

Abstract
A decline in estradiol (E2)-mediated cognitive benefits denotes a critical window for the therapeutic effects of E2, but the mechanism for closing of the critical window is unknown. We hypothesized that upregulating the expression of estrogen receptor α (ERα) or estrogen receptor β (ERβ) in the hippocampus of aged animals would restore the therapeutic potential of E2 treatments and rejuvenate E2-induced hippocampal plasticity. Female rats (15 months) were ovariectomized, and, 14 weeks later, adeno-associated viral vectors were used to express ERα, ERβ, or green fluorescent protein (GFP) in the CA1 region of the dorsal hippocampus. Animals were subsequently treated for 5 weeks with cyclic injections of 17β-estradiol-3-benzoate (EB, 10 μg) or oil vehicle. Spatial memory was examined 48 h after EB/oil treatment. EB treatment in the GFP (GFP + EB) and ERβ (ERβ + EB) groups failed to improve episodic spatial memory relative to oil-treated animals, indicating closing of the critical window. Expression of ERβ failed to improve cognition and was associated with a modest learning impairment. Cognitive benefits were specific to animals expressing ERα that received EB treatment (ERα + EB), such that memory was improved relative to ERα + oil and GFP + EB. Similarly, ERα + EB animals exhibited enhanced NMDAR-mediated synaptic transmission compared with the ERα + oil and GFP + EB groups. This is the first demonstration that the window for E2-mediated benefits on cognition and hippocampal E2 responsiveness can be reinstated by increased expression of ERα.
SIGNIFICANCE STATEMENT Estradiol is neuroprotective, promotes synaptic plasticity in the hippocampus, and protects against cognitive decline associated with aging and neurodegenerative diseases. However, animal models and clinical studies indicate a critical window for the therapeutic treatment such that the beneficial effects are lost with advanced age and/or with extended hormone deprivation. We used gene therapy to upregulate expression of the estrogen receptors ERα and ERβ and demonstrate that the window for estradiol's beneficial effects on memory and hippocampal synaptic function can be reinstated by enhancing the expression of ERα. Our findings suggest that the activity of ERα controls the therapeutic window by regulating synaptic plasticity mechanisms involved in memory.
Keywords: aging, ERα and ERβ, estrogen, hippocampus, learning and memory, NMDA receptor
Introduction
Beneficial effects of estrogen replacement on cognitive function during the perimenopausal period are widely accepted; however, prescribing estrogen therapy to women experiencing cognitive problems remains controversial (Rocca et al., 2011; Maki, 2013). This controversy is based on findings from the Women's Health Initiative Memory Study (WHIMS), which examined estrogen effects on women that were postmenopausal for ∼15 years. These results suggest that estrogen therapy did not improve cognitive function (Espeland et al., 2004; Shumaker et al., 2004). Animal and clinical studies have provided evidence for a critical time interval for therapy initiation (i.e., a therapeutic or critical window) such that advanced age and/or extended hormone deprivation result in a loss of the E2-mediated beneficial effects (Gibbs, 2000; Foster et al., 2003; Daniel et al., 2006; Rocca et al., 2011).
E2 induces changes in hippocampal anatomy, physiology, transcription, and Ca2 + signaling cascades that are opposite to that observed during aging, suggesting possible mechanisms for hormonal regulation of cognitive function (Foster, 2005; Aenlle and Foster, 2010). Hippocampal E2 responsiveness declines with advancing age or extended hormone deprivation (Foster, 2005, 2012a; Bean et al., 2014). NMDAR synaptic responses are increased at hippocampal CA1 synapses 48 h after E2 treatment (Woolley et al., 1997; Smith and McMahon, 2006; Snyder et al., 2011) and the ability of E2 to enhance NMDAR responses is reduced by extended hormone deprivation (Smith et al., 2010; Vedder et al., 2014). Episodic spatial memory depends on NMDAR function (Foster, 2012b). Therefore, a decline in NMDAR-mediated synaptic transmission is associated with age-related deficits in episodic memory (Foster, 2012b; Kumar and Foster, 2013; Lee et al., 2014). In female rats, delay-dependent spatial episodic memory is particularly sensitive to the history of E2 treatment (Gibbs, 2000; Markham et al., 2002; Markowska and Savonenko, 2002; Foster et al., 2003; Daniel et al., 2006). Therefore, the loss of NMDAR responsiveness to E2 may contribute to the loss of E2's beneficial effects on spatial episodic memory.
The mechanism for the closing of the therapeutic window is unknown, but may involve a shift in estrogen receptor (ER) expression (Foster, 2005, 2012a; Bean et al., 2014). Estrogen's actions are mediated through two classical nuclear ERs, ERα and ERβ. Expression of both ER subtypes declines in the hippocampus of aged rodent models (Adams et al., 2002; Mehra et al., 2005; Waters et al., 2011; Zhang et al., 2011). Studies using viral-vector-mediated expression of hippocampal ERα and ERβ suggest that ERα is critical in maintaining cognitive function in young and middle-aged animals (Foster et al., 2008; Witty et al., 2012; Han et al., 2013). These results suggest that closing of the therapeutic window may be due to decreased activity of ERα and/or ERβ and that increased receptor expression in the hippocampus could rejuvenate estrogen responsiveness and reopen the therapeutic window.
The current study explores the hypothesis that increased expression of ERα or ERβ will reinstate estrogen's beneficial effects, enhancing NMDAR synaptic responses and cognitive function at a time point when estrogen therapy alone has no effect (i.e., after the closing of the critical window). We used an adeno-associated virus (AAV) to upregulate ERα, ERβ, or green fluorescent protein (GFP) in the hippocampus of female rats (18.5 months) that had been ovariectomized 14 weeks earlier. After viral injections, rats were treated with cyclic injections of 17β-estradiol-3-benzoate (EB) or oil for 5 weeks. Learning and memory on the spatial water maze was assessed 48 h after an EB/oil treatment. After behavioral testing, cyclic injections continued until the hippocampus was collected for histology, Western blot analysis, or examination of NMDAR function in an in vitro hippocampal slice preparation. Animals that received both AAV-ERα and EB treatment had enhanced episodic spatial memory. Electrophysiological recordings revealed that the combined expression of ERα with EB treatment enhanced the NMDAR component of synaptic transmission. Together, our results provide strong evidence for the idea that the critical window depends on ERα activity, such that increasing ERα expression reinstates hippocampal responsiveness to E2 treatment.
Materials and Methods
Subjects
Eighty female Fischer 344 rats (14 months of age) were obtained from National Institute on Aging colony (Taconic) through the University of Florida Animal Care and Service Facility. Animals were housed 2 per cage and maintained on a 12:12 h light/dark cycle (lights on at 6:00 A.M.). All procedures involving animal subjects were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Florida and were performed in accordance with guidelines established by the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals. Food and water were provided ad libitum until surgery, after which animals were switched to a casein-based chow (Cincinnati Lab Supply); this diet has lower levels of phytoestrogens compared with soy-based rat chow. For the experimental timeline, please refer to Figure 1. Eight animals had to be removed from this study due to health concerns. The final six experimental groups included: ERα + EB (n = 13), ERβ + EB (n = 11), GFP + EB (n = 13), ERα + oil (n = 12), ERβ + oil (n = 11), and GFP + oil (n = 12).
Figure 1.

Experimental timeline. Female Fischer 344 rats were received at 14 months of age and lavaged to examine the estrous cycle before removal of the ovaries at 15 months. Five to 6 weeks after OVX, the animals were behaviorally tested in the spatial water maze and assigned to ER or EB/oil treatment groups. Eight weeks later, 14 weeks after OVX, stereotaxic surgery was used to deliver viral vectors encoding ERα, ERβ, or GFP bilaterally into the dorsal hippocampi. The animals were allowed 1 week to recover and were then given cyclic injections of EB or oil for 5 weeks. Final behavioral assessment in the water maze was at 20 months of age and initiated 48 h after the seventh cycle of EB/oil treatment. Cyclic EB/oil treatments continued (1–7 additional cycles) and rats were sacrificed (Sac) 48 h after a final EB/oil treatment for electrophysiology, histology, and Western blot analysis.
Endocrine status and efficacy of EB treatment
Vaginal lavage was performed each day for 2–3 weeks to confirm an estrous cycle before ovariectomy (OVX), after OVX for 1 week to validate removal of the ovaries and during EB/oil treatment to confirm efficacy of treatment. Every morning between 9:00 and 11:00 A.M., vaginal secretions were collected from each animal using a smooth-tipped glass eye dropper and 1 drop (∼20–30 μl) of sterile 0.9% saline. Vaginal secretions were placed on glass slides. The phase of the estrous cycle was recorded after viewing the unstained wet slide under low magnification on a light microscope. Determination of the estrous phase (proestrus, estrus, metestrus, and diestrus) was based on the cytology of the collected cells (nucleated epithelial cells, cornified squamous epithelial cells, and leukocytes) (Marcondes et al., 2002).
At the time rats were killed, 48 hours after the final EB treatment, blood was collected and centrifuged at 5000 rpm for 10 min at 4°C. The plasma was stored at −20°C for subsequent analysis. Plasma E2 levels were measured by using a mouse/rat estradiol ELISA kit (Calbiotech). In addition, 2-cm-long sections of the right uterine horn (cut at the base) were removed and weighed to confirm efficacy of EB treatments.
OVX
Female rats received bilateral OVX at 15 months of age using aseptic procedures as described previously (Kumar and Foster, 2002; Sharrow et al., 2002; Foster et al., 2003; Kumar and Foster, 2004). Briefly, isoflurane (Piramal Healthcare) was administered in O2 using an isoflurane anesthesia system (VetEquip). Induction of anesthesia was initiated within an induction chamber using a 4% concentration of isoflurane. After transfer to a nose cone, a 1.5–2% concentration was used for maintenance of the surgical plane of anesthesia. O2 was delivered at 1 L/min. The ovaries were removed and the overlying muscle was sutured and the skin stapled to close the incisions. Subcutaneous injections of buprenorphine 0.03 mg/kg and 5–10 ml of saline were given for pain and hydration, respectively.
Hippocampal AAV injections
Fourteen weeks after OVX and 8–9 weeks after pretest behavioral characterization (Fig. 1), AAV1 (2 μl; ∼3.0 × 1013 transducing units) encoding ERα, ERβ, or GFP was bilaterally injected into the CA1 region of the dorsal hippocampus (anterior/posterior −3.3 mm and medial/lateral ± 2 mm of bregma, dorsal/ventral −2.6 to −2.8 mm) using a Kopf stereotaxic frame. Techniques for hippocampal injection of virus have been described previously (Foster et al., 2008; Lee et al., 2012; Lee et al., 2014).
Spatial water maze
Five to 6 weeks after OVX, animals were pseudorandomly assigned to virus (i.e., ER expression) and treatment groups and characterized for visual and spatial discrimination on the spatial water maze (Pretest). A second series of spatial and cue discrimination testing occurred 5–6 weeks after the intrahippocampal injections of viral vectors and the initiation of EB or oil treatment (Final test). For Pretest characterization, rats were first trained on a cue version of the spatial water maze. Three days later, rats were trained in a 1 d spatial discrimination version using previously described methods (Foster et al., 2003; Foster and Kumar, 2007; Guidi and Foster, 2012; Kumar et al., 2012; Lee et al., 2012; Kumar and Foster, 2013; Guidi et al., 2014). Briefly, animals were first habituated to the pool by 3 30 s free swims with gentle guidance to a platform that extended 1 cm above the water line and supported a visible white flag. Cue training consisted of 5 blocks of 3 trials with all training occurring in 1 d. Release points and platform locations were randomized across trials. At the beginning of each trial, rats were placed into the water and given 60 s to find the escape platform. If the animal did not escape within the allotted time, it was gently guided to the platform. For spatial discrimination, the escape platform was hidden 1.5 cm beneath the water level and remained in the same location throughout the training. Large extra-maze cues were placed on black curtains that surrounded the pool. Training consisted of five blocks of three trials, followed by a 60 s free swim probe trial to test acquisition of a spatial search strategy. For probe trials, the platform was removed and the animal was released from the quadrant opposite the goal quadrant (Guidi and Foster, 2012). A refresher-training block followed the probe trial to reinforce the location of the submerged platform (Guidi and Foster, 2012). A final Pretest retention probe was administered 24 h after spatial training. For both cue and spatial training, the intertrial intervals were 20 s and interblock intervals were ∼15 min.
Due to poor 24 h retention observed during the Pretest retention probe trial (see Fig. 4D), the spatial training task was modified for the Final test under the experimental conditions. The acquisition probe was conducted immediately after the end of Block 4 and followed by a refresher block (Block 5). After the completion of Block 5, there was a 1 h delay before the retention probe, which was followed by a second refresher block (Block 6). Twenty-four hours later, the animals were subjected to a single training block (Block 7) before a final probe trial. After the final probe trial, the cue discrimination task was again administered to insure that virus or injection treatments did not result in visuomotor impairments. EthoVision video tracking software (Noldus) was used to record the distances traveled, the time spent in each quadrant, and the total time in the arena. For each probe trial, quadrant search time was used to calculate a discrimination index (DI) score using the formula (G − O)/(G + O), where G and O represent the percentage of time spent in the goal quadrant and quadrant opposite the goal, respectively.
