and the C3bBb complexes (Fig 5G). All gain‐of‐function mutations were located either close to the C3b‐binding site or at a location in Bb close to the C3b‐binding site for FH CCP3 (Marinozzi et al, 2014). In the latter case, the increased convertase stability (less FH decay activity) is likely to result if the mutant Bb is less susceptible to FH competition. A similar approach was very recently employed to characterize a large panel of 23 mutations in C3 identified in aHUS patients (Schramm et al, 2015) (Fig 5F). Functional assays showed that 17 of these had lower affinities for MCP and FH, which in many cases translated into decreased cofactor activity, and all mutations conferring these defects were part of the FH‐binding site or close to it. Interestingly, there was good correlation between how FH and MCP activity was affected by the various C3 mutations, suggesting that the two regulators have overlapping interaction areas on the C3b surface (Fig 5F). In contrast, the cofactor activity of CR1 was much less affected with this panel of C3 mutants suggesting a somewhat different mode of interaction with C3b.
Paroxysmal nocturnal hemoglobinuria (PNH) is an intravascular hemolytic anemia caused by complement‐mediated destruction of red blood cells. Both DAF and CD59 are tethered to the cell surface through a GPI anchor. Rare somatic mutations (1 in 1,000,000) in the PIGA gene on the X chromosome, encoding phosphatidyl‐inositol glycan class A, which is involved in GPI production, lead to loss of the two regulators and hence uncontrolled alternative pathway amplification and terminal pathway activation on erythrocytes. PNH is induced if PIGA mutations lead to GPI deficiency in hematopoietic stem cells, which subsequently acquire clonal dominance. The disease is chronic as a consequence of the constitutive activation of the AP, and patients suffer from anemia, thrombosis, and smooth muscle dystonia (Brodsky, 2014). PNH is effectively treated with the C5 antibody eculizumab that binds to the MG7 domain in C5, thus preventing cleavage by the C5 convertase (Fig 5H). The presence of a mutation of a single residue in C5 Arg885 to either histidine or cysteine causes the carrier to be non‐responsive to eculizumab. The Arg885His mutation is present in 3.5% of the Japanese population; however, it has not been reported in Caucasians (Nishimura et al, 2014). The observed effects are explained by our model of the C5‐convertase complex (Laursen et al, 2011) since Arg885 is located within the patch on the C5 MG7 domain predicted to be recognized by C3b and C4b in the AP and CP C5 convertases, respectively (Fig 5H). C5 Arg885His/Cys is not bound by eculizumab (Nishimura et al, 2014), and the mutated C5 can be cleaved by the convertases.
Conclusion and perspectives
For more than a century, complement has been recognized as a central component of the humoral immune system, and it has remained a field of intense study. Yet recent research has continued to uncover intriguing and fundamental new functions and structural details of this ancient network of PRMs, proteases and their substrates, regulators, and immune receptors. As discussed here, recent studies have uncovered a central role of MASP‐3, as well as the PRM CL‐K1, in human fetal development, underlying the etiology of the 3MC syndrome. Furthermore, early classical pathway components have been deemed central for correct synaptic pruning in the developing brain. It appears that the molecular recognition complexes, proteolytic activities, and associated signaling pathways including specific receptors can double up and function in both development and tissue remodeling and host defense. Considering that the associated host defense functions, if activated improperly or excessively, can cause host tissue damage, their functions during development may similarly be envisioned to contribute to untoward reactions during, for example, age‐ and disease‐associated neurodegeneration. These are areas of active investigation.
As discussed earlier, the common polymorphisms in complement proteins affect the extent of complement activation in an individual and their susceptibility to untoward inflammation or infection. The list of polymorphisms in complement genes is growing, and the implication in pathologies as well as the intricate way in which the combined effect of these polymorphisms may create a unique complement setting is becoming increasingly appreciated. Therefore, the analysis of an individual's genetic predisposition to complement‐associated diseases may prove essential in the prevention, management, and treatment of inflammatory and infectious diseases. Although significant insight into the molecular mechanisms of complement activation and regulation has been provided by the rapidly increasing number of crystal structures of complement proteins, a full molecular understanding of how such polymorphisms in complement proteins translate into disease requires high‐resolution structures of very large surface‐associated macromolecular complexes with the convertases and the C1 complex being prominent examples. The recent revolution in single particle cryo‐EM will significantly promote future studies of large, rare, and unstable complexes of complement proteins. The ultimate goal will be the visualization of the complement response onto bacteria or even human cells with cryo‐EM tomography as pioneered with the recent liposome‐bound C1 structure.
Finally, complement research appears to uncover ever‐expanding roles of the system in the traditional domain of host defense. Two of the most striking recent observations are the roles of complement in shaping T‐cell immunity and the function of complement as a sensor of extracellular to intracellular translocation of invading bacteria and viruses. In this light, undoubtedly, many more exciting discoveries lie ahead.
Acknowledgements
This work was supported by the LUNA Nanomedicine Center, The Lundbeck Foundation (GRA and ST), the Danish Science Research Council (GRA), and the Danish Council for Independent Research, Medical Sciences (ST). GRA received a Hallas‐Møller stipend from the Novo‐Nordisk Foundation. SD was supported by a Marie Curie International Outgoing Fellowship within the 7th European Community Framework Programme.
Conflict of interest
GRA declares collaboration with Alexion Pharmaceuticals.
The EMBO Journal (2015) 34: 2735–2757
See the Glossary for abbreviations used in this article.
