

Abstract
Angelman syndrome (AS) is a neurodevelopmental disorder associated with developmental delay, lack of speech, motor dysfunction, and epilepsy. In the majority of the patients, AS is caused by the deletion of small portions of maternal chromosome 15 harboring the UBE3A gene. This results in a lack of expression of the UBE3A gene because the paternal allele is genetically imprinted. The UBE3A gene encodes an enzyme termed ubiquitin ligase E3A (E6-AP) that targets proteins for degradation by the 26S proteasome. Because neurodegenerative disease and other neurodevelopmental disorders have been linked to oxidative stress, we asked whether mitochondrial reactive oxygen species (ROS) played a role in impaired synaptic plasticity and memory deficits exhibited by AS model mice. We discovered that AS mice have increased levels of superoxide in area CA1 of the hippocampus that is reduced by MitoQ 10-methanesuflonate (MitoQ), a mitochondria-specific antioxidant. In addition, we found that MitoQ rescued impairments in hippocampal synaptic plasticity and deficits in contextual fear memory exhibited by AS model mice. Our findings suggest that mitochondria-derived oxidative stress contributes to hippocampal pathophysiology in AS model mice and that targeting mitochondrial ROS pharmacologically could benefit individuals with AS.
SIGNIFICANCE STATEMENT Oxidative stress has been hypothesized to contribute to the pathophysiology of neurodevelopmental disorders, including autism spectrum disorders and Angelman syndrome (AS). Herein, we report that AS model mice exhibit elevated levels of mitochondria-derived reactive oxygen species in pyramidal neurons in hippocampal area CA1. Moreover, we demonstrate that the administration of MitoQ (MitoQ 10-methanesuflonate), a mitochondria-specific antioxidant, to AS model mice normalizes synaptic plasticity and restores memory. Finally, our findings suggest that antioxidants that target the mitochondria could be used therapeutically to ameliorate synaptic and cognitive deficits in individuals with AS.
Keywords: hippocampus, memory, mitochondria, neurodevelopmental disorders, oxidative stress, synpatic plasticity
Introduction
Angelman syndrome (AS) is a genetic neurodevelopmental disorder characterized by developmental delay, lack of speech, motor dysfunction, and epilepsy, along with multiple other features (Lossie et al., 2001; Williams et al., 2006). In the majority of the cases, AS is caused by deletion of the maternal chromosome 15q11-q13 region, which includes the UBE3A gene (Lossie et al., 2001). This results in the absence of UBE3A expression in the brain of AS patients because the process of genomic imprinting normally results in the silencing of the paternal UBE3A allele (Chamberlain and Lalande, 2010). UBE3A encodes for an ubiquitin E3 ligase, termed E6-AP, which covalently attaches polyubiquitin chains to proteins to signal for their recognition and degradation by the 26S proteasome (Knoll et al., 1989; Kishino et al., 1997; Matsuura et al., 1997; Sutcliffe et al., 1997).
It was shown that AS model mice, which display endophenotypes consistent with the human disorder, exhibit mitochondrial dysfunction and altered mitochondrial morphology in the hippocampus (Su et al., 2011). Mitochondria are a prominent source of reactive oxygen species (ROS) and other neurodevelopmental disorders such as autism spectrum disorder (ASD) have been linked to oxidative stress (for review, see Chauhan and Chauhan, 2006; Kern and Jones, 2006). For example, mitochondrial dysfunction and altered expression of electron transport chain (ETC) genes have been observed in autism (Anitha et al., 2013; Gu et al., 2013). Therefore, we investigated whether levels of mitochondrial ROS were altered in the hippocampus of AS model mice and, if so, whether they contributed to impairments in hippocampal synaptic plasticity and memory deficits displayed by these mice.
Herein, we show that there are increased levels of mitochondrial superoxide in the hippocampus of AS model mice, which can be reduced by treatment with MitoQ 10-methanesuflonate (MitoQ), a mitochondria-targeted antioxidant that crosses the blood–brain barrier and selectively accumulates in mitochondria (McManus et al., 2011). We also found that MitoQ rescued impairments in hippocampal long-term potentiation (LTP) and contextual fear memory in the AS model mice. Together, these findings indicate that increased levels of mitochondrial ROS contribute significantly to hippocampal pathophysiology in AS model mice and suggest that therapeutically targeting mitochondrial ROS could be beneficial for individuals with AS.
Materials and Methods
Mice.
AS model mice on a C57BL/6 background were generated and genotyped as described previously (Jiang et al., 1998). All mice were housed under standardized conditions in the Transgenic Mouse Facility of New York University (New York, NY) that were compliant with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male and female AS model mice and wild-type (WT) littermates 12–18 weeks of age were used for the experiments.
Drug preparation and treatment.