Figure 4.

Water maze Pretest indicates no initial difference in performance of the treatment groups. Rats were pseudorandomly assigned to ER and EB/oil treatment groups. For this and all subsequent figures, circle = ERα, triangle = ERβ, square = GFP, open = EB, and filled = oil. Five to 6 weeks after OVX and before virus injections, animals were tested on the cue task (A) and spatial discrimination task (B). The symbols indicate the mean escape distance (±SEM) for each training block. Individual DI scores and mean (bar) for the acquisition (C) and 24 h retention probe (D) trials. No group differences were observed. Across all groups, animals exhibited acquisition of a spatial discrimination with impaired retention 24 h later.
Estradiol treatments
Subcutaneous injection of EB elevates blood E2 levels shortly after injection and E2 levels stabilize into the physiological range over the next 24–48 h (Woolley and McEwen, 1993). In addition, cyclic injections of EB given 48 h before behavioral testing improve memory retention in the water maze task for young and middle-aged OVX female rats (Sandstrom and Williams, 2001) and middle-aged mice (Aenlle et al., 2009). Therefore, aged female rats (∼300 g) received either EB (10 μg in 100 μl of oil vehicle, ∼33 μg/kg) or oil (100 μl) injected subcutaneously at the nape of the neck on 2 contiguous days of a 5 d cycle. The injections were initiated 1 week after viral injection and continued for 7 cycles before behavior testing (Final test). After behavioral testing, cyclic EB/oil treatments continued (1–7 more cycles) and rats were pseudorandomly killed for electrophysiology, histology, and Western blot analysis 48 h after the final EB/oil treatment (Fig. 1).
Hippocampal slice preparation and electrophysiological recordings
Methods for hippocampal slice preparation and electrophysiological recordings have been published previously (Bodhinathan et al., 2010; Kumar et al., 2012; Kumar and Foster, 2013, 2014; Lee et al., 2014). Briefly, 48 h after the final EB/oil treatment, the rats were deeply anesthetized using isoflurane and decapitated with a guillotine (MyNeuroLab). The brains were quickly removed, the hippocampi dissected, and slices (∼400 μm) were cut parallel to the alvear fiber using a tissue chopper (Mickle Laboratory Engineering). The slices were placed in a recording chamber (Warner Instruments) and continuously bathed with oxygenated artificial CSF (aCSF) containing the following (in mm): 124 NaCl, 2 KCl, 1.25 KH2PO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 10 d-glucose at the rate of 2 ml/min and 30 ± 0.5°C. The aCSF had been previously bubbled with 95% O2 and 5% CO2 for 20 min and the pH was adjusted to 7.4.
Field EPSPs (fEPSPs) were recorded from the stratum radiatum of the CA1 region of the hippocampus using glass micropipettes (4–6 MΩ) filled with aCSF and placed 1 mm away from the stimulating electrode as described previously (Bodhinathan et al., 2010; Kumar and Foster, 2013; Lee et al., 2014). A concentric bipolar stimulating electrode (outer pole: stainless steel, 200 μm diameter; inner pole: platinum/iridium, 25 μm diameter) was localized to the middle of the stratum radiatum to stimulate CA3 inputs onto CA1. Using a SD9 stimulator (Grass Instruments), field potentials (0.033 Hz) were induced by diphasic stimulus pulses (100 μs). Signals were then amplified, filtered (1 Hz and 1 kHz) and saved on a computer for later offline analysis.
Methods to isolate the NMDAR-mediated component of the fEPSP (NMDAR-fEPSP) have been described previously (Bodhinathan et al., 2010; Kumar and Foster, 2013; Lee et al., 2014; Guidi et al., 2015). After recording of the total-fEPSP, slices were subsequently bathed in aCSF containing low extracellular Mg2+ (0.5 mm), 6,7-dinitroquinoxaline-2,3-dione (DNQX, 30 μm, Sigma-Aldrich), and picrotoxin (PTX, 10 μm; Tocris Bioscience). The DNQX was initially dissolved in dimethylsulfoxide (Sigma-Aldrich) before dilution in aCSF and PTX was initially dissolved in ethanol before dilution in aCSF. Input–output curves for the total-fEPSP and NMDAR-fEPSP slope were constructed for increasing stimulation strength. EPSP slope (mV/msec) was calculated as the difference between two cursors, separated by 1 ms, and placed on the middle portion of the descending phase of the EPSP.
Immunohistochemistry
Forty-eight hours after the last cycle of EB/oil treatment, 3 animals from each group were perfused using 4% paraformaldehyde (PFA) in 1× PBS. Brains were harvested, postfixed in 4% PFA for 2 h, and placed into 30% sucrose in PBS at 4°C until fully infiltrated. The brains were then embedded in Tissue-Tek O.C.T. Compound (Ted Pella) before sectioning. To determine viral vector expression specificity, sections (16 μm) were incubated with primary antibodies against the cmyc-tag (A21281, 1:2000 dilution; Invitrogen), NeuN (MABN140, 1:500 dilution; Millipore), glial fibrillary acidic protein (GFAP, Z0334, 1:500; Dako), or ERα (MC20, 1:200 dilution; Santa Cruz Biotechnology) overnight at 4°C, washed 3 × 15 min in PBS, and then incubated in Alexa Fluor 594 or Alexa Fluor 488 secondary antibody (1:500 dilution; Invitrogen) for 1 h at room temperature. Sections were washed and counterstained with DAPI (0.1 μg/ml in PBS). Stained slides were viewed on a Leica DM2500 upright fluorescent microscope equipped with an Optronics camera and images were captured using MagnaFire software.
Western blot analysis
The CA1 regions from dorsal hippocampi were isolated, flash frozen in liquid nitrogen, and stored at −80°C. Tissues were sonicated and lysed in radio-immunoprecipitation assay buffer (Thermo Scientific) supplemented with phosphatase inhibitors, protease inhibitors, and EDTA (Thermo Scientific) and centrifuged at 20,000 × g for 10 min at 4°C. Protein concentrations were measured using a Pierce BCA protein assay. Lysates were denatured in Laemmli buffer with 2-mercaptoethanol (Bio-Rad) and boiled at 100°C for 5 min before electrophoresis. Aliquots (15–30 μg) and Kaleidoscope protein standards (Bio-Rad) were separated on 7.5% extracellular signal-regulated kinase (ERK) or 4%–15% Tris-HCl gels (Bio-Rad).
For the initial testing of virus expression in cell cultures (see Fig. 3B), Western blot analysis used a film method to image and quantify ERα and ERβ protein levels; all other figures for Western blot analysis used the new Li-Cor Odyssey system for analysis and quantification. For the film method, the gels were transferred to PVDF membranes (GE Healthcare). The immunoblots were blocked in Tris-buffered saline (TBS) with 5% nonfat powdered milk and 0.1% Tween 20 for 1 h at room temperature, followed by overnight incubation at 4°C with diluted primary antibodies for ERα (Ab17, RB-1521, 1:1000 dilution; Thermo Scientific), ERβ (H-150, sc-8974, 1:100; Santa Cruz Biotechnology), or cmyc-tag (A21281, 1:2000 dilution; Invitrogen) diluted in blocking buffer solution. Membranes were subsequently incubated with horseradish peroxidase-conjugated secondary antibody directed against the primary antibody (Cell Signaling Technology). Membranes were reacted with an enhanced chemiluminescent substrate (, catalog #RPN2132; GE Healthcare) for visualization. Blots were exposed to BioMaxMR film (Kodak), developed on a film processor (SRX-101A; Konica Medical), and scanned using a GS-800 Calibrated Densitometer (Bio-Rad). Protein levels were quantified using ImageJ software.
Figure 3.

Analysis of functionality and in vivo expression of viral vectors. A, Plasmids expressing cmyc-tagged ERα, cmyc-tagged ERβ, or GFP were cotransfected with an ERE-SEAP reporter plasmid in HEK293T cells with the addition of 1 or 10 nm EB treatment or vehicle. SEAP reporter levels were measured 24 h after application of treatments and compared with control groups. These results confirm functionality of the encoded ERs in vitro. Asterisks indicate a significant (p < 0.05) increase in SEAP expression relative to control. B, Western analysis of HEK293T cells treated with AAV-ERα, AAV-ERβ, or the GFP control virus. Top, For cells treated with AAV-ERα, antibodies for ERα (Ab17) and cmyc-tag (A21281) detected the 67 kDa full-length protein for ERα. Bottom, For cells treated with AAV-ERβ, antibodies for ERβ (H-150) and cmyc-tag (A21281) detected a band at ∼56 kDa. Note that HEK293T cells treated with the GFP virus exhibit a band at ∼56 kDa, which is increased in cultures treated with AAV-ERβ-cmyc. C, D, Immunofluorescent staining for the cmyc-tag, verified hippocampal expression of ERα and ERβ 4 weeks after injection of ER-encoding vectors. AAV-GFP was mixed with the ER virus in a 1:4 ratio to visualize distribution of expression vectors.
For membranes imaged and quantified with the Odyssey infrared scanner (Li-Cor), proteins were transferred to a supported nitrocellulose membrane (catalog #162-0070; Bio-Rad), blocked with Odyssey blocking buffer (catalog #927-50000; Li-Cor) for 1 h, and probed overnight in 4°C with antibodies for ERα (Ab17, RB-1521, 1:1000, Thermo Scientific or MC20, sc-542, 1:500, Santa Cruz Biotechnology), ERβ (D7N, 51-7700, 1:500; Invitrogen), PSD95 (MAB1598, 1:2000; Millipore), synaptophysin (#4329, 1:1000; Cell Signaling Technology), NR2A (M264, 1:1000; Sigma-Aldrich), NR2B (05-920, 1:1000; Millipore), pNR2B (S1303, GTX62089, 1:1000; GeneTex), GluR1 (sc-13185, 1:1000; Santa Cruz Biotechnology), pGluR1 (S831, P1160-831, 1:1000; Phospho-Solutions), total ERK (1:10,000, catalog #9107; Cell Signaling Technology), phosphorylated ERK (pERK, 1:2000, #9101; Cell Signaling Technology), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:10,000; EnCor Biotechnology). Membranes were incubated with an IRDye 800CW and 680LT secondary antibodies (1:25,000 and 1:20,000, respectively) for 1 h at room temperature, washed with TBST 3 times for 10 min each, and rinsed 3 times with TBS before imaging. Protein expression was normalized to the expression of GAPDH. For statistical analysis, the GAPDH normalized values were normalized to expression in GFP + oil animals.
Statistical analyses
For behavioral and electrophysiological studies, repeated-measures ANOVA and two-way ANOVA for main effects of ER group and treatment were used to identify significant main effects and interactions; Fisher's PLSD were used for post hoc comparisons (p < 0.05). For completeness, the F-score statistics are provided for ANOVAs in which the p-values are <0.2. For main effects or interactions that exhibited a tendency toward significance (0.1 > p > 0.05), post hoc ANOVAs within each treatment or ER group were used to localize specific differences (Hsu, 1996; Huck, 2009). One-group/one-sample t tests (p < 0.05; Gehring, 1978) were used to determine whether DI scores, calculated from the quadrant search behavior, were different from what was expected by chance (i.e., a DI score = 0).
Construction of AAV vectors
CTR4-ESR1-cmyc and CTR0-ESR2-cmyc.
cDNAs for human ESR1 (ERα) and ESR2 (ERβ) were subcloned into a rAAV expression plasmid under the control of the hybrid promoter, chicken β actin (CBA), with a woodchuck hepatitis virus posttranscriptional-regulatory element and bovine growth hormone polyadenylation signal. In addition, the hybrid CBA promoter contained a cytomegalovirus (CMV) enhancer. The cmyc epitope tag DNA sequence was added to the 3′ end of the ER cDNA by PCR. Figure 2 illustrates the plasmid maps for CTR4-ESR1-cmyc and CTR0-ESR2-cmyc. The integrity of the resulting ESR1 and ESR2 plasmid vectors was confirmed by sequencing. Plasmids were subsequently packaged by cotransfecting with AAV1 helper plasmid (pDP1rs) in HEK293T cells. Seventy-two hours after transfection, cells were harvested and lysed in the presence of 0.5% sodium deoxycholate and 50 U/ml benzonase (Sigma-Aldrich) by freeze-thaw cycles. The virus was isolated using a discontinuous iodixanol gradient and affinity purified with an Amicon Ultra filter device 100,000 MVCO (Millipore). Vectors were titered using qPCR (Bio-Rad CFX384). Genomic titers were ∼3 × 1013 vectors genome per milliliter for each vector.
Figure 2.