References
- Agarwal S, Ferreira VP, Cortes C, Pangburn MK, Rice PA, Ram S (2010) An evaluation of the role of properdin in alternative pathway activation on Neisseria meningitidis and Neisseria gonorrhoeae . J Immunol 185: 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcorlo M, Tortajada A, Rodriguez de Cordoba S, Llorca O (2013) Structural basis for the stabilization of the complement alternative pathway C3 convertase by properdin. Proc Natl Acad Sci USA 110: 13504–13509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleshin AE, DiScipio RG, Stec B, Liddington RC (2012) Crystal structure of C5b‐6 suggests structural basis for priming assembly of the membrane attack complex. J Biol Chem 287: 19642–19652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper CA, Boenisch T, Watson L (1972) Genetic polymorphism in human glycine‐rich beta‐glycoprotein. J Exp Med 135: 68–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, Lambris JD, Huber‐Lang M (2008) Interaction between the coagulation and complement system. Adv Exp Med Biol 632: 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, Bruckner UB, Nilsson B, Gebhard F, Lambris JD, Huber‐Lang M (2010) Molecular intercommunication between the complement and coagulation systems. J Immunol 185: 5628–5636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews PW, Knowles BB, Parkar M, Pym B, Stanley K, Goodfellow PN (1985) A human cell‐surface antigen defined by a monoclonal antibody and controlled by a gene on human chromosome 1. Ann Hum Genet 49: 31–39 [DOI] [PubMed] [Google Scholar]
- Arlaud GJ, Gaboriaud C, Thielens NM, Rossi V, Bersch B, Hernandez JF, Fontecilla‐Camps JC (2001) Structural biology of C1: dissection of a complex molecular machinery. Immunol Rev 180: 136–145 [DOI] [PubMed] [Google Scholar]
- Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, Cook HT, Kemper C (2013) C3a modulates IL‐1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 122: 3473–3481 [DOI] [PubMed] [Google Scholar]
- Axelgaard E, Jensen L, Dyrlund TF, Nielsen HJ, Enghild JJ, Thiel S, Jensenius JC (2013) Investigations on collectin liver 1. J Biol Chem 288: 23407–23420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic G, Yatime L, Klos A, Andersen GR (2013a) Human C3a and C3a desArg anaphylatoxins have conserved structures, in contrast to C5a and C5a desArg. Protein Sci 22: 204–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic G, Yatime L, Sim RB, Vorup‐Jensen T, Andersen GR (2013b) Structural insight on the recognition of surface‐bound opsonins by the integrin I domain of complement receptor 3. Proc Natl Acad Sci USA 110: 16426–16431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bally I, Rossi V, Lunardi T, Thielens NM, Gaboriaud C, Arlaud GJ (2009) Identification of the C1q‐binding sites of human C1r and C1s: a refined three‐dimensional model of the C1 complex of complement. J Biol Chem 284: 19340–19348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bally I, Ancelet S, Moriscot C, Gonnet F, Mantovani A, Daniel R, Schoehn G, Arlaud GJ, Thielens NM (2013) Expression of recombinant human complement C1q allows identification of the C1r/C1s‐binding sites. Proc Natl Acad Sci USA 110: 8650–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberg CE, Mackay CR, Lee H, Zahra D, Jackson J, Lim YS, Whitfeld PL, Craig S, Corsini E, Lu B, Gerard C, Gerard NP (2010) The C5a receptor (C5aR) C5L2 is a modulator of C5aR‐mediated signal transduction. J Biol Chem 285: 7633–7644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow PN, Hageman GS, Lea SM (2008) Complement factor H: using atomic resolution structure to illuminate disease mechanisms. Adv Exp Med Biol 632: 117–142 [PMC free article] [PubMed] [Google Scholar]
- Barnum SR (1995) Complement biosynthesis in the central nervous system. Crit Rev Oral Biol Med 6: 132–146 [DOI] [PubMed] [Google Scholar]
- Berends ET, Kuipers A, Ravesloot MM, Urbanus RT, Rooijakkers SH (2014) Bacteria under stress by complement and coagulation. FEMS Microbiol Rev 38: 1146–1171 [DOI] [PubMed] [Google Scholar]
- Blackmore TK, Sadlon TA, Ward HM, Lublin DM, Gordon DL (1996) Identification of a heparin binding domain in the seventh short consensus repeat of complement factor H. J Immunol 157: 5422–5427 [PubMed] [Google Scholar]
- Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrin D, Stehle T (2015) Structural basis for sialic acid‐mediated self‐recognition by complement factor H. Nat Chem Biol 11: 77–82 [DOI] [PubMed] [Google Scholar]
- Blom AM, Villoutreix BO, Dahlback B (2004) Complement inhibitor C4b‐binding protein‐friend or foe in the innate immune system? Mol Immunol 40: 1333–1346 [DOI] [PubMed] [Google Scholar]
- Bokisch VA, Muller‐Eberhard HJ (1970) Anaphylatoxin inactivator of human plasma: its isolation and characterization as a carboxypeptidase. J Clin Invest 49: 2427–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH (1992) Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum 35: 630–640 [DOI] [PubMed] [Google Scholar]
- Brier S, Pflieger D, Le Mignon M, Bally I, Gaboriaud C, Arlaud GJ, Daniel R (2010) Mapping surface accessibility of the C1r/C1s tetramer by chemical modification and mass spectrometry provides new insights into assembly of the human C1 complex. J Biol Chem 285: 32251–32263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann CR, Jensen L, Dagnaes‐Hansen F, Holm IE, Endo Y, Fujita T, Thiel S, Jensenius JC, Degn SE (2013) Mitochondria and the lectin pathway of complement. J Biol Chem 288: 8016–8027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky RA (2014) Paroxysmal nocturnal hemoglobinuria. Blood 124: 2804–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DR (1985) Immunoglobulin G: functional sites. Mol Immunol 22: 161–206 [DOI] [PubMed] [Google Scholar]
- Carroll MC (2000) The role of complement in B cell activation and tolerance. Adv Immunol 74: 61–88 [DOI] [PubMed] [Google Scholar]
- Carroll MC, Isenman DE (2012) Regulation of humoral immunity by complement. Immunity 37: 199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CB, Wallis R (2004) Two mechanisms for mannose‐binding protein modulation of the activity of its associated serine proteases. J Biol Chem 279: 26058–26065 [DOI] [PubMed] [Google Scholar]
- Chen X, Yu Y, Mi LZ, Walz T, Springer TA (2012) Molecular basis for complement recognition by integrin alphaXbeta2. Proc Natl Acad Sci USA 109: 4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenoweth DE, Goodman MG, Weigle WO (1982) Demonstration of a specific receptor for human C5a anaphylatoxin on murine macrophages. J Exp Med 156: 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianflone K, Xia Z, Chen LY (2003) Critical review of acylation‐stimulating protein physiology in humans and rodents. Biochim Biophys Acta 1609: 127–143 [DOI] [PubMed] [Google Scholar]
- Clark SJ, Higman VA, Mulloy B, Perkins SJ, Lea SM, Sim RB, Day AJ (2006) His‐384 allotypic variant of factor H associated with age‐related macular degeneration has different heparin binding properties from the non‐disease‐associated form. J Biol Chem 281: 24713–24720 [DOI] [PubMed] [Google Scholar]
- Cook WJ, Galakatos N, Boyar WC, Walter RL, Ealick SE (2010) Structure of human desArg‐C5a. Acta Crystallogr D Biol Crystallogr 66: 190–197 [DOI] [PubMed] [Google Scholar]
- de Cordoba SR, Tortajada A, Harris CL, Morgan BP (2012) Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology 217: 1034–1046 [DOI] [PubMed] [Google Scholar]
- de Cordoba SR (2015) Complement genetics and susceptibility to inflammatory disease. Lessons from genotype‐phenotype correlations. Immunobiology doi:10.1016/j.imbio.2015.05.015 [DOI] [PubMed] [Google Scholar]
- Cortes C, Ohtola JA, Saggu G, Ferreira VP (2012) Local release of properdin in the cellular microenvironment: role in pattern recognition and amplification of the alternative pathway of complement. Front Immunol 3: 412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesio CL, Jiang W (2006) Mannan‐binding lectin‐associated serine protease 3 cleaves synthetic peptides and insulin‐like growth factor‐binding protein 5. Arch Biochem Biophys 449: 164–170 [DOI] [PubMed] [Google Scholar]
- Coulthard LG, Woodruff TM (2015) Is the complement activation product C3a a proinflammatory molecule? Re‐evaluating the evidence and the Myth. J Immunol 194: 3542–3548 [DOI] [PubMed] [Google Scholar]
- Cui W, Lapointe M, Gauvreau D, Kalant D, Cianflone K (2009) Recombinant C3adesArg/acylation stimulating protein (ASP) is highly bioactive: a critical evaluation of C5L2 binding and 3T3‐L1 adipocyte activation. Mol Immunol 46: 3207–3217 [DOI] [PubMed] [Google Scholar]
- Dahl MR, Thiel S, Matsushita M, Fujita T, Willis AC, Christensen T, Vorup‐Jensen T, Jensenius JC (2001) MASP‐3 and its association with distinct complexes of the mannan‐binding lectin complement activation pathway. Immunity 15: 127–135 [DOI] [PubMed] [Google Scholar]
- Davis AE III (1988) C1 inhibitor and hereditary angioneurotic edema. Annu Rev Immunol 6: 595–628 [DOI] [PubMed] [Google Scholar]
- Degn SE, Hansen AG, Steffensen R, Jacobsen C, Jensenius JC, Thiel S (2009) MAp44, a human protein associated with pattern recognition molecules of the complement system and regulating the lectin pathway of complement activation. J Immunol 183: 7371–7378 [DOI] [PubMed] [Google Scholar]
- Degn SE, Thiel S, Nielsen O, Hansen AG, Steffensen R, Jensenius JC (2011) MAp19, the alternative splice product of the MASP2 gene. J Immunol Methods 373: 89–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degn SE, Thiel S (2013) Humoral pattern recognition and the complement system. Scand J Immunol 78: 181–193 [DOI] [PubMed] [Google Scholar]
- Degn SE, Jensen L, Olszowski T, Jensenius JC, Thiel S (2013a) Co‐complexes of MASP‐1 and MASP‐2 associated with the soluble pattern‐recognition molecules drive lectin pathway activation in a manner inhibitable by MAp44. J Immunol 191: 1334–1345 [DOI] [PubMed] [Google Scholar]
- Degn SE, Thiel S, Jensenius JC (2013b) Recombinant expression of the autocatalytic complement protease MASP‐1 is crucially dependent on co‐expression with its inhibitor, C1 inhibitor. Protein Expr Purif 88: 173–182 [DOI] [PubMed] [Google Scholar]
- Degn SE, Jensenius JC, Thiel S (2014a) The pro‐factor D cleaving activity of MASP‐1/‐3 is not required for alternative pathway function. J Immunol 192: 5447–5448 [DOI] [PubMed] [Google Scholar]
- Degn SE, Kjaer TR, Kidmose RT, Jensen L, Hansen AG, Tekin M, Jensenius JC, Andersen GR, Thiel S (2014b) Complement activation by ligand‐driven juxtaposition of discrete pattern recognition complexes. Proc Natl Acad Sci USA 111: 13445–13450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJ, van de Winkel JG, Wilson IA, Koster AJ, Taylor RP, Saphire EO, Burton DR, Schuurman J, Gros P, Parren PW (2014) Complement is activated by IgG hexamers assembled at the cell surface. Science 343: 1260–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragon‐Durey MA, Fremeaux‐Bacchi V, Loirat C, Blouin J, Niaudet P, Deschenes G, Coppo P, Fridman WH, Weiss L (2004) Heterozygous and homozygous factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol 15: 787–795 [DOI] [PubMed] [Google Scholar]
- Duncan RC, Bergstrom F, Coetzer TH, Blom AM, Wijeyewickrema LC, Pike RN (2012) Multiple domains of MASP‐2, an initiating complement protease, are required for interaction with its substrate C4. Mol Immunol 49: 593–600 [DOI] [PubMed] [Google Scholar]
- Eberhardt HU, Buhlmann D, Hortschansky P, Chen Q, Bohm S, Kemper MJ, Wallich R, Hartmann A, Hallstrom T, Zipfel PF, Skerka C (2013) Human factor H‐related protein 2 (CFHR2) regulates complement activation. PLoS ONE 8: e78617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Elsen JM, Isenman DE (2011) A crystal structure of the complex between human complement receptor 2 and its ligand C3d. Science 332: 608–611 [DOI] [PubMed] [Google Scholar]
- Esparza‐Gordillo J, Soria JM, Buil A, Almasy L, Blangero J, Fontcuberta J, de Cordoba SR (2004) Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics 56: 77–82 [DOI] [PubMed] [Google Scholar]
- Fang Y, Xu C, Fu YX, Holers VM, Molina H (1998) Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen‐specific IgG response. J Immunol 160: 5273–5279 [PubMed] [Google Scholar]
- Farzan M, Schnitzler CE, Vasilieva N, Leung D, Kuhn J, Gerard C, Gerard NP, Choe H (2001) Sulfated tyrosines contribute to the formation of the C5a docking site of the human C5a anaphylatoxin receptor. J Exp Med 193: 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon DT, Austen KF, Ruddy S (1973) Formation of a hemolytically active cellular intermediate by the interaction between properdin factors B and D and the activated third component of complement. J Exp Med 138: 1305–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon DT, Austen KF (1975) Properdin: binding to C3b and stabilization of the C3b‐dependent C3 convertase. J Exp Med 142: 856–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon DT (1980) Identification of the membrane glycoprotein that is the C3b receptor of the human erythrocyte, polymorphonuclear leukocyte, B lymphocyte, and monocyte. J Exp Med 152: 20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira VP, Herbert AP, Hocking HG, Barlow PN, Pangburn MK (2006) Critical role of the C‐terminal domains of factor H in regulating complement activation at cell surfaces. J Immunol 177: 6308–6316 [DOI] [PubMed] [Google Scholar]
- Ferreira VP, Pangburn MK, Cortes C (2010) Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol 47: 2187–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forneris F, Ricklin D, Wu J, Tzekou A, Wallace RS, Lambris JD, Gros P (2010) Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science 330: 1816–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredslund F, Laursen NS, Roversi P, Jenner L, Oliveira CL, Pedersen JS, Nunn MA, Lea SM, Discipio R, Sottrup‐Jensen L, Andersen GR (2008) Structure of and influence of a tick complement inhibitor on human complement component 5. Nat Immunol 9: 753–760 [DOI] [PubMed] [Google Scholar]
- Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs‐Mecarelli L, Fremeaux‐Bacchi V, Roumenina LT (2013) Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 122: 282–292 [DOI] [PubMed] [Google Scholar]
- Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, Pandey MK, Kohl J, Zipfel PF, Weber BH, Skerka C (2010) An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age‐related macular degeneration (AMD). Hum Mol Genet 19: 4694–4704 [DOI] [PubMed] [Google Scholar]
- Gadjeva M, Verschoor A, Brockman MA, Jezak H, Shen LM, Knipe DM, Carroll MC (2002) Macrophage‐derived complement component C4 can restore humoral immunity in C4‐deficient mice. J Immunol 169: 5489–5495 [DOI] [PubMed] [Google Scholar]
- Ghannam A, Fauquert JL, Thomas C, Kemper C, Drouet C (2014) Human complement C3 deficiency: Th1 induction requires T cell‐derived complement C3a and CD46 activation. Mol Immunol 58: 98–107 [DOI] [PubMed] [Google Scholar]