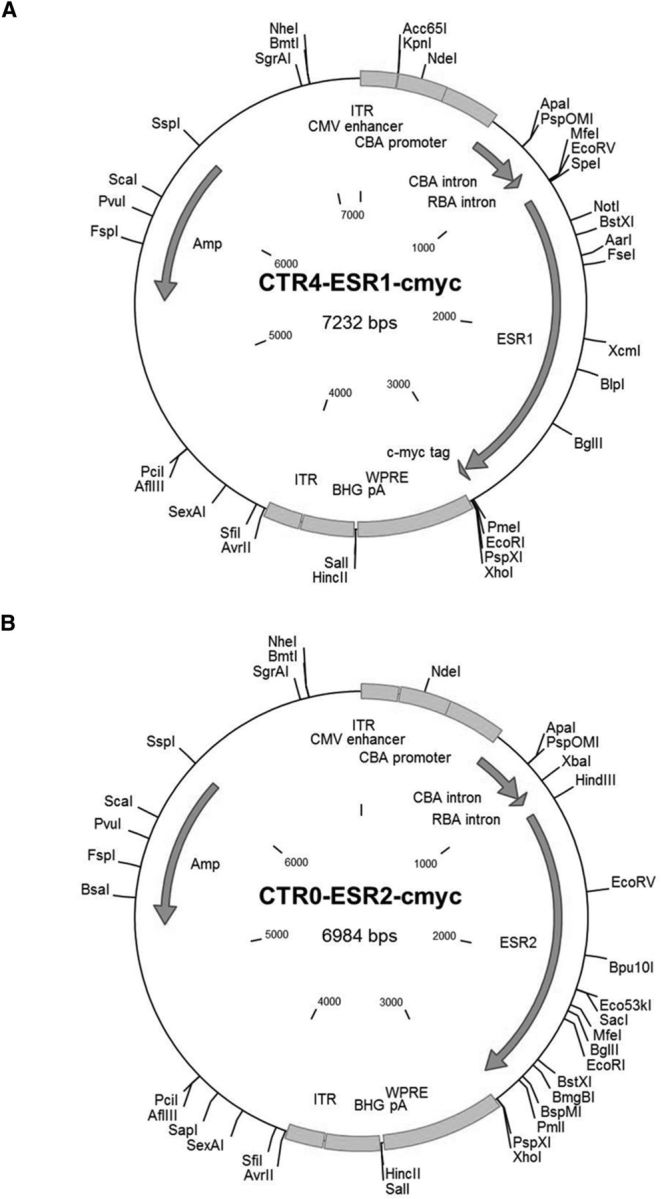
Plasmid maps. ESR1 cDNA for ERα expression (A) and ESR2 cDNA for ERβ expression (B) were cloned into vectors containing a CBA promoter with a CMV enhancer element. Plasmids were packaged into an AAV1 capsid before stereotaxic injections into the dorsal hippocampus of aged OVX female rats.
pcDNA-ERE-TA-SEAP.
Additional plasmids were created to assess the function of the ERα and ERβ vectors. DNA for secreted embryonic alkaline phosphatase (SEAP; gift from Dr. Bradley Fletcher, University of Florida) and pcDNA plasmid vector were cut with restriction enzyme NOT1 and then ligated using a rapid ligation kit. The resulting plasmid was cloned, cut with BglII and EcoRI, and ligated with DNA containing the minimal TA promoter and the estrogen response element (ERE). The final plasmid was cloned, purified, and subsequently verified by sequencing. Additional control plasmids were created by removing the ERE sequence (negative control) and by replacing the TA promoter with a constitutively active CMV promoter (positive control).
Transfections.
HEK293T cells were maintained in DMEM containing 10% charcoal-filtered serum (Cocalico Biologicals) and 1% penicillin/streptomycin. The day before transfection, cells were plated onto 24-well plates and grown overnight in DMEM containing 10% charcoal-filtered serum. On the day of transfection, the medium was removed and replaced with 0.4 ml of fresh medium. Transfection mixture for each well was prepared by adding 800 ng of ERE-TA-SEAP vector (or control vector) and 800 ng of either CTR4-ESR1-cmyc (ERα), CTR0-ESR2-cmyc (ERβ), or AAV-GFP plasmids to 50 μl of Opti-MEM 1, mixing, and then adding Lipofectamine 2000 (Life Technologies) according to the manufacturer's protocol. This mixture was incubated at room temperature for 20 min and the mixture was added into each well. After a 5 h incubation at 37°C, the cultures were rinsed and allowed to grow overnight at 37°C in fresh medium containing 10% charcoal-filtered serum. EB (1 or 10 nm) or vehicle was added to the culture medium. After overnight incubation, 50 μl of culture media was collected from each culture well, centrifuged at 12,000 rpm for 10 s, and the cleared supernatant was transferred into a fresh microcentrifuge tube. Levels of SEAP in the medium were measured using a Phospha-Light chemiluminescent assay kit (Applied Biosystems) according to the manufacturer's protocol. Samples were run in triplicate, and light emission from the processed samples and SEAP standards were measured using a Bio Tek luminometer with a delay of 2 s and integration time of 5 s. The amount of SEAP in the culture medium samples was calculated from the standard curve. Western blot analysis was also used to verify increased expression of the cmyc-tagged ERs in cell culture.
Results
Functional characterization of ER-containing plasmids and virus
To test the functionality of the ER expression plasmids, HEK293T cells were cotransfected with the CTR4-ESR1-cmyc (ERα), CTR0-ESR2-cmyc (ERβ), or AAV-GFP plasmids and a vector encoding the SEAP reporter under the control of an ERE and a minimal promoter (reporter construct: ERE-TA-SEAP). The SEAP level was influenced differentially by the receptor subtype and dose of EB (Fig. 3A). An ANOVA on the SEAP levels indicated a significant difference across groups (F(8,24) = 19.98, p < 0.001) and post hoc tests confirmed that, relative to the control group, expression of ERα alone could drive transcription (Foster et al., 2008). SEAP levels were further increased to an apparent asymptote by addition of 1 or 10 nm EB. For ERβ-transfected cells, SEAP expression was not altered in the absence of EB treatment and expression increased dose dependently after EB treatment. These results indicate that the receptors are functional and confirm that ERα is more effective than ERβ in inducing transcription (Foster, 2012a; Han et al., 2013).
To verify successful packaging of the plasmids into the AAV capsids, cmyc-tagged ERα or ERβ expression was confirmed in cell lysate by Western analysis (Fig. 3B). A band of cmyc-tag was localized at the appropriate atomic mass for ERα (∼67 kDa) and ERβ (∼56 kDa). In addition, young female rats were injected with AAV-GFP mixed with the cmyc-tagged ER vector at a 1:4 ratio. Immunofluorescent chemistry for the cmyc tag and visualization of GFP (Fig. 3C,D) confirmed protein expression in the dorsal hippocampus 4 weeks after injection. Similar to our previous results, expression was largely limited to region CA1 of the hippocampus (Foster et al., 2008; Han et al., 2013). Together, these results established the efficacy of our expression vectors.
Effects of ERα and ERβ expression on spatial learning and memory
After OVX and before virus injections (Fig. 1), cognitive function was assessed on the water maze (Pretest). Repeated-measures ANOVA for the cue task indicated an effect of training on escape path length (F(4,264) = 19.22, p < 0.0001; Fig. 4A) with no effect of ER group or treatment group assignment. For the spatial task, again, there was an effect of training for path length (F(4,264) = 23.448, p < 0.0001; Fig. 4B) in the absence of difference for the preassigned ER and treatment grouping. There was considerable variability in probe trial performance (Fig. 4C,D); however, ANOVAs indicated no effect of ER or treatment assignment and no interaction on the immediate acquisition (Probe 1). Similarly, no ER, treatment, or ER × treatment interaction differences were observed in the 24 h retention (Probe 2) DI scores for the preassigned groups. The results indicate no group differences before virus injections and EB/oil treatment. A one-group t test comparing the DI scores to that expected by chance (i.e., a DI score of 0) indicated that, across all animals, the DI scores were above chance for the acquisition probe trial (t(71) = 3.93, p < 0.0005), indicating that animals were able to acquire a spatial search strategy. For the 24 h retention probe trial, a one-group t test on the DI scores indicated that the animals performed below chance (t(71) = −5.9, p < 0.0001), indicating that, over the 24 h period, animals abandoned the spatial search strategy focused on the goal quadrant.
Fourteen weeks after OVX, virus encoding ERα, ERβ, or GFP was delivered to the hippocampus by stereotaxic surgery. One week after the viral vector injections, animals were treated with EB or oil delivered by subcutaneous injection on 2 adjoining days in a 5 d cyclic regimen (Fig. 1). Treatments were continued for 5 weeks before behavior testing. Cognitive performance on the spatial version of the water maze was again assessed (Final test). At this time, animals were 20 months of age, 6 weeks after virus injections, and testing began 48 h after the previous EB/oil treatment. Due to poor retention during the pretest (Fig. 4D), the protocol was altered such that the retention probe trial was delivered 1 h after training Block 4; this was followed by a refresher training block. An ANOVA on the swim speed, averaged across Blocks 1–5, indicated no effect of ER group and no ER group × treatment interaction (Fig. 5A). There was a trend (F(1,66) = 3.33, p = 0.073) for a treatment effect due to slower swim speed in EB-treated animals, which is consistent with previous work (Foster et al., 2003; Talboom et al., 2008). A repeated-measures ANOVA for escape path length (distance) across training Blocks 1–5 for the spatial task indicated significant main effects of training (F(4,264) = 24.69, p < 0.0001), a tendency for an effect of ER group (F(2,66) = 2.81, p = 0.068) in the absence of an effect of treatment (F(1,66) = 2.08, p = 0.15), and no ER group × treatment interaction (Fig. 5B). Post hoc tests for ER group effects collapsed across treatments indicated that animals expressing ERβ exhibited increased distance to escape relative to animals expressing ERα (p < 0.05; Fig. 5C). Post hoc tests also indicated a tendency (p = 0.098) for poorer performance of ERβ-expressing animals relative to GFP controls. The difference was mainly due to increased path length for the ERβ-treated animals in Blocks 2 (F(2,66) = 3.36, p < 0.05) and 5 (F(2,66) = 3.09, p = 0.052). Post hoc tests indicated a significant increased escape distance for ERβ relative to ERα on Block 2 and a significant increase in distance for ERβ relative to ERα and GFP on Block 5. Despite the group difference during training, no differences were observed for the initial training block and Block 4 immediately preceding the acquisition probe trial. Therefore, initial performance was the same and all groups appear to learn, although there may have been differences in the rate of learning.
Figure 5.
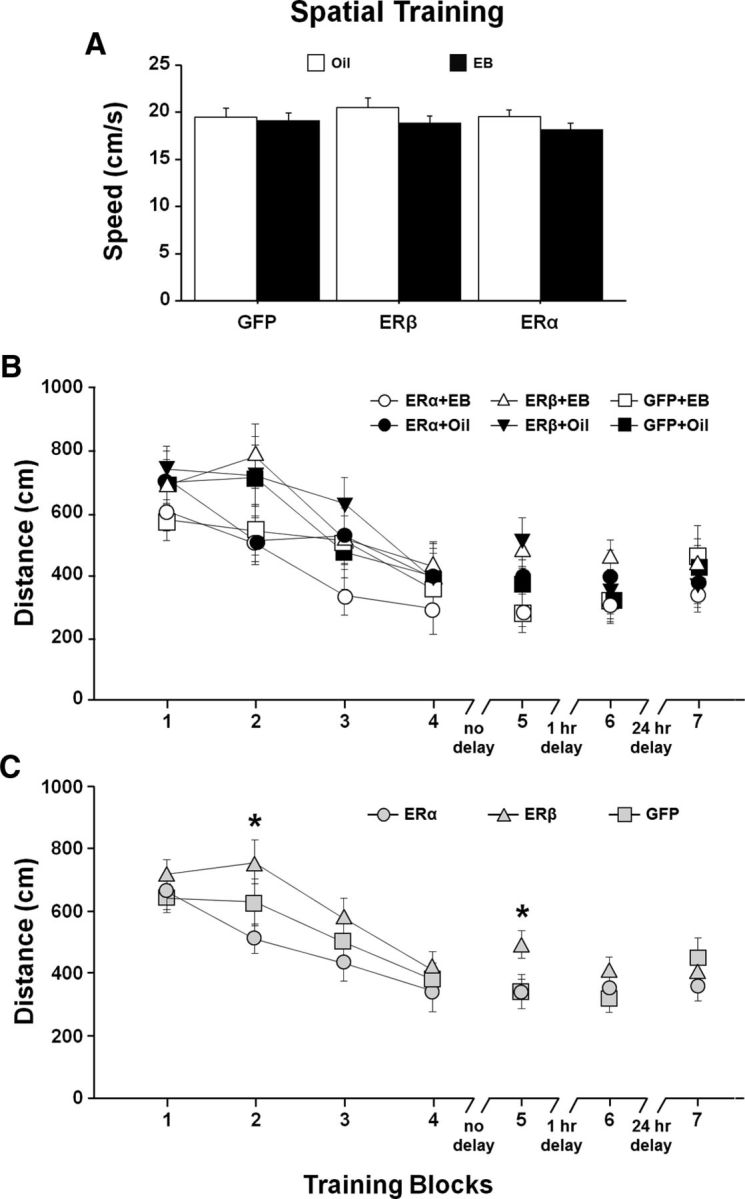
Spatial discrimination performance after differential expression of ER and EB/oil treatment. A, Mean swim speed (+ SEM) across training Blocks 1–5 for EB (filled bars)- and oil (open bars)-treated animals. Symbols indicate the mean (±SEM). B, Escape path length to the submerged platform for the ER and treatment groups. C, Mean (±SEM) escape path length collapsed across EB/oil treatments (gray) for each ER group, illustrating the shortest and longest escape distances in ERα and ERβ groups, respectively. The asterisks indicate trial blocks during acquisition training with significant (p < 0.05) differences between ERβ and other groups.
An ANOVA of the DI scores for the acquisition probe trial indicated no effect of ER group or treatment and no ER group × treatment interaction (F(2,66) = 2.22, p = 0.12; Fig. 6A). In contrast, an ANOVA on the DI scores for the 1 h retention probe trial indicated an effect of treatment (F(1,66) = 6.74, p < 0.05) and a tendency for an interaction of ER group and treatment (F(2,66) = 2.85, p = 0.065). The treatment effect was due to an increase in the retention DI scores of EB-treated rats (Fig. 6B) and subsequent ANOVAs indicated an effect of EB only for the ERα group (F(2,34) = 4.83, p < 0.05), such that ERα + EB exhibited increased retention relative to ERα + oil. Importantly, EB treatment had no effect in the GFP or ERβ groups, consistent with the idea that a long duration of estrogen deprivation results in a decrease in E2's ability to improve memory (i.e., closing of the critical window). ANOVAs for ER group effects within each treatment condition indicated that the tendency for the ER group by treatment interaction was due to an ER group effect for EB-treated animals (F(1,23) = 17.07, p < 0.001). Post hoc comparisons confirmed superior performance by the ERα + EB group such that the DI scores for animals expressing ERα + EB were greater than DI scores for the GFP + EB group (p < 0.05), with a tendency (p = 0.09) for better performance by the ERα + EB group relative to the ERβ + EB group. No effect of ER group, treatment, or interaction was observed for the escape distance on the refresher block (Block 6) administered after the retention probe trial (Fig. 5B).
Figure 6.
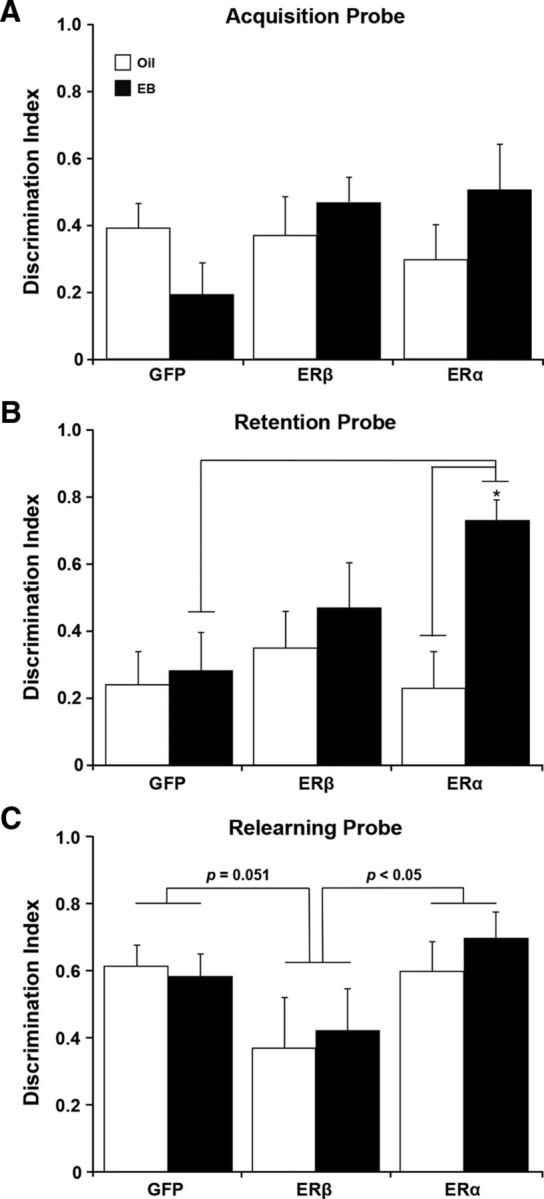
Expression of AAV-ERα in conjunction with EB treatment improved retention of spatial information for platform location. A–C, Bars represent the mean (±SEM) DI score calculated from performance on the probe trials for rats injected with AAV vectors encoding ERα, ERβ, or GFP and subsequently treated with EB (filled bars) or oil (open bars). A, Acquisition probe trial directly followed Block 4 of the spatial discrimination task. B, Retention probe trial followed a 1 h delay between Blocks 5 and 6. Superior performance was seen in EB-treated rats injected with the AAV expressing ERα. Asterisk indicates significant difference (p < 0.05). C, The probe trial immediately followed a refresher block the day after spatial training. The p-values above the bars indicate for differences between the ERα and GFP groups and animals with viral-mediated expression of ERβ.
The next day, animals were given a block of training (Block 7), which was immediately followed by a final probe trial. No effect of ER group, treatment (F(1,66) = 2.38, p = 0.13), or interaction was observed for the escape distance during the training block (Fig. 5B). An ANOVA on the DI scores for the final probe trial indicated an effect of ER group (F(2,66) = 3.41, p < 0.05; Fig. 6C) in the absence of a treatment effect or an interaction. Post hoc tests indicated better performance for ERα (p < 0.05) and a tendency (p = 0.051) for better performance by GFP-treated groups relative to the ERβ-treated groups. For the final cue discrimination test, an ANOVA on average swim speed indicated a tendency (F(1,66) = 2.90, p = 0.09) for a slower swim speed of EB-treated animals in the absence of an effect of ER group or an interaction. For escape distance, an ANOVA indicated a training effect (F(4,264) = 14.44, p < 0.0001) in the absence of a main effect of ER group, treatment, or interactions; though the ER group × training interaction was (F(8,264) = 1.47, p < 0.17). Together, these results suggest that differences in spatial learning/memory were not likely due to differences in sensory-motor function (Fig. 7).
Figure 7.

Expression of ERα, ERβ, or GFP and EB/oil treatments did not influence sensory-motor abilities examined as average swim speed (A) or distance (B) to find the visible platform across the training blocks on a cue discrimination task administered after the completion of the probe trial on day 2.
Overall, these results indicate that a long duration of E2 deprivation limits subsequent beneficial effects of E2 treatment on memory. Expression of ERβ was ineffective in improving cognition and associated with impaired learning. Finally, increasing the expression of ERα in hippocampal CA1 neurons restores the cognitive enhancing effects of E2 in aged female rats.
Influence of increased ERα expression and treatment on synaptic transmission
To examine the possibility that modification of NMDAR function underlies the reopening of the critical window, a subset of ERα + EB (n = 5/10 animals/slices), GFP + EB (n = 4/8 animals/slices), ERα + oil (n = 4/8 animals/slices), and GFP + oil (n = 4/8 animals/slices) animals were used for electrophysiological examination. After behavioral characterization, cyclic injections of EB/oil were continued (range 1–7 cycles, mean 4.12 ± 0.44 cycles) and hippocampal slices were prepared 48 h after the final EB treatment. Input–output curves for the total-fEPSP (Fig. 8A) and NMDAR–fEPSP (Fig. 8B) slope (mV/msec) were constructed from extracellular field recordings from the CA1 region of hippocampal slices. An ANOVA on the total-fEPSP response, repeated across stimulation intensities, indicated a main effect of stimulus intensity (F(7,210) = 29.73, p < 0.0001), a tendency (F(1,30) = 4.17, p = 0.05) for a treatment effect due to increased responses for EB-treated animals, and an interaction of stimulus intensity and treatment (F(7,210) = 3.31, p < 0.005) in the absence of an ER group effect (Fig. 8A).
Figure 8.

Expression of ERα in conjunction with EB treatment enhanced the NMDAR-synaptic response in the hippocampus of aged female rats. Input–output curves are illustrated for the mean slope of the total-fEPSP (A) and NMDAR-fEPSP (B). EB treatment was associated with an increase in the total-fEPSP response, which was particularly evident for animals that received AAV-ERα. After collection of the total-fEPSP, the NMDAR component of the synaptic response was isolated. In contrast to the total-fEPSP, the EB-mediated increase in NMDAR-fEPSP was specific to animals that received AAV-ERα. The insets in A and B show representative traces of synaptic responses from animals injected with AAV-GFP (top) and AAV-ERα (bottom) and treated with EB (black line) or oil (gray line). C, The ratio of the NMDAR-fEPSP/total-fEPSP for the highest stimulation intensities (32–40 V) was increased in the ERα + EB group. Asterisks indicate significant (p > 0.05) treatment effect, with greater NMDAR-fEPSP/total-fEPSP ratio in the ERα + EB group relative to the ERα + oil group. Pound signs indicate a significant ER group effect with a greater NMDAR-fEPSP/total-fEPSP ratio in ERα + EB group relative to GFP + EB group. The inset shows the total-fEPSP and isolation of NMDAR-fEPSP from the same slice.
After collection of the total-fEPSP responses, the recording medium was switched to a medium which permitted the isolation of the NMDAR component of synaptic transmission (Bodhinathan et al., 2010; Kumar and Foster, 2013; Lee et al., 2014; Guidi et al., 2015). An ANOVA, repeated across stimulation intensities, indicated a main effect of stimulus intensity (F(7,210) = 36.20, p < 0.0001), ER group (F(1,30) = 5.62, p < 0.05), and treatment (F(1,30) = 7.56, p < 0.05) and an ER group × treatment interaction (F(1,30) = 5.88, p < 0.05) on the NMDAR response (Fig. 8B). The increase in the NMDAR-mediated synaptic response was specific to ERα + EB animals. ANOVAs within each ER group indicated a treatment effect (F(1,16) = 8.63, p < 0.01) only in the ERα-expressing group with increased NMDAR-fEPSPs in ERα + EB relative to ERα + oil. Similarly, an effect of ER expression was observed for animals treated with EB (F(1,16) = 7.09, p < 0.05) such that the NMDAR-fEPSP was increased in ERα + EB relative to GFP + EB.
Finally, to determine the relative influence of enhanced expression of ERα and EB treatment, the ratio of NMDAR response/total-fEPSP response was calculated for the highest three stimulation levels (32, 36, and 40 V; Fig. 8C). The ratio was increased with increasing stimulation intensity (F(2,60) = 11.73, p < 0.0001) in the absence of an ER group or treatment effect (F(1,30) = 1. 95, p = 0.17). However, an interaction was observed for the ER group × treatment condition (F(1,30) = 7.62, p < 0.01). Post hoc ANOVA's examining treatment effects in each ER group indicated a treatment effect only for animals expressing ERα such that an increase in the NMDAR response/total response ratio was observed in the ERα + EB group relative to ERα + oil group (F(1,16) = 8.22, p < 0.05). Post hoc ANOVAs examining ER group effects in the separate treatment groups indicated that, for animals treated with EB, the ratio was increased in the ERα + EB relative to GFP + EB (F(1,16) = 7.01, p < 0.05).
Western analysis and immunohistochemistry
Western blot analysis and immunofluorescent chemistry were used to confirm viral-mediated ER expression for tissue harvested 48 h after the last injection. Immunofluorescent chemistry (n = 3 from each group) confirmed enhanced expression of the virally delivered ER throughout the dorsal hippocampus, which was largely localized to the CA1 region of the hippocampus (Fig. 9A). The anterior/posterior spread of ER expression was ∼2800 μm throughout the dorsal hippocampus, which is consistent with our previous work using AAV (Lee et al., 2012). Using a neuronal marker (NeuN) and an astrocyte marker (GFAP), we confirmed that virally mediated expression of ERα was restricted to neurons in the CA1 region (Fig. 9B,C). Western blot analysis (n = 4–8 per group) of ER expression established ERα immunoreactivity at ∼67 kDa (Fig. 9D). Quantification of human ERα was accomplished using the Ab17 antibody raised against an N-terminal sequence of human ERα. The results indicated a significant increase in expression of human ERα for animals injected with AAV-ERα relative to AAV-GFP (F(1,14) = 14.38, p < 0.005) in the absence of a treatment and treatment × ER group interaction. The endogenous rat ERα was examined using the MC-20 antibody raised against the last 20 aa of the mouse ERα; this sequence is identical to the C terminus of the rat ERα, but differs from the C terminus of the human ERα by 6 aa (Bollig-Fischer et al., 2012). Again, a band was observed at ∼67 kDa (Fig. 9E); however, no difference in endogenous expression was observed as a result of ERα or GFP expression, treatment, or the interaction. Immunoreactivity for the ERβ antibody was observed at ∼56 kDa and two other bands were observed at ∼63 and ∼61 kDa from tissue lysate isolated from the CA1 region of the dorsal hippocampus (Fig. 9F). Analysis of the ∼56 kDa ERβ band indicated an effect of ERβ or GFP virus injection (F(1,20) = 24.38, p < 0.0001) in the absence of an effect of treatment or an interaction. No effect of virus injection, treatment, or the interaction was observed for the 63 and 61 kDa bands, which may represent nonspecific binding, although ERβ variants within this range have been described previously (Solakidi et al., 2005; Alvarez-Delgado et al., 2010).