- Gingras AR, Girija UV, Keeble AH, Panchal R, Mitchell DA, Moody PC, Wallis R (2011) Structural basis of mannan‐binding lectin recognition by its associated serine protease MASP‐1: implications for complement activation. Structure 19: 1635–1643 [DOI] [PubMed] [Google Scholar]
- Gladman DD, Ibanez D, Urowitz MB (2002) Systemic lupus erythematosus disease activity index 2000. J Rheumatol 29: 288–291 [PubMed] [Google Scholar]
- Goicoechea de Jorge E, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, Hakobyan S, Morgan BP, Harris CL, Pickering MC, Lea SM (2013) Dimerization of complement factor H‐related proteins modulates complement activation in vivo . Proc Natl Acad Sci USA 110: 4685–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DL, Kaufman RM, Blackmore TK, Kwong J, Lublin DM (1995) Identification of complement regulatory domains in human factor H. J Immunol 155: 348–356 [PubMed] [Google Scholar]
- Guo RF, Ward PA (2005) Role of C5A in inflammatory responses. Annu Rev Immunol 23: 821–852 [DOI] [PubMed] [Google Scholar]
- Hadders MA, Beringer DX, Gros P (2007) Structure of C8alpha‐MACPF reveals mechanism of membrane attack in complement immune defense. Science 317: 1552–1554 [DOI] [PubMed] [Google Scholar]
- Hadders MA, Bubeck D, Roversi P, Hakobyan S, Forneris F, Morgan BP, Pangburn MK, Llorca O, Lea SM, Gros P (2012) Assembly and regulation of the membrane attack complex based on structures of C5b6 and sC5b9. Cell Rep 1: 200–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE (2004) The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol 138: 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harboe M, Garred P, Karlstrom E, Lindstad JK, Stahl GL, Mollnes TE (2009) The down‐stream effects of mannan‐induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol Immunol 47: 373–380 [DOI] [PubMed] [Google Scholar]
- Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP (2012) The complotype: dictating risk for inflammation and infection. Trends Immunol 33: 513–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt K, Kraiczy P, Wallich R, Brade V, Skerka C, Zipfel PF (2007) Binding of human factor H‐related protein 1 to serum‐resistant Borrelia burgdorferi is mediated by borrelial complement regulator‐acquiring surface proteins. J Infect Dis 196: 124–133 [DOI] [PubMed] [Google Scholar]
- Hawlisch H, Wills‐Karp M, Karp CL, Kohl J (2004) The anaphylatoxins bridge innate and adaptive immune responses in allergic asthma. Mol Immunol 41: 123–131 [DOI] [PubMed] [Google Scholar]
- Hawlisch H, Kohl J (2006) Complement and Toll‐like receptors: key regulators of adaptive immune responses. Mol Immunol 43: 13–21 [DOI] [PubMed] [Google Scholar]
- Haynes T, Luz‐Madrigal A, Reis ES, Echeverri Ruiz NP, Grajales‐Esquivel E, Tzekou A, Tsonis PA, Lambris JD, Del Rio‐Tsonis K (2013) Complement anaphylatoxin C3a is a potent inducer of embryonic chick retina regeneration. Nat Commun 4: 2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Atkinson C, Qiao F, Cianflone K, Chen X, Tomlinson S (2009) A complement‐dependent balance between hepatic ischemia/reperfusion injury and liver regeneration in mice. J Clin Invest 119: 2304–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebecker M, Jozsi M (2012) Factor H‐related protein 4 activates complement by serving as a platform for the assembly of alternative pathway C3 convertase via its interaction with C3b protein. J Biol Chem 287: 19528–19536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, Carroll MC (2013) Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity 38: 1164–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesters BA, Myers RC, Carroll MC (2014) Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol 14: 495–504 [DOI] [PubMed] [Google Scholar]
- Heinen S, Hartmann A, Lauer N, Wiehl U, Dahse HM, Schirmer S, Gropp K, Enghardt T, Wallich R, Halbich S, Mihlan M, Schlotzer‐Schrehardt U, Zipfel PF, Skerka C (2009) Factor H‐related protein 1 (CFHR‐1) inhibits complement C5 convertase activity and terminal complex formation. Blood 114: 2439–2447 [DOI] [PubMed] [Google Scholar]
- Hellwage J, Jokiranta TS, Koistinen V, Vaarala O, Meri S, Zipfel PF (1999) Functional properties of complement factor H‐related proteins FHR‐3 and FHR‐4: binding to the C3d region of C3b and differential regulation by heparin. FEBS Lett 462: 345–352 [DOI] [PubMed] [Google Scholar]
- Helmy KY, Katschke KJ Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, Scales SJ, Ghilardi N, van Lookeren Campagne M (2006) CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 124: 915–927 [DOI] [PubMed] [Google Scholar]
- Henriksen ML, Brandt J, Andrieu JP, Nielsen C, Jensen PH, Holmskov U, Jorgensen TJ, Palarasah Y, Thielens NM, Hansen S (2013a) Heteromeric complexes of native collectin kidney 1 and collectin liver 1 are found in the circulation with MASPs and activate the complement system. J Immunol 191: 6117–6127 [DOI] [PubMed] [Google Scholar]
- Henriksen ML, Brandt J, Iyer SS, Thielens NM, Hansen S (2013b) Characterization of the interaction between collectin 11 (CL‐11, CL‐K1) and nucleic acids. Mol Immunol 56: 757–767 [DOI] [PubMed] [Google Scholar]
- Herbert AP, Deakin JA, Schmidt CQ, Blaum BS, Egan C, Ferreira VP, Pangburn MK, Lyon M, Uhrin D, Barlow PN (2007) Structure shows that a glycosaminoglycan and protein recognition site in factor H is perturbed by age‐related macular degeneration‐linked single nucleotide polymorphism. J Biol Chem 282: 18960–18968 [DOI] [PubMed] [Google Scholar]
- Hillebrandt S, Wasmuth HE, Weiskirchen R, Hellerbrand C, Keppeler H, Werth A, Schirin‐Sokhan R, Wilkens G, Geier A, Lorenzen J, Kohl J, Gressner AM, Matern S, Lammert F (2005) Complement factor 5 is a quantitative trait gene that modifies liver fibrogenesis in mice and humans. Nat Genet 37: 835–843 [DOI] [PubMed] [Google Scholar]
- Hofmeyer T, Schmelz S, Degiacomi MT, Dal Peraro M, Daneschdar M, Scrima A, van den Heuvel J, Heinz DW, Kolmar H (2013) Arranged sevenfold: structural insights into the C‐terminal oligomerization domain of human C4b‐binding protein. J Mol Biol 425: 1302–1317 [DOI] [PubMed] [Google Scholar]
- Honore C, Rorvig S, Hummelshoj T, Skjoedt MO, Borregaard N, Garred P (2010) Tethering of Ficolin‐1 to cell surfaces through recognition of sialic acid by the fibrinogen‐like domain. J Leukoc Biol 88: 145–158 [DOI] [PubMed] [Google Scholar]
- Huber‐Lang MS, Sarma JV, McGuire SR, Lu KT, Padgaonkar VA, Younkin EM, Guo RF, Weber CH, Zuiderweg ER, Zetoune FS, Ward PA (2003) Structure‐function relationships of human C5a and C5aR. J Immunol 170: 6115–6124 [DOI] [PubMed] [Google Scholar]
- Huber‐Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA (2006) Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 12: 682–687 [DOI] [PubMed] [Google Scholar]
- Hughes‐Jones NC, Gardner B (1979) Reaction between the isolated globular sub‐units of the complement component C1q and IgG‐complexes. Mol Immunol 16: 697–701 [DOI] [PubMed] [Google Scholar]
- Iwaki D, Kanno K, Takahashi M, Endo Y, Lynch NJ, Schwaeble WJ, Matsushita M, Okabe M, Fujita T (2006) Small mannose‐binding lectin‐associated protein plays a regulatory role in the lectin complement pathway. J Immunol 177: 8626–8632 [DOI] [PubMed] [Google Scholar]