Figure 9.

Western blots and histology were used to confirm increased ER expression in neurons of the dorsal hippocampus. Immunofluorescent chemistry (A–C) showing expression of ERα within the CA1 region of the hippocampus. A, Merged image shows expression of AAV-ERα tagged with cmyc (green), and ERα (red). B, C, Cmyc (green) tagged to ERα was expressed mainly in neurons immunostained with a neural marker (NeuN, red; B), but not in astrocytes immunostained with the glial marker, glial fibrillary acidic protein (GFAP, red; C). Images A–C were counterstained with the nuclear marker DAPI (blue). D, E, Western blots using an antibody selective against the human ERα (D) or rat ERα (E) confirmed a band at ∼67 kDa. Animals injected with virus carrying ERα exhibited increased expression of the human ERα and no difference was observed for endogenous ERα. F, G, An antibody against ERβ confirmed that the ERβ vector increased the expression of human ERβ (∼56 kDa; F) in the absence of a change in the 63 and 61 kDa bands (G). For these and subsequent Western blots, the numbers in or above the bars indicate the number of samples used in the analysis.
Western blot analysis (n = 3–7 per group) was used to assess possible molecular changes in the CA1 region of the hippocampus for animals expressing ERα or GFP and treated 48 h earlier with EB or oil. We examined the expression of proteins that respond to E2 treatment in young animals and are thought to contribute to E2-dependent synaptic plasticity. Previous work suggested a correlation between E2-mediated increased NMDAR synaptic transmission and an increase in dendritic spines (Woolley et al., 1997; Smith and McMahon, 2005). Further, the synaptic proteins postsynaptic density 95 (PSD95) and synaptophysin are increased after E2 treatment in young animals (Brake et al., 2001; Frick et al., 2002; Akama and McEwen, 2003; Spencer et al., 2008a; Waters et al., 2009). However, no effect of ER group, treatment, or interaction was observed for PSD95 and no effect of ER group, treatment, or interaction was observed for synaptophysin (Fig. 10A,B). E2 treatment in younger animals increases markers of glutamate receptors, GluR1, NR2B, and NR2A (Cyr et al., 2001; Adams et al., 2004; Morissette et al., 2008; Waters et al., 2009; Qu et al., 2013). No effect of ER group, treatment, or interaction was observed for the NR2B and NR2A subunits of NMDARs and the GluR1 glutamate receptor subunit (Fig. 11A–C).
Figure 10.
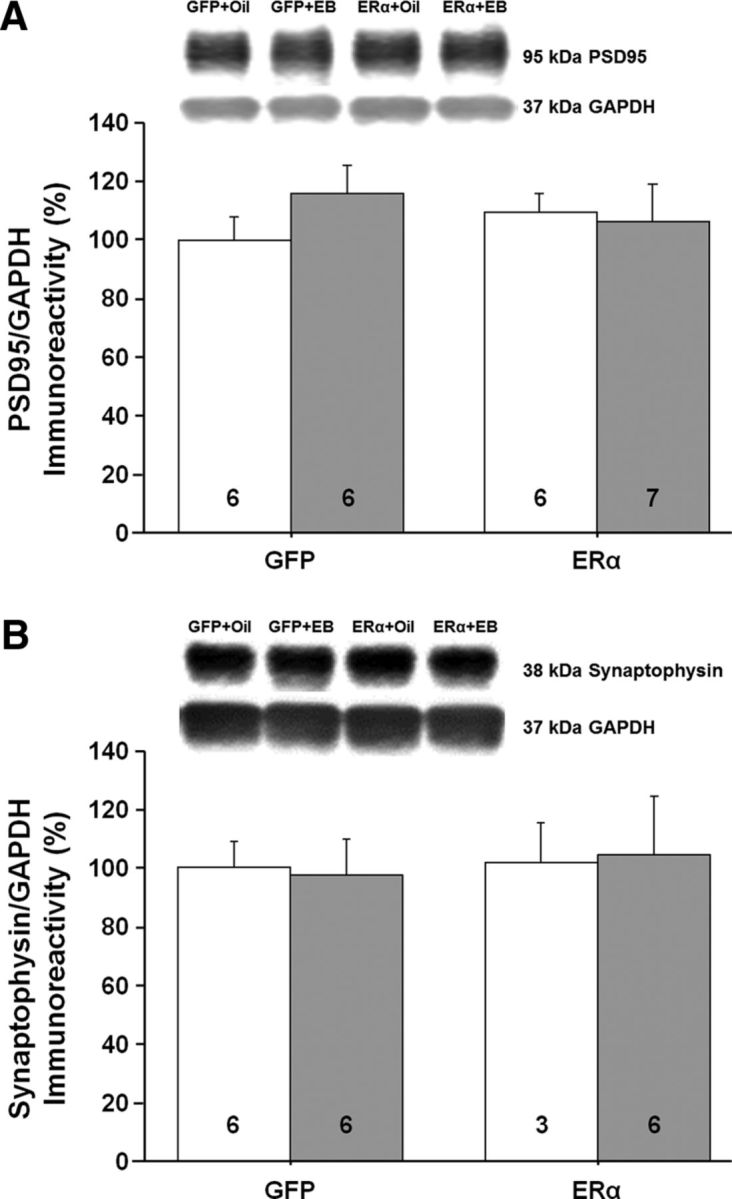
Western blots to assess the possible growth of dendritic spines in the CA1 region of the hippocampus for animals expressing ERα or GFP. The bars represent the means (±SEM) for the expression of the synaptic proteins PSD95 (A) and synaptophysin (B).
Figure 11.
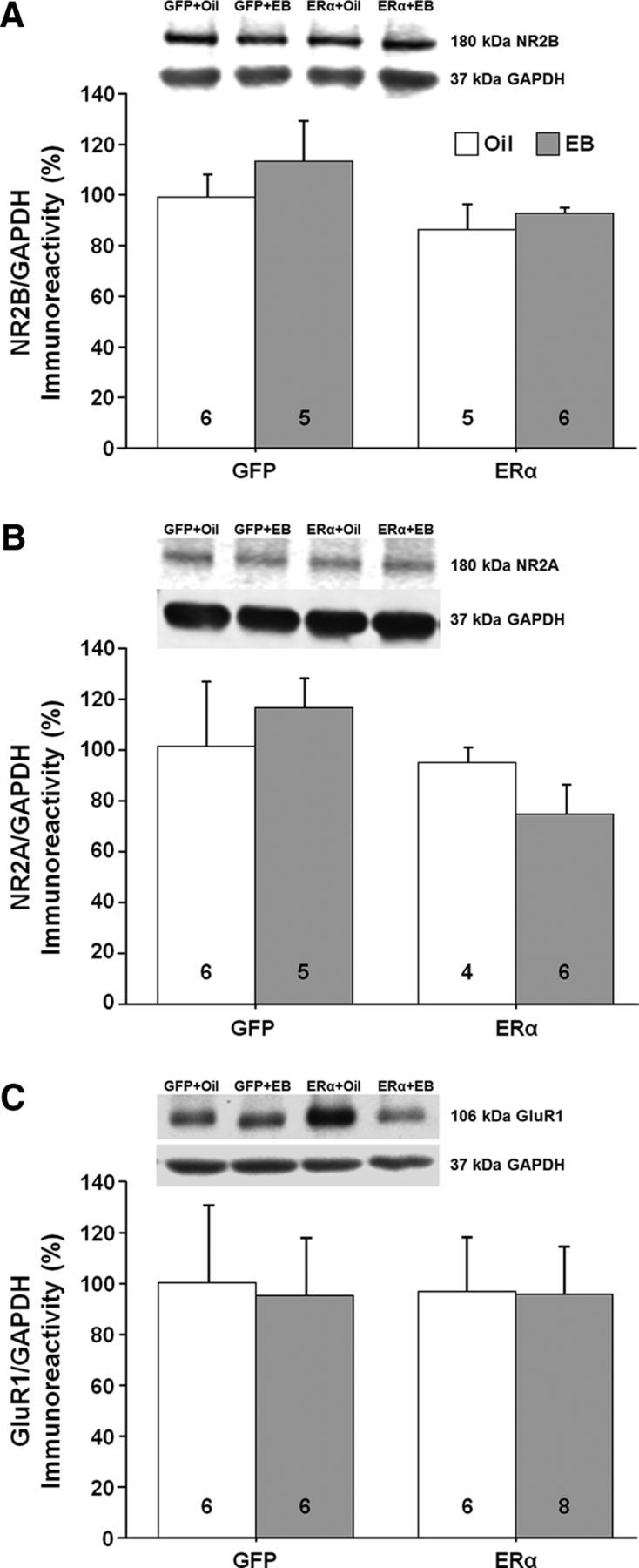
Western blots of glutamate receptor expression in the CA1 region of the hippocampus for animals expressing ERα or GFP. The bars represent the means (±SEM) for the expression of the NR2B subunit (A), the NR2A subunit (B), and the GluR1 subunit (C).
E2 treatment activates signaling cascades (Foster, 2005; Zadran et al., 2009; Kumar et al., 2015), including ERK, which can increase synaptic transmission, possibly through the phosphorylation of glutamate receptors (Xu et al., 2010; Logan et al., 2011; Raval et al., 2012). No effect of ER group or treatment was observed for the phosphorylation state of NR2B(S1303), GluR1(S831), ERK1, or ERK2 (Fig. 12A–C).
Figure 12.
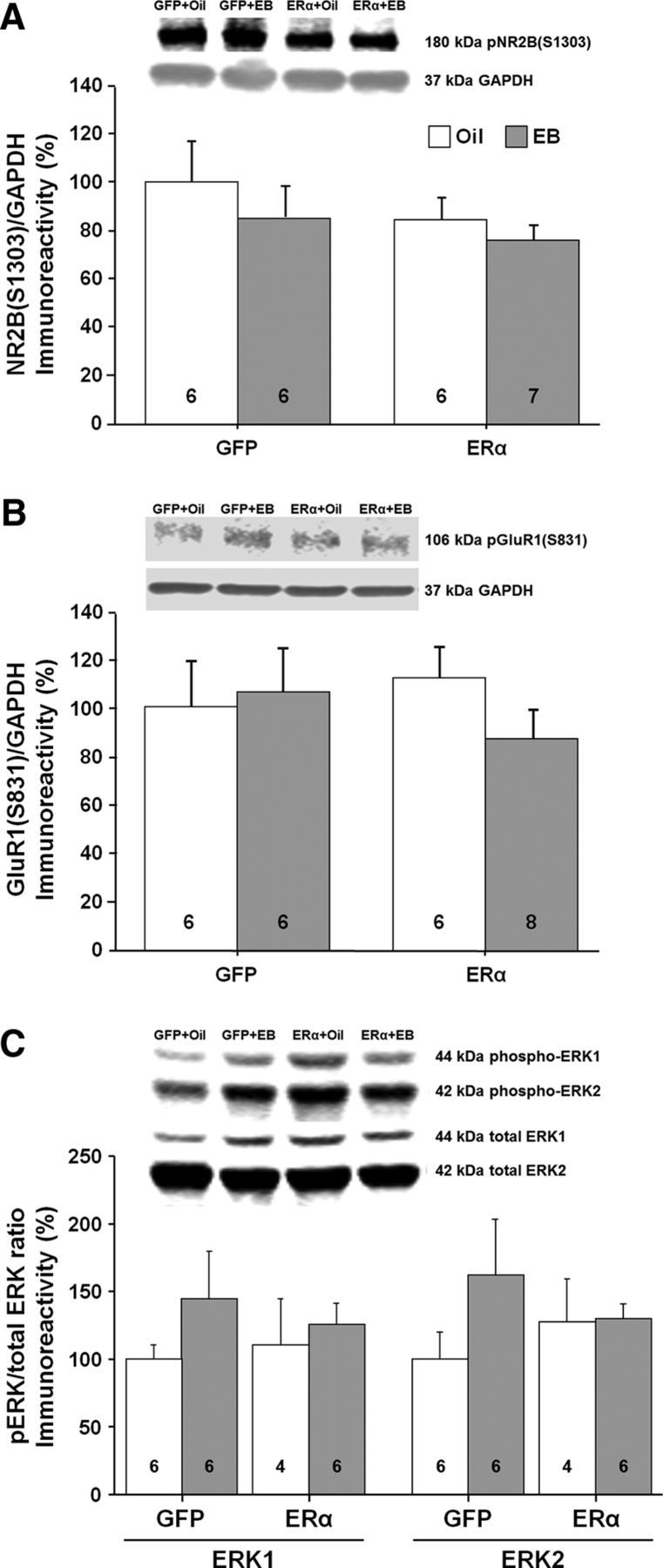
Western blots to assess possible changes in phosphorylation and kinase activity in the CA1 region of the hippocampus for animals expressing ERα or GFP. The bars represent the means (±SEM) for the phosphorylation of NR2B at S1303 (A), GluR1 at S831 (B), and ERK1/2 (C).