- Iwaki D, Kanno K, Takahashi M, Endo Y, Matsushita M, Fujita T (2011) The role of mannose‐binding lectin‐associated serine protease‐3 in activation of the alternative complement pathway. J Immunol 187: 3751–3758 [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Huizinga EG, Raaijmakers HC, Roos A, Daha MR, Nilsson‐Ekdahl K, Nilsson B, Gros P (2005) Structures of complement component C3 provide insights into the function and evolution of immunity. Nature 437: 505–511 [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Christodoulidou A, McCarthy A, Lambris JD, Gros P (2006) Structure of C3b reveals conformational changes that underlie complement activity. Nature 444: 213–216 [DOI] [PubMed] [Google Scholar]
- Jozsi M, Zipfel PF (2008) Factor H family proteins and human diseases. Trends Immunol 29: 380–387 [DOI] [PubMed] [Google Scholar]
- Kang YH, Tan LA, Carroll MV, Gentle ME, Sim RB (2009) Target pattern recognition by complement proteins of the classical and alternative pathways. Adv Exp Med Biol 653: 117–128 [DOI] [PubMed] [Google Scholar]
- Kawasaki N, Kawasaki T, Yamashina I (1983) Isolation and characterization of a mannan‐binding protein from human serum. J Biochem 94: 937–947 [DOI] [PubMed] [Google Scholar]
- Kemper C, Mitchell LM, Zhang L, Hourcade DE (2008) The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci USA 105: 9023–9028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Nan R, Gor J, Mulloy B, Perkins SJ (2012) Bivalent and co‐operative binding of complement factor H to heparan sulfate and heparin. Biochem J 444: 417–428 [DOI] [PubMed] [Google Scholar]
- Kidmose RT, Laursen NS, Dobo J, Kjaer TR, Sirotkina S, Yatime L, Sottrup‐Jensen L, Thiel S, Gal P, Andersen GR (2012) Structural basis for activation of the complement system by component C4 cleavage. Proc Natl Acad Sci USA 109: 15425–15430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YU, Carroll MC, Isenman DE, Nonaka M, Pramoonjago P, Takeda J, Inoue K, Kinoshita T (1992) Covalent binding of C3b to C4b within the classical complement pathway C5 convertase. Determination of amino acid residues involved in ester linkage formation. J Biol Chem 267: 4171–4176 [PubMed] [Google Scholar]
- Kjaer TR, Hansen AG, Sorensen UB, Nielsen O, Thiel S, Jensenius JC (2011) Investigations on the pattern recognition molecule M‐ficolin: quantitative aspects of bacterial binding and leukocyte association. J Leukoc Biol 90: 425–437 [DOI] [PubMed] [Google Scholar]
- Kjaer TR, Thiel S, Andersen GR (2013) Toward a structure‐based comprehension of the lectin pathway of complement. Mol Immunol 56: 413–422 [DOI] [PubMed] [Google Scholar]
- Kjaer TR, Le LTM, Pedersen JS, Sander B, Golas M, Jensenius JC, Andersen GR, Thiel S (2015) Structural insights into the initiating complex of the lectin pathway of complement activation. Structure 23: 342–351 [DOI] [PubMed] [Google Scholar]
- Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J (2009) The role of the anaphylatoxins in health and disease. Mol Immunol 46: 2753–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klos A, Wende E, Wareham KJ, Monk PN (2013) International Union of Basic and Clinical Pharmacology. [corrected]. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol Rev 65: 500–543 [DOI] [PubMed] [Google Scholar]
- Krarup A, Sorensen UB, Matsushita M, Jensenius JC, Thiel S (2005) Effect of capsulation of opportunistic pathogenic bacteria on binding of the pattern recognition molecules mannan‐binding lectin, L‐ficolin, and H‐ficolin. Infect Immun 73: 1052–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krisinger MJ, Goebeler V, Lu Z, Meixner SC, Myles T, Pryzdial EL, Conway EM (2012) Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood 120: 1717–1725 [DOI] [PubMed] [Google Scholar]
- Kuhn S, Zipfel PF (1996) Mapping of the domains required for decay acceleration activity of the human factor H‐like protein 1 and factor H. Eur J Immunol 26: 2383–2387 [DOI] [PubMed] [Google Scholar]
- Kunert A, Losse J, Gruszin C, Huhn M, Kaendler K, Mikkat S, Volke D, Hoffmann R, Jokiranta TS, Seeberger H, Moellmann U, Hellwage J, Zipfel PF (2007) Immune evasion of the human pathogen Pseudomonas aeruginosa: elongation factor Tuf is a factor H and plasminogen binding protein. J Immunol 179: 2979–2988 [DOI] [PubMed] [Google Scholar]
- La Bonte LR, Pavlov VI, Tan YS, Takahashi K, Takahashi M, Banda NK, Zou C, Fujita T, Stahl GL (2012) Mannose‐binding lectin‐associated serine protease‐1 is a significant contributor to coagulation in a murine model of occlusive thrombosis. J Immunol 188: 885–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix M, Dumestre‐Perard C, Schoehn G, Houen G, Cesbron JY, Arlaud GJ, Thielens NM (2009) Residue Lys57 in the collagen‐like region of human L‐ficolin and its counterpart Lys47 in H‐ficolin play a key role in the interaction with the mannan‐binding lectin‐associated serine proteases and the collectin receptor calreticulin. J Immunol 182: 456–465 [DOI] [PubMed] [Google Scholar]
- Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, Morgan BP, Sivasankar B, Mortellaro A (2013) Cutting edge: the NLRP3 inflammasome links complement‐mediated inflammation and IL‐1beta release. J Immunol 191: 1006–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen NS, Andersen KR, Braren I, Spillner E, Sottrup‐Jensen L, Andersen GR (2011) Substrate recognition by complement convertases revealed in the C5‐cobra venom factor complex. EMBO J 30: 606–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen NS, Magnani F, Gottfredsen RH, Petersen SV, Andersen GR (2012) Structure, function and control of complement C5 and its proteolytic fragments. Curr Mol Med 12: 1083–1097 [DOI] [PubMed] [Google Scholar]
- Law SK, Dodds AW (1997) The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci 6: 263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Atmaca‐Sonmez P, Othman M, Branham KE, Khanna R, Wade MS, Li Y, Liang L, Zareparsi S, Swaroop A, Abecasis GR (2006) CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age‐related macular degeneration. Nat Genet 38: 1049–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Coulthard LG, Wu MC, Taylor SM, Woodruff TM (2013) C5L2: a controversial receptor of complement anaphylatoxin, C5a. FASEB J 27: 855–864 [DOI] [PubMed] [Google Scholar]
- Linton SM, Morgan BP (1999) Complement activation and inhibition in experimental models of arthritis. Mol Immunol 36: 905–914 [DOI] [PubMed] [Google Scholar]
- Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S, Arstila TP, Pekkarinen PT, Ma M, Cope A, Reinheckel T, Rodriguez de Cordoba S, Afzali B, Atkinson JP, Kemper C (2013) Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39: 1143–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszewski MK, Atkinson JP (2015) Complement regulators in human disease: lessons from modern genetics. J Intern Med 277: 294–305 [DOI] [PubMed] [Google Scholar]
- Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, Kelly ME, Chatterjee Bhowmick D, Murano I, Cohen P, Banks AS, Khandekar MJ, Dietrich A, Flier JS, Cinti S, Bluher M, Danial NN, Berggren PO, Spiegelman BM (2014) Adipsin is an adipokine that improves beta cell function in diabetes. Cell 158: 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makou E, Herbert AP, Barlow PN (2013) Functional anatomy of complement factor H. Biochemistry 52: 3949–3962 [DOI] [PubMed] [Google Scholar]