Hormone treatment efficacy
To determine the efficacy of our EB treatments, blood E2 levels were measured and the right uterine horns were weighed. ANOVAs indicated a main effect of EB treatment on plasma E2 levels (F(1,66) = 18.011, p < 0.0001; ERα + EB: 65.8 ± 22.9 pg/ml mean ± SEM, ERβ + EB: 45.0 ± 20.2, GFP + EB: 53.0 ± 18.3, ERα + oil: 3.0 ± 0.7, ERβ + oil: 1.0 ± 0.3, or GFP + oil: 2.8 ± 0.8) and uterine weight (F(1,66) = 212.26, p < 0.0001; ERα + EB: 0.33 ± 0.02 g mean ± SEM, ERβ + EB: 0.29 ± 0.01, GFP + EB: 0.33 ± 0.03, ERα + oil: 0.11 ± 0.02, ERβ + oil: 0.09 ± 0.01, or GFP + oil: 0.09 ± 0.01). No effect of ER group or interaction of ER group and treatment was observed for plasma E2 or uterine weight.
Discussion
Critical window for cognitive function
Consistent with other studies using water escape tasks (O'Neal et al., 1996; Bimonte and Denenberg, 1999; Markham et al., 2002; Foster et al., 2003; Daniel et al., 2005), EB's beneficial effect on cognitive function is specific to delay-dependent retention of novel spatial information (i.e., spatial episodic or working memory). However, E2 effects on episodic memory weaken with advanced age or with extended hormone deprivation, denoting closing of the therapeutic window (Gibbs, 2000; Markowska and Savonenko, 2002; Foster et al., 2003; Daniel et al., 2006). In the current study, EB failed to improve memory in aged OVX animals expressing GFP or ERβ, indicating that these animals were beyond the critical window. In contrast, upregulation of ERα combined with EB treatment improved memory. Therefore, this is the first demonstration that E2-mediated beneficial effects can be reinstated by upregulation of ERα, but not ERβ, in the hippocampus.
Interestingly, hippocampal expression of ERβ resulted in a learning impairment, observed for escape path length during the initial training and the DI score of the final probe trial. These results provide support for a differential role of ERα and ERβ in hippocampal function (Foster, 2012a; Han et al., 2013; Bean et al., 2014). Studies using ERα knock-out (ERαKO) mice demonstrate that ERα is important for maintaining memory in adults (Fugger et al., 2000; Han et al., 2013). Similar to the current study, viral expression of hippocampal ERβ in ERβKO mice impairs spatial learning (Han et al., 2013). The impairment may result from ERβ inhibition of ERα-mediated transcription rendering the hippocampus more vulnerable to the stressors of aging (Foster, 2005, 2012a; Bean et al., 2014).
The required combination of ERα + EB for cognitive benefits is in contrast to our previous work showing that increased ERα expression alone can improve cognition in young ERαKO mice and middle-aged female rats (Foster et al., 2008; Witty et al., 2012). The difference may be due to a decrease in local E2 availability or reduced ligand-independent ERα-mediated transcription in older animals. Synthesis of E2 in the hippocampus declines after OVX (Barker and Galea, 2009). Indeed, a decline in local E2 synthesis may explain why higher levels of plasma E2 are required to improve cognition with advancing age (Foster et al., 2003; Talboom et al., 2008). In addition, during aging, there is a shift in kinase/phosphatase activity, favoring phosphatases (Norris et al., 1998; Sharrow et al., 2002; Jackson et al., 2009). This shift is thought to reduce ERα phosphorylation, diminishing ligand-independent ERα-mediated transcription in older animals (Foster, 2005). In this case, transcription may be enhanced by the combination of ERα upregulation and EB treatment. Regardless of the exact mechanism, the current study supports the idea that ERα activity is an essential component for regulating the critical window.
Mechanisms for rejuvenating the hippocampus
E2 treatment induces plasticity in the hippocampus and E2 responsiveness declines with age. In older animals, E2 fails to induce the growth of spines (Adams et al., 2001b; Akama and McEwen, 2003; Williams et al., 2011) and the transcription of genes related to synaptic function and maintenance of cell health (Aenlle and Foster, 2010). Importantly, the current study supports the idea that the closing of the critical window is associated with an inability of E2 to enhance NMDAR-mediated synaptic transmission (Smith et al., 2010; Vedder et al., 2014). EB induced a robust increase in NMDAR synaptic transmission in animals expressing ERα, but not in GFP controls, again suggesting that GFP animals are beyond the critical window.
It is possible that memory formation during training acted to increase NMDAR responses. In several neural systems, learning induces an increase in the fast component of synaptic transmission, which can be recorded in vitro 1–7 d after training (Foster et al., 1996; McKernan and Shinnick-Gallagher, 1997; Power et al., 1997; Barkai and Saar, 2001; Foster and Dumas, 2001; Sacchetti et al., 2001; Schroeder and Shinnick-Gallagher, 2005; Mitsushima et al., 2011). In contrast, NMDAR-mediated synaptic transmission is decreased or unchanged after learning (Zinebi et al., 2003; Quinlan et al., 2004; Mitsushima et al., 2011). It has been suggested that the decline in NMDAR function may preserve changes in the fast synaptic transmission by shifting the threshold for synaptic plasticity (Quinlan et al., 2004). Importantly, previous research indicates that the E2-mediated increase in NMDAR synaptic responses are observed in the absence of behavioral training (Woolley et al., 1997; Smith and McMahon, 2006; Snyder et al., 2011).
Alternatively, it is possible that the combination of ERα + EB increased NMDAR function and it is this increase that mediated improvements in learning and memory. Work with mutant mice and pharmacological studies in rats demonstrate that spatial episodic memory, but not spatial reference memory, depends on hippocampal NMDAR function (Foster, 2012b) and a decline in NMDAR function during aging is associated with impaired episodic, but not impaired reference memory (Foster, 2012b; Kumar and Foster, 2013; Lee et al., 2014; Guidi et al., 2015). Overexpression of the NR2B subunit in the hippocampus enhances learning and memory (Tang et al., 1999; White and Youngentob, 2004; Brim et al., 2013). Moreover, viral-mediated upregulation of antioxidant genes in the hippocampus enhances NMDAR function by altering an age-related shift in redox state (Lee et al., 2012; Lee et al., 2014). In this case, virus expression of antioxidant enzymes rejuvenates the hippocampus, increasing hippocampal NMDAR function in older animals, and it is an increase in NMDAR function that underlies enhanced episodic memory.
The mechanism for the increase in NMDAR function is an important question that remains to be elucidated. A shift in NR2 subunit composition, favoring NR2B would be expected to increase NMDAR responses and could influence the threshold for synaptic modifiability during learning (Zinebi et al., 2003; Skibinska et al., 2005; Sun et al., 2005; Lebel et al., 2006; Baez et al., 2013; Laeremans et al., 2015). Aging and E2 deprivation are associated with a decline in NR2B mRNA, decreased NR2B ligand-specific binding in the hippocampus, and loss in dendritic spines containing NR2B subunits (Adams et al., 2001a; Cyr et al., 2001; Morissette et al., 2008; Guidi et al., 2015); however, we did not observe a shift in the expression of the different NMDA subunits, consistent with previous work in young animals (Snyder et al., 2011).
Before closing of the critical window, E2 can increase the number of dendritic spines and expression of a number of synaptic molecular markers, including glutamate receptor subunits, suggesting new functional synapses (Frick et al., 2002; Jelks et al., 2007; Spencer et al., 2008b; Waters et al., 2009; Smith et al., 2010; Qu et al., 2013). Therefore, one possibility is that the increase in NMDAR response was due to E2-induced synaptogenesis. However, we did not observe an increase in the expression of glutamate receptor subunits (GluR1, NR2A, or NR2B) or the synaptic markers PSD95 and synaptophysin in the ERα + EB condition.
It is possible that the increase in the NMDAR response was due to a shift in NMDAR trafficking. Glutamate receptor location and function is regulated by kinase/phosphatase activity. E2 alters kinase/phosphatase activity to increase synaptic transmission (Sawai et al., 2002; Sharrow et al., 2002; Foster, 2005; Zadran et al., 2009; Kumar et al., 2015). Therefore, the increase in synaptic responses may have resulted from enzyme activity, including phosphorylation of glutamate receptors (Xu et al., 2010; Logan et al., 2011; Raval et al., 2012). However, the well characterized NR2B S1303 (Chen and Roche, 2007) and GluR1 S831 (Lee et al., 2010) phosphorylation sites were not altered in the ERα + EB group, indicating that receptor phosphorylation does not mediate increased synaptic transmission observed 48 h after E2 treatment (Snyder et al., 2011). Furthermore, pERK, which provides an indication of E2-induced kinase activity, was not altered. Although it is possible that other phosphorylation sites may have influenced glutamate receptors, the idea that the increase in synaptic transmission is due to maintenance of elevated kinase activity and phosphorylation of glutamate receptors seems doubtful.
Genomic actions of ERs, including ligand-independent ERα-mediated transcription, can influence transcription to regulate synaptic function (Murphy and Segal, 1997; Lee et al., 2004; Aenlle et al., 2009; Aenlle and Foster, 2010; Han et al., 2013; Bean et al., 2014). The fact that ERα + EB effects were observed 48 h after treatment suggests a possible transcriptional mechanism. E2 influences transcription of hippocampal genes for growth, inflammation, oxidative stress, mitochondrial function, and synaptic plasticity. In aging male rats, impaired cognition and decreased NMDAR synaptic function is related to oxidative stress and intracellular redox state (Bodhinathan et al., 2010; Foster, 2012b; Kumar and Foster, 2013; Lee et al., 2014). E2 regulates oxidative stress though multiple mechanisms, including transcription (Behl et al., 1995; Kiray et al., 2004; Zhang et al., 2009; Aenlle and Foster, 2010; Irwin et al., 2012; López-Grueso et al., 2014). However, it remains for future studies to determine whether the increase in NMDAR function was related to E2-induced transcription and modulation of the redox state.
In conclusion, our current findings demonstrate that increasing the expression of functional ERα in aged rats after an extended period of ovarian hormone depletion can restore the critical window for estrogen's positive effects on hippocampal synaptic transmission and spatial memory. These results establish ERα activity as an integral part of the mechanism regulating the critical window. The molecular studies indicate that the beneficial effects were not associated with an increased expression of synaptic marker proteins, NMDAR subunits, or the continuous activation of kinase signaling cascades. These results leave open the question of whether the beneficial effects are due to altered transcription or redox state. Future studies should explore interventions or methodologies that facilitate ERα activation specifically in the brain or augment NMDAR synaptic responses as a strategy to detour cognitive decline in aging women.
Footnotes
This work was supported by the National Institute on Aging–National Institutes of Health (Grants R01AG037984, R37AG036800, and R01AG49711) and the Evelyn F. McKnight Brain Research Foundation. We thank Bradley Fletcher for providing the SEAP DNA; Barrett Severance, Semir Karic, and Erica Wilner for helping in behavioral characterization of animals; and Olga Tchigrinova and Carolina Ceballos-Diaz for technical support in Western blot analysis.
The authors declare no competing financial interests.