- Manuelian T, Hellwage J, Meri S, Caprioli J, Noris M, Heinen S, Jozsi M, Neumann HPH, Remuzzi G, Zipfel PF (2003) Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest 111: 1181–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, Cayla M, Tabarin F, Jablonski M, Hue C, Smith RJ, Noris M, Halbwachs‐Mecarelli L, Donadelli R, Fremeaux‐Bacchi V, Roumenina LT (2014) Complement factor B mutations in atypical hemolytic uremic syndrome‐disease‐relevant or benign? J Am Soc Nephrol 25: 2053–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Leffler J, Blom AM (2012) Annexin A2 and A5 serve as new ligands for C1q on apoptotic cells. J Biol Chem 287: 33733–33744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastellos D, Papadimitriou JC, Franchini S, Tsonis PA, Lambris JD (2001) A novel role of complement: mice deficient in the fifth component of complement (C5) exhibit impaired liver regeneration. J Immunol 166: 2479–2486 [DOI] [PubMed] [Google Scholar]
- Matsushita M, Thiel S, Jensenius JC, Terai I, Fujita T (2000) Proteolytic activities of two types of mannose‐binding lectin‐associated serine protease. J Immunol 165: 2637–2642 [DOI] [PubMed] [Google Scholar]
- Matsushita M (2013) Ficolins in complement activation. Mol Immunol 55: 22–26 [DOI] [PubMed] [Google Scholar]
- Matthews KW, Mueller‐Ortiz SL, Wetsel RA (2004) Carboxypeptidase N: a pleiotropic regulator of inflammation. Mol Immunol 40: 785–793 [DOI] [PubMed] [Google Scholar]
- McRae JL, Duthy TG, Griggs KM, Ormsby RJ, Cowan PJ, Cromer BA, McKinstry WJ, Parker MW, Murphy BF, Gordon DL (2005) Human factor H‐related protein 5 has cofactor activity, inhibits C3 convertase activity, binds heparin and C‐reactive protein, and associates with lipoprotein. J Immunol 174: 6250–6256 [DOI] [PubMed] [Google Scholar]
- Medicus RG, Gotze O, Muller‐Eberhard HJ (1976) Alternative pathway of complement: recruitment of precursor properdin by the labile C3/C5 convertase and the potentiation of the pathway. J Exp Med 144: 1076–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof ME, Kinoshita T, Nussenzweig V (1984) Inhibition of complement activation on the surface of cells after incorporation of decay‐accelerating factor (DAF) into their membranes. J Exp Med 160: 1558–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megyeri M, Mako V, Beinrohr L, Doleschall Z, Prohaszka Z, Cervenak L, Zavodszky P, Gal P (2009) Complement protease MASP‐1 activates human endothelial cells: PAR4 activation is a link between complement and endothelial function. J Immunol 183: 3409–3416 [DOI] [PubMed] [Google Scholar]
- Meri S, Pangburn MK (1990) A mechanism of activation of the alternative complement pathway by the classical pathway: protection of C3b from inactivation by covalent attachment to C4b. Eur J Immunol 20: 2555–2561 [DOI] [PubMed] [Google Scholar]
- Meri S, Waldmann H, Lachmann PJ (1991) Distribution of protectin (CD59), a complement membrane attack inhibitor, in normal human tissues. Lab Invest 65: 532–537 [PubMed] [Google Scholar]
- Metlay JP, Witmer‐Pack MD, Agger R, Crowley MT, Lawless D, Steinman RM (1990) The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J Exp Med 171: 1753–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BP, Gasque P (1997) Extrahepatic complement biosynthesis: where, when and why? Clin Exp Immunol 107: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BP, Berg CW, Harris CL (2005) “Homologous restriction” in complement lysis: roles of membrane complement regulators. Xenotransplantation 12: 258–265 [DOI] [PubMed] [Google Scholar]
- Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, Kavanagh D, Mertens HD, Svergun DI, Johansson CM, Uhrin D, Barlow PN, Hannan JP (2011) Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol 18: 463–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen S, Kidmose RT, Petersen SV, Szilagyi A, Prohaszka Z, Andersen GR (2015) Structural basis for the function of complement component C4 within the classical and lectin pathways of complement. J Immunol 194: 5488–5496 [DOI] [PubMed] [Google Scholar]
- Moskovich O, Fishelson Z (2007) Live cell imaging of outward and inward vesiculation induced by the complement c5b‐9 complex. J Biol Chem 282: 29977–29986 [DOI] [PubMed] [Google Scholar]
- Mukherjee P, Thomas S, Pasinetti GM (2008) Complement anaphylatoxin C5a neuroprotects through regulation of glutamate receptor subunit 2 in vitro and in vivo . J Neuroinflammation 5: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller‐Eberhard HJ, Polley MJ, Calcott MA (1967) Formation and functional significance of a molecular complex derived from the second and the fourth component of human complement. J Exp Med 125: 359–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B, Georgiou T, Machet D, Hill P, McRae J (2002) Factor H‐related protein‐5: a novel component of human glomerular immune deposits. Am J Kidney Dis 39: 24–27 [DOI] [PubMed] [Google Scholar]
- Nayak A, Pednekar L, Reid KB, Kishore U (2012) Complement and non‐complement activating functions of C1q: a prototypical innate immune molecule. Innate Immun 18: 350–363 [DOI] [PubMed] [Google Scholar]
- Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, Noji H, Kitamura K, Eto T, Takahashi T, Masuko M, Matsumoto T, Wano Y, Shichishima T, Shibayama H, Hase M, Li L, Johnson K, Lazarowski A, Tamburini P et al (2014) Genetic variants in C5 and poor response to eculizumab. N Engl J Med 370: 632–639 [DOI] [PubMed] [Google Scholar]
- Noris M, Remuzzi G (2009) Atypical hemolytic‐uremic syndrome. N Engl J Med 361: 1676–1687 [DOI] [PubMed] [Google Scholar]
- Oppermann M, Manuelian T, Jozsi M, Brandt E, Jokiranta TS, Heinen S, Meri S, Skerka C, Gotze O, Zipfel PF (2006) The C‐terminus of complement regulator Factor H mediates target recognition: evidence for a compact conformation of the native protein. Clin Exp Immunol 144: 342–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paidassi H, Tacnet‐Delorme P, Garlatti V, Darnault C, Ghebrehiwet B, Gaboriaud C, Arlaud GJ, Frachet P (2008) C1q binds phosphatidylserine and likely acts as a multiligand‐bridging molecule in apoptotic cell recognition. J Immunol 180: 2329–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangburn MK, Schreiber RD, Müller‐Eberhard HJ (1981) Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b‐like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med 154: 856–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins SJ, Fung KW, Khan S (2014) Molecular interactions between complement factor H and Its heparin and heparan sulfate ligands. Front Immunol 5: 126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan TG, Grigorova I, Okada T, Cyster JG (2007) Subcapsular encounter and complement‐dependent transport of immune complexes by lymph node B cells. Nat Immunol 8: 992–1000 [DOI] [PubMed] [Google Scholar]
- Phan TG, Green JA, Gray EE, Xu Y, Cyster JG (2009) Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat Immunol 10: 786–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AE, Toth J, Dodds AW, Girija UV, Furze CM, Pala E, Sim RB, Reid KB, Schwaeble WJ, Schmid R, Keeble AH, Wallis R (2009) Analogous interactions in initiating complexes of the classical and lectin pathways of complement. J Immunol 182: 7708–7717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ (2000) Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol 76: 227–324 [DOI] [PubMed] [Google Scholar]
- Podack ER, Kolb WP, Muller‐Eberhard HJ (1978) The C5b‐6 complex: formation, isolation, and inhibition of its activity by lipoprotein and the S‐protein of human serum. J Immunol 120: 1841–1848 [PubMed] [Google Scholar]
- Poursharifi P, Lapointe M, Fisette A, Lu H, Roy C, Munkonda MN, Fairlie DP, Cianflone K (2014) C5aR and C5L2 act in concert to balance immunometabolism in adipose tissue. Mol Cell Endocrinol 382: 325–333 [DOI] [PubMed] [Google Scholar]
- Prosser BE, Johnson S, Roversi P, Herbert AP, Blaum BS, Tyrrell J, Jowitt TA, Clark SJ, Tarelli E, Uhrín D, Barlow PN, Sim RB, Day AJ, Lea SM (2007) Structural basis for complement factor H linked age‐related macular degeneration. J Exp Med 204: 2277–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez‐Ortiz ZG, Pendergraft WF 3rd, Prasad A, Byrne MH, Iram T, Blanchette CJ, Luster AD, Hacohen N, El Khoury J, Means TK (2013) The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol 14: 917–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynes M, Aubert JP, Cohen JH, Audouin J, Tricottet V, Diebold J, Kazatchkine MD (1985) Human follicular dendritic cells express CR1, CR2, and CR3 complement receptor antigens. J Immunol 135: 2687–2694 [PubMed] [Google Scholar]
- Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber‐Lang M, Mackay CR, Zetoune FS, Gerard NP, Cianflone K, Kohl J, Gerard C, Sarma JV, Ward PA (2008) Functional roles for C5a receptors in sepsis. Nat Med 14: 551–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Latz E, Ontiveros F, Kono H (2010) The sterile inflammatory response. Annu Rev Immunol 28: 321–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez de Cordoba S, Rubinstein P (1984) Genetic polymorphism of human factor H (beta 1H). J Immunol 132: 1906–1908 [PubMed] [Google Scholar]
- Rodriguez E, Rallapalli PM, Osborne AJ, Perkins SJ (2014) New functional and structural insights from updated mutational databases for complement factor H, Factor I, membrane cofactor protein and C3. Biosci Rep 34: e00146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooijakkers SH, Wu J, Ruyken M, van Domselaar R, Planken KL, Tzekou A, Ricklin D, Lambris JD, Janssen BJ, van Strijp JA, Gros P (2009) Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat Immunol 10: 721–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooryck C, Diaz‐Font A, Osborn DP, Chabchoub E, Hernandez‐Hernandez V, Shamseldin H, Kenny J, Waters A, Jenkins D, Kaissi AA, Leal GF, Dallapiccola B, Carnevale F, Bitner‐Glindzicz M, Lees M, Hennekam R, Stanier P, Burns AJ, Peeters H, Alkuraya FS et al (2011) Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat Genet 43: 197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GD (2000) Regulation of the adhesion versus cytotoxic functions of the Mac‐1/CR3/alphaMbeta2‐integrin glycoprotein. Crit Rev Immunol 20: 197–222 [PubMed] [Google Scholar]
- Rossi V, Cseh S, Bally I, Thielens NM, Jensenius JC, Arlaud GJ (2001) Substrate specificities of recombinant mannan‐binding lectin‐associated serine proteases‐1 and ‐2. J Biol Chem 276: 40880–40887 [DOI] [PubMed] [Google Scholar]
- Rossi V, Teillet F, Thielens NM, Bally I, Arlaud GJ (2005) Functional characterization of complement proteases C1s/mannan‐binding lectin‐associated serine protease‐2 (MASP‐2) chimeras reveals the higher C4 recognition efficacy of the MASP‐2 complement control protein modules. J Biol Chem 280: 41811–41818 [DOI] [PubMed] [Google Scholar]
- Roversi P, Johnson S, Caesar JJ, McLean F, Leath KJ, Tsiftsoglou SA, Morgan BP, Harris CL, Sim RB, Lea SM (2011) Structural basis for complement factor I control and its disease‐associated sequence polymorphisms. Proc Natl Acad Sci USA 108: 12839–12844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruseva MM, Takahashi M, Fujita T, Pickering MC (2014) C3 dysregulation due to factor H deficiency is mannan‐binding lectin‐associated serine proteases (MASP)‐1 and MASP‐3 independent in vivo . Clin Exp Immunol 176: 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saggu G, Cortes C, Emch HN, Ramirez G, Worth RG, Ferreira VP (2013) Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O). J Immunol 190: 6457–6467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayah S, Jauneau AC, Patte C, Tonon MC, Vaudry H, Fontaine M (2003) Two different transduction pathways are activated by C3a and C5a anaphylatoxins on astrocytes. Brain Res Mol Brain Res 112: 53–60 [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 74: 691–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz‐Jakobsen JA, Yatime L, Larsen C, Petersen SV, Klos A, Andersen GR (2014) Structural and functional characterization of human and murine C5a anaphylatoxins. Acta Crystallogr D Biol Crystallogr 70: 1704–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CQ, Herbert AP, Kavanagh D, Gandy C, Fenton CJ, Blaum BS, Lyon M, Uhrin D, Barlow PN (2008) A new map of glycosaminoglycan and C3b binding sites on factor H. J Immunol 181: 2610–2619 [DOI] [PubMed] [Google Scholar]
- Schramm EC, Roumenina LT, Rybkine T, Chauvet S, Vieira‐Martins P, Hue C, Maga T, Valoti E, Wilson V, Jokiranta S, Smith RJ, Noris M, Goodship T, Atkinson JP, Fremeaux‐Bacchi V (2015) Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood 194: 5129–5138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, Goldstein JI, Triebwasser M, Anderson HE, Zerbib J, Kavanagh D, Souied E, Katsanis N, Daly MJ, Atkinson JP, Raychaudhuri S (2013) Rare variants in CFI, C3 and C9 are associated with high risk of advanced age‐related macular degeneration. Nat Genet 45: 1366–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman L, Hansen S (2012) Structure and function of collectin liver 1 (CL‐L1) and collectin 11 (CL‐11, CL‐K1). Immunobiology 217: 851–863 [DOI] [PubMed] [Google Scholar]
- Shinjyo N, Stahlberg A, Dragunow M, Pekny M, Pekna M (2009) Complement‐derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem Cells 27: 2824–2832 [DOI] [PubMed] [Google Scholar]
- Sirmaci A, Walsh T, Akay H, Spiliopoulos M, Sakalar YB, Hasanefendioglu‐Bayrak A, Duman D, Farooq A, King MC, Tekin M (2010) MASP1 mutations in patients with facial, umbilical, coccygeal, and auditory findings of Carnevale, Malpuech, OSA, and Michels syndromes. Am J Hum Genet 87: 679–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjoedt MO, Hummelshoj T, Palarasah Y, Honore C, Koch C, Skjodt K, Garred P (2010) A novel mannose‐binding lectin/ficolin‐associated protein is highly expressed in heart and skeletal muscle tissues and inhibits complement activation. J Biol Chem 285: 8234–8243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M, Santiago‐Schwarz F, Al‐Abed Y, Diamond B (2012) C1q limits dendritic cell differentiation and activation by engaging LAIR‐1. Proc Natl Acad Sci USA 109: E3160–E3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Barres BA, Stevens B (2012) The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 35: 369–389 [DOI] [PubMed] [Google Scholar]
- Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, Lambris JD (2003) The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med 198: 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto R, Yae Y, Akaiwa M, Kitajima S, Shibata Y, Sato H, Hirata J, Okochi K, Izuhara K, Hamasaki N (1998) Cloning and characterization of the Hakata antigen, a member of the ficolin/opsonin p35 lectin family. J Biol Chem 273: 20721–20727 [DOI] [PubMed] [Google Scholar]
- Swierzko A, Lukasiewicz J, Cedzynski M, Maciejewska A, Jachymek W, Niedziela T, Matsushita M, Lugowski C (2012) New functional ligands for ficolin‐3 among lipopolysaccharides of Hafnia alvei. Glycobiology 22: 267–280 [DOI] [PubMed] [Google Scholar]
- Szalai AJ, Agrawal A, Greenhough TJ, Volanakis JE (1999) C‐reactive protein: structural biology and host defense function. Clin Chem Lab Med 37: 265–270 [DOI] [PubMed] [Google Scholar]