References
- Adams MM, Oung T, Morrison JH, Gore AC. Length of postovariectomy interval and age, but not estrogen replacement, regulate N-methyl-D-aspartate receptor mRNA levels in the hippocampus of female rats. Exp Neurol. 2001a;170:345–356. doi: 10.1006/exnr.2001.7716. [DOI] [PubMed] [Google Scholar]
- Adams MM, Shah RA, Janssen WG, Morrison JH. Different modes of hippocampal plasticity in response to estrogen in young and aged female rats. Proc Natl Acad Sci U S A. 2001b;98:8071–8076. doi: 10.1073/pnas.141215898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MM, Fink SE, Shah RA, Janssen WG, Hayashi S, Milner TA, McEwen BS, Morrison JH. Estrogen and aging affect the subcellular distribution of estrogen receptor-alpha in the hippocampus of female rats. J Neurosci. 2002;22:3608–3614. doi: 10.1523/JNEUROSCI.22-09-03608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MM, Fink SE, Janssen WG, Shah RA, Morrison JH. Estrogen modulates synaptic N-methyl-D-aspartate receptor subunit distribution in the aged hippocampus. J Comp Neurol. 2004;474:419–426. doi: 10.1002/cne.20148. [DOI] [PubMed] [Google Scholar]
- Aenlle KK, Foster TC. Aging alters the expression of genes for neuroprotection and synaptic function following acute estradiol treatment. Hippocampus. 2010;20:1047–1060. doi: 10.1002/hipo.20703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aenlle KK, Kumar A, Cui L, Jackson TC, Foster TC. Estrogen effects on cognition and hippocampal transcription in middle-aged mice. Neurobiol Aging. 2009;30:932–945. doi: 10.1016/j.neurobiolaging.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akama KT, McEwen BS. Estrogen stimulates postsynaptic density-95 rapid protein synthesis via the Akt/protein kinase B pathway. J Neurosci. 2003;23:2333–2339. doi: 10.1523/JNEUROSCI.23-06-02333.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Delgado C, Mendoza-Rodríguez CA, Picazo O, Cerbón M. Different expression of alpha and beta mitochondrial estrogen receptors in the aging rat brain: interaction with respiratory complex V. Exp Gerontol. 2010;45:580–585. doi: 10.1016/j.exger.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Baez MV, Oberholzer MV, Cercato MC, Snitcofsky M, Aguirre AI, Jerusalinsky DA. NMDA receptor subunits in the adult rat hippocampus undergo similar changes after 5 minutes in an open field and after LTP induction. PLoS One. 2013;8:e55244. doi: 10.1371/journal.pone.0055244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkai E, Saar D. Cellular correlates of olfactory learning in the rat piriform cortex. Rev Neurosci. 2001;12:111–120. doi: 10.1515/revneuro.2001.12.2.111. [DOI] [PubMed] [Google Scholar]
- Barker JM, Galea LA. Sex and regional differences in estradiol content in the prefrontal cortex, amygdala and hippocampus of adult male and female rats. Gen Comp Endocrinol. 2009;164:77–84. doi: 10.1016/j.ygcen.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Bean LA, Ianov L, Foster TC. Estrogen receptors, the hippocampus, and memory. Neuroscientist. 2014;20:534–545. doi: 10.1177/1073858413519865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl C, Widmann M, Trapp T, Holsboer F. 17-beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Commun. 1995;216:473–482. doi: 10.1006/bbrc.1995.2647. [DOI] [PubMed] [Google Scholar]
- Bimonte HA, Denenberg VH. Estradiol facilitates performance as working memory load increases. Psychoneuroendocrinology. 1999;24:161–173. doi: 10.1016/S0306-4530(98)00068-7. [DOI] [PubMed] [Google Scholar]
- Bodhinathan K, Kumar A, Foster TC. Intracellular redox state alters NMDA receptor response during aging through Ca2 + /calmodulin-dependent protein kinase II. J Neurosci. 2010;30:1914–1924. doi: 10.1523/JNEUROSCI.5485-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollig-Fischer A, Thakur A, Sun Y, Wu J, Liao DJ. The predominant proteins that react to the MC-20 estrogen receptor alpha antibody differ in molecular weight between the mammary gland and uterus in the mouse and rat. Int J Biomed Sci. 2012;8:51–63. [PMC free article] [PubMed] [Google Scholar]
- Brake WG, Alves SE, Dunlop JC, Lee SJ, Bulloch K, Allen PB, Greengard P, McEwen BS. Novel target sites for estrogen action in the dorsal hippocampus: an examination of synaptic proteins. Endocrinology. 2001;142:1284–1289. doi: 10.1210/endo.142.3.8036. [DOI] [PubMed] [Google Scholar]
- Brim BL, Haskell R, Awedikian R, Ellinwood NM, Jin L, Kumar A, Foster TC, Magnusson KR. Memory in aged mice is rescued by enhanced expression of the GluN2B subunit of the NMDA receptor. Behav Brain Res. 2013;238:211–226. doi: 10.1016/j.bbr.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Roche KW. Regulation of NMDA receptors by phosphorylation. Neuropharmacology. 2007;53:362–368. doi: 10.1016/j.neuropharm.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr M, Ghribi O, Thibault C, Morissette M, Landry M, Di Paolo T. Ovarian steroids and selective estrogen receptor modulators activity on rat brain NMDA and AMPA receptors. Brain Res Brain Res Rev. 2001;37:153–161. doi: 10.1016/S0165-0173(01)00115-1. [DOI] [PubMed] [Google Scholar]
- Daniel JM, Hulst JL, Lee CD. Role of hippocampal M2 muscarinic receptors in the estrogen-induced enhancement of working memory. Neuroscience. 2005;132:57–64. doi: 10.1016/j.neuroscience.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Daniel JM, Hulst JL, Berbling JL. Estradiol replacement enhances working memory in middle-aged rats when initiated immediately after ovariectomy but not after a long-term period of ovarian hormone deprivation. Endocrinology. 2006;147:607–614. doi: 10.1210/en.2005-0998. [DOI] [PubMed] [Google Scholar]
- Espeland MA, Rapp SR, Shumaker SA, Brunner R, Manson JE, Sherwin BB, Hsia J, Margolis KL, Hogan PE, Wallace R, Dailey M, Freeman R, Hays J, Women's Health Initiative Memory Study Conjugated equine estrogens and global cognitive function in postmenopausal women: Women's Health Initiative Memory Study. JAMA. 2004;291:2959–2968. doi: 10.1001/jama.291.24.2959. [DOI] [PubMed] [Google Scholar]
- Foster TC. Interaction of rapid signal transduction cascades and gene expression in mediating estrogen effects on memory over the life span. Front Neuroendocrinol. 2005;26:51–64. doi: 10.1016/j.yfrne.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Foster TC. Role of estrogen receptor alpha and beta expression and signaling on cognitive function during aging. Hippocampus. 2012a;22:656–669. doi: 10.1002/hipo.20935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC. Dissecting the age-related decline on spatial learning and memory tasks in rodent models: N-methyl-D-aspartate receptors and voltage-dependent Ca(2)( +) channels in senescent synaptic plasticity. Prog Neurobiol. 2012b;96:283–303. doi: 10.1016/j.pneurobio.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC, Dumas TC. Mechanism for increased hippocampal synaptic strength following differential experience. J Neurophysiol. 2001;85:1377–1383. doi: 10.1152/jn.2001.85.4.1377. [DOI] [PubMed] [Google Scholar]
- Foster TC, Kumar A. Susceptibility to induction of long-term depression is associated with impaired memory in aged Fischer 344 rats. Neurobiol Learn Mem. 2007;87:522–535. doi: 10.1016/j.nlm.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC, Gagne J, Massicotte G. Mechanism of altered synaptic strength due to experience: relation to long-term potentiation. Brain Res. 1996;736:243–250. doi: 10.1016/0006-8993(96)00707-X. [DOI] [PubMed] [Google Scholar]
- Foster TC, Sharrow KM, Kumar A, Masse J. Interaction of age and chronic estradiol replacement on memory and markers of brain aging. Neurobiol Aging. 2003;24:839–852. doi: 10.1016/S0197-4580(03)00014-9. [DOI] [PubMed] [Google Scholar]
- Foster TC, Rani A, Kumar A, Cui L, Semple-Rowland SL. Viral vector-mediated delivery of estrogen receptor-alpha to the hippocampus improves spatial learning in estrogen receptor-alpha knockout mice. Mol Ther. 2008;16:1587–1593. doi: 10.1038/mt.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick KM, Fernandez SM, Bulinski SC. Estrogen replacement improves spatial reference memory and increases hippocampal synaptophysin in aged female mice. Neuroscience. 2002;115:547–558. doi: 10.1016/S0306-4522(02)00377-9. [DOI] [PubMed] [Google Scholar]
- Fugger HN, Foster TC, Gustafsson J, Rissman EF. Novel effects of estradiol and estrogen receptor alpha and beta on cognitive function. Brain Res. 2000;883:258–264. doi: 10.1016/S0006-8993(00)02993-0. [DOI] [PubMed] [Google Scholar]
- Gehring RE. Basic behavioral statistics. Boston: Houghton Mifflin; 1978. [Google Scholar]
- Gibbs RB. Long-term treatment with estrogen and progesterone enhances acquisition of a spatial memory task by ovariectomized aged rats. Neurobiol Aging. 2000;21:107–116. doi: 10.1016/s0197-4580(00)00103-2. [DOI] [PubMed] [Google Scholar]
- Guidi M, Foster TC. Behavioral model for assessing cognitive decline. Methods Mol Biol. 2012;829:145–153. doi: 10.1007/978-1-61779-458-2_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi M, Kumar A, Rani A, Foster TC. Assessing the emergence and reliability of cognitive decline over the life span in Fisher 344 rats using the spatial water maze. Front Aging Neurosci. 2014;6:2. doi: 10.3389/fnagi.2014.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi M, Rani A, Karic S, Severance B, Kumar A, Foster TC. Contribution of N-methyl-D-aspartate receptors to attention and episodic spatial memory during senescence. Neurobiol Learn Mem. 2015;125:36–46. doi: 10.1016/j.nlm.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Aenlle KK, Bean LA, Rani A, Semple-Rowland SL, Kumar A, Foster TC. Role of estrogen receptor alpha and beta in preserving hippocampal function during aging. J Neurosci. 2013;33:2671–2683. doi: 10.1523/JNEUROSCI.4937-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J. Multiple comparisons: theory and methods. Boca Raton, FL: Chapman and Hall/CRC; 1996. [Google Scholar]
- Huck SW. Statistical misconceptions. New York: Psychology; 2009. [Google Scholar]
- Irwin RW, Yao J, To J, Hamilton RT, Cadenas E, Brinton RD. Selective oestrogen receptor modulators differentially potentiate brain mitochondrial function. J Neuroendocrinol. 2012;24:236–248. doi: 10.1111/j.1365-2826.2011.02251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TC, Rani A, Kumar A, Foster TC. Regional hippocampal differences in AKT survival signaling across the lifespan: implications for CA1 vulnerability with aging. Cell Death Differ. 2009;16:439–448. doi: 10.1038/cdd.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelks KB, Wylie R, Floyd CL, McAllister AK, Wise P. Estradiol targets synaptic proteins to induce glutamatergic synapse formation in cultured hippocampal neurons: critical role of estrogen receptor-alpha. J Neurosci. 2007;27:6903–6913. doi: 10.1523/JNEUROSCI.0909-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiray M, Uysal N, Sönmez A, Açikgöz O, Gönenç S. Positive effects of deprenyl and estradiol on spatial memory and oxidant stress in aged female rat brains. Neurosci Lett. 2004;354:225–228. doi: 10.1016/j.neulet.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC. 17beta-estradiol benzoate decreases the AHP amplitude in CA1 pyramidal neurons. J Neurophysiol. 2002;88:621–626. doi: 10.1152/jn.2002.88.2.621. [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC. Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J Neurophysiol. 2004;91:2437–2444. doi: 10.1152/jn.01148.2003. [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC. Linking redox regulation of NMDAR synaptic function to cognitive decline during aging. J Neurosci. 2013;33:15710–15715. doi: 10.1523/JNEUROSCI.2176-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Foster TC. Interaction of DHPG-LTD and synaptic-LTD at senescent CA3-CA1 hippocampal synapses. Hippocampus. 2014;24:466–475. doi: 10.1002/hipo.22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Rani A, Tchigranova O, Lee WH, Foster TC. Influence of late-life exposure to environmental enrichment or exercise on hippocampal function and CA1 senescent physiology. Neurobiol Aging. 2012;33:828.e821–828.e817. doi: 10.1016/j.neurobiolaging.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Bean LA, Rani A, Jackson T, Foster TC. Contribution of estrogen receptor subtypes, ERalpha, ERbeta, and GPER1 in rapid estradiol-mediated enhancement of hippocampal synaptic transmission in mice. Hippocampus. 2015;25:1556–1566. doi: 10.1002/hipo.22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laeremans A, Sabanov V, Ahmed T, Nys J, Van de Plas B, Vinken K, Woolley DG, Gantois I, D'Hooge R, Arckens L, Balschun D. Distinct and simultaneously active plasticity mechanisms in mouse hippocampus during different phases of Morris water maze training. Brain Struct Funct. 2015;220:1273–1290. doi: 10.1007/s00429-014-0722-z. [DOI] [PubMed] [Google Scholar]
- Lebel D, Sidhu N, Barkai E, Quinlan EM. Learning in the absence of experience-dependent regulation of NMDAR composition. Learn Mem. 2006;13:566–570. doi: 10.1101/lm.276606. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Campomanes CR, Sikat PT, Greenfield AT, Allen PB, McEwen BS. Estrogen induces phosphorylation of cyclic AMP response element binding (pCREB) in primary hippocampal cells in a time-dependent manner. Neuroscience. 2004;124:549–560. doi: 10.1016/j.neuroscience.2003.11.035. [DOI] [PubMed] [Google Scholar]
- Lee WH, Kumar A, Rani A, Herrera J, Xu J, Someya S, Foster TC. Influence of viral vector-mediated delivery of superoxide dismutase and catalase to the hippocampus on spatial learning and memory during aging. Antioxidants and Redox Signaling. 2012;16:339–350. doi: 10.1089/ars.2011.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WH, Kumar A, Rani A, Foster TC. Role of antioxidant enzymes in redox regulation of N-methyl-D-aspartate receptor function and memory in middle-aged rats. Neurobiol Aging. 2014;35:1459–1468. doi: 10.1016/j.neurobiolaging.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan SM, Sarkar SN, Zhang Z, Simpkins JW. Estrogen-induced signaling attenuates soluble Abeta peptide-mediated dysfunction of pathways in synaptic plasticity. Brain Res. 2011;1383:1–12. doi: 10.1016/j.brainres.2011.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Grueso R, Gambini J, Abdelaziz KM, Monleón D, Díaz A, El Alami M, Bonet-Costa V, Borrás C, Viña J. Early, but not late onset estrogen replacement therapy prevents oxidative stress and metabolic alterations caused by ovariectomy. Antioxidants and Redox Signaling. 2014;20:236–246. doi: 10.1089/ars.2012.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki PM. Critical window hypothesis of hormone therapy and cognition: a scientific update on clinical studies. Menopause. 2013;20:695–709. doi: 10.1097/GME.0b013e3182960cf8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcondes FK, Bianchi FJ, Tanno AP. Determination of the estrous cycle phases of rats: some helpful considerations. Braz J Biol. 2002;62:609–614. doi: 10.1590/S1519-69842002000400008. [DOI] [PubMed] [Google Scholar]
- Markham JA, Pych JC, Juraska JM. Ovarian hormone replacement to aged ovariectomized female rats benefits acquisition of the morris water maze. Horm Behav. 2002;42:284–293. doi: 10.1006/hbeh.2002.1819. [DOI] [PubMed] [Google Scholar]
- Markowska AL, Savonenko AV. Effectiveness of estrogen replacement in restoration of cognitive function after long-term estrogen withdrawal in aging rats. J Neurosci. 2002;22:10985–10995. doi: 10.1523/JNEUROSCI.22-24-10985.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Mehra RD, Sharma K, Nyakas C, Vij U. Estrogen receptor alpha and beta immunoreactive neurons in normal adult and aged female rat hippocampus: a qualitative and quantitative study. Brain Res. 2005;1056:22–35. doi: 10.1016/j.brainres.2005.06.073. [DOI] [PubMed] [Google Scholar]
- Mitsushima D, Ishihara K, Sano A, Kessels HW, Takahashi T. Contextual learning requires synaptic AMPA receptor delivery in the hippocampus. Proc Natl Acad Sci U S A. 2011;108:12503–12508. doi: 10.1073/pnas.1104558108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morissette M, Le Saux M, Di Paolo T. Effect of oestrogen receptor alpha and beta agonists on brain N-methyl-D-aspartate receptors. J Neuroendocrinol. 2008;20:1006–1014. doi: 10.1111/j.1365-2826.2008.01754.x. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Segal M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc Natl Acad Sci U S A. 1997;94:1482–1487. doi: 10.1073/pnas.94.4.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Halpain S, Foster TC. Alterations in the balance of protein kinase/phosphatase activities parallel reduced synaptic strength during aging. J Neurophysiol. 1998;80:1567–1570. doi: 10.1152/jn.1998.80.3.1567. [DOI] [PubMed] [Google Scholar]
- O'Neal MF, Means LW, Poole MC, Hamm RJ. Estrogen affects performance of ovariectomized rats in a two-choice water-escape working memory task. Psychoneuroendocrinology. 1996;21:51–65. doi: 10.1016/0306-4530(95)00032-1. [DOI] [PubMed] [Google Scholar]
- Power JM, Thompson LT, Moyer JR, Jr, Disterhoft JF. Enhanced synaptic transmission in CA1 hippocampus after eyeblink conditioning. J Neurophysiol. 1997;78:1184–1187. doi: 10.1152/jn.1997.78.2.1184. [DOI] [PubMed] [Google Scholar]
- Qu N, Wang L, Liu ZC, Tian Q, Zhang Q. Oestrogen receptor alpha agonist improved long-term ovariectomy-induced spatial cognition deficit in young rats. Int J Neuropsychopharmacol. 2013;16:1071–1082. doi: 10.1017/S1461145712000958. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Lebel D, Brosh I, Barkai E. A molecular mechanism for stabilization of learning-induced synaptic modifications. Neuron. 2004;41:185–192. doi: 10.1016/S0896-6273(03)00874-2. [DOI] [PubMed] [Google Scholar]
- Raval AP, Sick JT, Gonzalez GJ, Defazio RA, Dong C, Sick TJ. Chronic nicotine exposure inhibits estrogen-mediated synaptic functions in hippocampus of female rats. Neurosci Lett. 2012;517:41–46. doi: 10.1016/j.neulet.2012.04.015. [DOI] [PubMed] [Google Scholar]
- Rocca WA, Grossardt BR, Shuster LT. Oophorectomy, menopause, estrogen treatment, and cognitive aging: clinical evidence for a window of opportunity. Brain Res. 2011;1379:188–198. doi: 10.1016/j.brainres.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchetti B, Lorenzini CA, Baldi E, Bucherelli C, Roberto M, Tassoni G, Brunelli M. Long-lasting hippocampal potentiation and contextual memory consolidation. Eur J Neurosci. 2001;13:2291–2298. doi: 10.1046/j.0953-816x.2001.01609.x. [DOI] [PubMed] [Google Scholar]
- Sandstrom NJ, Williams CL. Memory retention is modulated by acute estradiol and progesterone replacement. Behav Neurosci. 2001;115:384–393. doi: 10.1037/0735-7044.115.2.384. [DOI] [PubMed] [Google Scholar]
- Sawai T, Bernier F, Fukushima T, Hashimoto T, Ogura H, Nishizawa Y. Estrogen induces a rapid increase of calcium-calmodulin-dependent protein kinase II activity in the hippocampus. Brain Res. 2002;950:308–311. doi: 10.1016/S0006-8993(02)03186-4. [DOI] [PubMed] [Google Scholar]
- Schroeder BW, Shinnick-Gallagher P. Fear learning induces persistent facilitation of amygdala synaptic transmission. Eur J Neurosci. 2005;22:1775–1783. doi: 10.1111/j.1460-9568.2005.04343.x. [DOI] [PubMed] [Google Scholar]
- Sharrow KM, Kumar A, Foster TC. Calcineurin as a potential contributor in estradiol regulation of hippocampal synaptic function. Neuroscience. 2002;113:89–97. doi: 10.1016/S0306-4522(02)00151-3. [DOI] [PubMed] [Google Scholar]
- Shumaker SA, Legault C, Kuller L, Rapp SR, Thal L, Lane DS, Fillit H, Stefanick ML, Hendrix SL, Lewis CE, Masaki K, Coker LH, Women's Health Initiative Memory Study Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women's Health Initiative Memory Study. JAMA. 2004;291:2947–2958. doi: 10.1001/jama.291.24.2947. [DOI] [PubMed] [Google Scholar]
- Skibinska A, Lech M, Kossut M. Differential regulation of cortical NMDA receptor subunits by sensory learning. Brain Res. 2005;1065:26–36. doi: 10.1016/j.brainres.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Smith CC, McMahon LL. Estrogen-induced increase in the magnitude of long-term potentiation occurs only when the ratio of NMDA transmission to AMPA transmission is increased. J Neurosci. 2005;25:7780–7791. doi: 10.1523/JNEUROSCI.0762-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, McMahon LL. Estradiol-induced increase in the magnitude of long-term potentiation is prevented by blocking NR2B-containing receptors. J Neurosci. 2006;26:8517–8522. doi: 10.1523/JNEUROSCI.5279-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Vedder LC, Nelson AR, Bredemann TM, McMahon LL. Duration of estrogen deprivation, not chronological age, prevents estrogen's ability to enhance hippocampal synaptic physiology. Proc Natl Acad Sci U S A. 2010;107:19543–19548. doi: 10.1073/pnas.1009307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder MA, Cooke BM, Woolley CS. Estradiol potentiation of NR2B-dependent EPSCs is not due to changes in NR2B protein expression or phosphorylation. Hippocampus. 2011;21:398–408. doi: 10.1002/hipo.20756. [DOI] [PubMed] [Google Scholar]
- Solakidi S, Psarra AM, Sekeris CE. Differential subcellular distribution of estrogen receptor isoforms: localization of ERalpha in the nucleoli and ERbeta in the mitochondria of human osteosarcoma SaOS-2 and hepatocarcinoma HepG2 cell lines. Biochim Biophys Acta. 2005;1745:382–392. doi: 10.1016/j.bbamcr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Spencer JL, Waters EM, Milner TA, McEwen BS. Estrous cycle regulates activation of hippocampal Akt, LIM kinase, and neurotrophin receptors in C57BL/6 mice. Neuroscience. 2008a;155:1106–1119. doi: 10.1016/j.neuroscience.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JL, Waters EM, Romeo RD, Wood GE, Milner TA, McEwen BS. Uncovering the mechanisms of estrogen effects on hippocampal function. Front Neuroendocrinol. 2008b;29:219–237. doi: 10.1016/j.yfrne.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Mercado E, 3rd, Wang P, Shan X, Lee TC, Salvi RJ. Changes in NMDA receptor expression in auditory cortex after learning. Neurosci Lett. 2005;374:63–68. doi: 10.1016/j.neulet.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Talboom JS, Williams BJ, Baxley ER, West SG, Bimonte-Nelson HA. Higher levels of estradiol replacement correlate with better spatial memory in surgically menopausal young and middle-aged rats. Neurobiol Learn Mem. 2008;90:155–163. doi: 10.1016/j.nlm.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Vedder LC, Bredemann TM, McMahon LL. Estradiol replacement extends the window of opportunity for hippocampal function. Neurobiol Aging. 2014;35:2183–2192. doi: 10.1016/j.neurobiolaging.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters EM, Mitterling K, Spencer JL, Mazid S, McEwen BS, Milner TA. Estrogen receptor alpha and beta specific agonists regulate expression of synaptic proteins in rat hippocampus. Brain Res. 2009;1290:1–11. doi: 10.1016/j.brainres.2009.06.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters EM, Yildirim M, Janssen WG, Lou WY, McEwen BS, Morrison JH, Milner TA. Estrogen and aging affect the synaptic distribution of estrogen receptor beta-immunoreactivity in the CA1 region of female rat hippocampus. Brain Res. 2011;1379:86–97. doi: 10.1016/j.brainres.2010.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TL, Youngentob SL. The effect of NMDA-NR2B receptor subunit over-expression on olfactory memory task performance in the mouse. Brain Res. 2004;1021:1–7. doi: 10.1016/j.brainres.2004.05.114. [DOI] [PubMed] [Google Scholar]
- Williams TJ, Mitterling KL, Thompson LI, Torres-Reveron A, Waters EM, McEwen BS, Gore AC, Milner TA. Age- and hormone-regulation of opioid peptides and synaptic proteins in the rat dorsal hippocampal formation. Brain Res. 2011;1379:71–85. doi: 10.1016/j.brainres.2010.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty CF, Foster TC, Semple-Rowland SL, Daniel JM. Increasing hippocampal estrogen receptor alpha levels via viral vectors increases MAP kinase activation and enhances memory in aging rats in the absence of ovarian estrogens. PLoS One. 2012;7:e51385. doi: 10.1371/journal.pone.0051385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. J Comp Neurol. 1993;336:293–306. doi: 10.1002/cne.903360210. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. J Neurosci. 1997;17:1848–1859. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Ye Y, Li T, Chen L, Tian D, Luo Q, Lu M. Bisphenol-A rapidly promotes dynamic changes in hippocampal dendritic morphology through estrogen receptor-mediated pathway by concomitant phosphorylation of NMDA receptor subunit NR2B. Toxicol Appl Pharmacol. 2010;249:188–196. doi: 10.1016/j.taap.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Zadran S, Qin Q, Bi X, Zadran H, Kim Y, Foy MR, Thompson R, Baudry M. 17-Beta-estradiol increases neuronal excitability through MAP kinase-induced calpain activation. Proc Natl Acad Sci U S A. 2009;106:21936–21941. doi: 10.1073/pnas.0912558106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QG, Raz L, Wang R, Han D, De Sevilla L, Yang F, Vadlamudi RK, Brann DW. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J Neurosci. 2009;29:13823–13836. doi: 10.1523/JNEUROSCI.3574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QG, Han D, Wang RM, Dong Y, Yang F, Vadlamudi RK, Brann DW. C terminus of Hsc70-interacting protein (CHIP)-mediated degradation of hippocampal estrogen receptor-alpha and the critical period hypothesis of estrogen neuroprotection. Proc Natl Acad Sci U S A. 2011;108:E617–E624. doi: 10.1073/pnas.1104391108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinebi F, Xie J, Liu J, Russell RT, Gallagher JP, McKernan MG, Shinnick-Gallagher P. NMDA currents and receptor protein are downregulated in the amygdala during maintenance of fear memory. J Neurosci. 2003;23:10283–10291. doi: 10.1523/JNEUROSCI.23-32-10283.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]