- Takahashi M, Ishida Y, Iwaki D, Kanno K, Suzuki T, Endo Y, Homma Y, Fujita T (2010) Essential role of mannose‐binding lectin‐associated serine protease‐1 in activation of the complement factor D. J Exp Med 207: 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Chang WC, Takahashi M, Pavlov V, Ishida Y, La Bonte L, Shi L, Fujita T, Stahl GL, Van Cott EM (2011) Mannose‐binding lectin and its associated proteases (MASPs) mediate coagulation and its deficiency is a risk factor in developing complications from infection, including disseminated intravascular coagulation. Immunobiology 216: 96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata Y, Kinoshita T, Kozono H, Takeda J, Tanaka E, Hong K, Inoue K (1987) Covalent association of C3b with C4b within C5 convertase of the classical complement pathway. J Exp Med 165: 1494–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam JC, Bidgood SR, McEwan WA, James LC (2014) Intracellular sensing of complement C3 activates cell autonomous immunity. Science 345: 1256070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedder TF, Fearon DT, Gartland GL, Cooper MD (1983) Expression of C3b receptors on human be cells and myelomonocytic cells but not natural killer cells. J Immunol 130: 1668–1673 [PubMed] [Google Scholar]
- Tegla CA, Cudrici C, Patel S, Trippe R III, Rus V, Niculescu F, Rus H (2011) Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol Res 51: 45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teillet F, Dublet B, Andrieu JP, Gaboriaud C, Arlaud GJ, Thielens NM (2005) The two major oligomeric forms of human mannan‐binding lectin: chemical characterization, carbohydrate‐binding properties, and interaction with MBL‐associated serine proteases. J Immunol 174: 2870–2877 [DOI] [PubMed] [Google Scholar]
- Teillet F, Gaboriaud C, Lacroix M, Martin L, Arlaud GJ, Thielens NM (2008) Crystal structure of the CUB1‐EGF‐CUB2 domain of human MASP‐1/3 and identification of its interaction sites with mannan‐binding lectin and ficolins. J Biol Chem 283: 25715–25724 [DOI] [PubMed] [Google Scholar]
- Timmann C, Leippe M, Horstmann RD (1991) Two major serum components antigenically related to complement factor H are different glycosylation forms of a single protein with no factor H‐like complement regulatory functions. J Immunol 146: 1265–1270 [PubMed] [Google Scholar]
- Torreira E, Tortajada A, Montes T, Rodriguez de Cordoba S, Llorca O (2009) 3D structure of the C3bB complex provides insights into the activation and regulation of the complement alternative pathway convertase. Proc Natl Acad Sci USA 106: 882–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou K, Hughes TR, Triantafilou M, Morgan BP (2013) The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci 126: 2903–2913 [DOI] [PubMed] [Google Scholar]
- Tschopp J, French LE (1994) Clusterin: modulation of complement function. Clin Exp Immunol 97(Suppl 2): 11–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura M, Ishida C, Sagara Y, Miyazaki T, Murakami K, Shiraki H, Okochi K, Maeda Y (2001) Detection of serum thermolabile beta‐2 macroglycoprotein (Hakata antigen) by enzyme‐linked immunosorbent assay using polysaccharide produced by Aerococcus viridans. Clin Diagn Lab Immunol 8: 454–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman Girija U, Gingras AR, Marshall JE, Panchal R, Sheikh MA, Gal P, Schwaeble WJ, Mitchell DA, Moody PC, Wallis R (2013) Structural basis of the C1q/C1s interaction and its central role in assembly of the C1 complex of complement activation. Proc Natl Acad Sci USA 110: 13916–13920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis R, Mitchell DA, Schmid R, Schwaeble WJ, Keeble AH (2010) Paths reunited: Initiation of the classical and lectin pathways of complement activation. Immunobiology 215: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Blair E, Simpson CV, O'Hara M, Blackburn PE, Rot A, Graham GJ, Nibbs RJ (2004) The chemokine receptor D6 constitutively traffics to and from the cell surface to internalize and degrade chemokines. Mol Biol Cell 15: 2492–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler JM (1989) Regulation of C5 convertase activity by properdin, factors B and H. Immunol Res 8: 305–315 [DOI] [PubMed] [Google Scholar]
- Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, Cano M, Brandstatter H, Tsimikas S, Skerka C, Superti‐Furga G, Handa JT, Zipfel PF, Witztum JL, Binder CJ (2011) Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 478: 76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieme RJ, Demeulenaere L (1967) Genetically determined electrophoretic variant of the human complement component C'3. Nature 214: 1042–1043 [DOI] [PubMed] [Google Scholar]
- Wiesmann C, Katschke KJ, Yin J, Helmy KY, Steffek M, Fairbrother WJ, McCallum SA, Embuscado L, DeForge L, Hass PE, van Lookeren Campagne M (2006) Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature 444: 217–220 [DOI] [PubMed] [Google Scholar]
- Wilken HC, Gotze O, Werfel T, Zwirner J (1999) C3a(desArg) does not bind to and signal through the human C3a receptor. Immunol Lett 67: 141–145 [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Arumugam TV, Shiels IA, Reid RC, Fairlie DP, Taylor SM (2003) A potent human C5a receptor antagonist protects against disease pathology in a rat model of inflammatory bowel disease. J Immunol 171: 5514–5520 [DOI] [PubMed] [Google Scholar]
- Wu J, Wu YQ, Ricklin D, Janssen BJ, Lambris JD, Gros P (2009) Structure of complement fragment C3b‐factor H and implications for host protection by complement regulators. Nat Immunol 10: 728–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yongqing T, Wilmann PG, Reeve SB, Coetzer TH, Smith AI, Whisstock JC, Pike RN, Wijeyewickrema LC (2013) The x‐ray crystal structure of mannose‐binding lectin‐associated serine proteinase‐3 reveals the structural basis for enzyme inactivity associated with the Carnevale, Mingarelli, Malpuech, and Michels (3MC) syndrome. J Biol Chem 288: 22399–22407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa S, Nagasawa K, Yae Y, Niho Y, Okochi K (1997) A thermolabile beta 2‐macroglycoprotein (TMG) and the antibody against TMG in patients with systemic lupus erythematosus. Clin Chim Acta 264: 219–225 [DOI] [PubMed] [Google Scholar]
- Zaferani A, Vives RR, van der Pol P, Navis GJ, Daha MR, van Kooten C, Lortat‐Jacob H, Seelen MA, van den Born J (2012) Factor h and properdin recognize different epitopes on renal tubular epithelial heparan sulfate. J Biol Chem 287: 31471–31481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbin MA (2004) Current concepts in the pathogenesis of age‐related macular degeneration. Arch Ophthalmol 122: 598–614 [DOI] [PubMed] [Google Scholar]
- Zhan X, Larson DE, Wang C, Koboldt DC, Sergeev YV, Fulton RS, Fulton LL, Fronick CC, Branham KE, Bragg‐Gresham J, Jun G, Hu Y, Kang HM, Liu D, Othman M, Brooks M, Ratnapriya R, Boleda A, Grassmann F, von Strachwitz C et al (2013) Identification of a rare coding variant in complement 3 associated with age‐related macular degeneration. Nat Genet 45: 1375–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Takahashi K, Alicot EM, Vorup‐Jensen T, Kessler B, Thiel S, Jensenius JC, Ezekowitz RA, Moore FD, Carroll MC (2006) Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol 177: 4727–4734 [DOI] [PubMed] [Google Scholar]
- Zipfel PF, Lauer N, Skerka C (2010) The role of complement in AMD. Adv Exp Med Biol 703: 9–24 [DOI] [PubMed] [Google Scholar]