For electrophysiology and biochemistry, MitoQ was dissolved in DMSO (1 mm) and bath applied in artificial CSF (ACSF; 500 nm). In the in vivo experiments [dihydroethidium (DHE) staining and behavior], either MitoQ or decyl-tetraphenyl-phosphonium (decyl-TPP) was dissolved in DMSO and mixed with sterile saline solution for a final dilution of 0.5 mg/ml. Mice then were injected intraperitoneally with either 5 mg/kg MitoQ or decyl-TPP [in the text, this group is referred to as (veh)]. The injection regimen used for the behavioral studies is described in Figure 4A–C. The injections were always performed at least 4 h before testing.
Figure 4.

Contextual fear memory deficits are rescued in AS model mice treated with MitoQ. A–C, Schematics illustrating the MitoQL injection (5 mg/kg) regimen used for the contextual fear test (A) and the rotarod task (C), and the MitoQS regimen used for the rotarod test (B). D, Acquisition curves for contextual fear learning are similar among all groups. E, Consolidation of contextual fear memory is impaired in the AS model mice and normalized to WT levels after treatment with MitoQ. Results are displayed as the percentage of freezing during the entire test, minute by minute. ***p < 0.001 with a RM-ANOVA, post hoc Tukey's multiple-comparison tests. F, Consolidation of contextual fear memory. The results are displayed as the average percentage of freezing during the entire 5 min test. ***p < 0.001, ANOVA, post hoc Tukey's multiple-comparison tests. G, AS mice are significantly impaired on the rotarod task, which is not normalized with either MitoQL or MitoQS treatment, respectively. ***p < 0.001, RM-ANOVA, post hoc Tukey's multiple comparison tests. WT + veh, n = 15 mice; WT + MitoQ, n = 14 mice; AS + veh, n = 14 mice; AS + MitoQ, n = 13 mice; WT + MitoQL, n = 6 mice; AS + MitoQL, n = 6 mice; WT + MitoQS, n = 15 mice; AS + MitoQS, n = 15 mice.
Hippocampal slice preparation.
Brains from AS model mice and their wild-type littermates (12–18 weeks of age) were quickly removed, and either transverse hippocampal slices (400 μm) or parasagittal cerebellar slices (400 μm) were isolated with a VT1200 Vibratome (Leica) in the following ice-cold cutting solution (in mm): 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 MgCl2, 5 glucose, and 0.6 ascorbate. Slices were allowed to recover for 20 min at room temperature in a 50:50 solution of ACSF containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 d-glucose, 2 CaCl2, and 1 MgCl2 ACSF, followed by additional recovery for 30 min in room temperature ACSF before being transferred for electrophysiology or mitochondrial superoxide detection.
Detection of superoxide.
For the detection of mitochondrial superoxide, we used MitoSOX Red (Invitrogen; 5 μm in DMSO) in acute brain slices and DHE (ThermoFisher Scientific; 27 mg/kg) administered with intraperitoneal injections. In the acute brain slices experiments, hippocampal and cerebellar slices were incubated for 4–5 h in carboxygenated ACSF at 32°C. Subsequently, either MitoQ (500 nm) or decyl-TPP (500 nm; in the text this group is referred to as veh) were bath applied for 30 min before the addition of MitoSOX Red (5 μm) for an additional 10 min. Immediately following this treatment, slices were fixed with ice-cold 4% paraformaldehyde (PFA) in PBS overnight (ON) at 4°C and cut into 40 μm sections. The in vivo experiments were performed as described previously (Hu et al., 2006). Briefly, mice received two intraperitoneal injections of DHE (final volume of 200 μl) with a 30 min interval. Eighteen hours after the final injection of DHE, mice were anesthetized and perfused with 4% PFA. Brains were postfixed in 4% PFA ON at 4°C and cut into 30 μm sections. The slices obtained from these procedures were mounted onto presubbed slides with Vectashield mounting medium with DAPI (Vector Laboratories). The hippocampal CA1 and cerebellar regions in the sections were imaged using a Leica TCS SP5 confocal microscope. All parameters (pinhole, contrast, gain, offset) were held constant for all sections from the same experiment.
Electrophysiology.
Electrophysiology was performed according to standard techniques, as described previously (Kaphzan et al., 2013). Briefly, bipolar stimulating electrodes (92:8 Pt:Y) were placed at the border of area CA3 and area CA1 along the Schaffer collateral pathway. ACSF-filled glass recording electrodes (1–3 MΩ) were placed in stratum radiatum of area CA1. Stable baseline synaptic transmission was established for at least 60 min. LTP was induced with two trains of high-frequency stimulation (HFS; 100 Hz for 1 s, with a 20 s interval). Either MitoQ (500 nm) or decyl-TPP (500 nm; in the text this group is referred to as veh) was bath applied for 40 min before, during, and 40 min after HFS.
Contextual fear conditioning.
Contextual fear conditioning was performed according to standard techniques, as described previously (Kaphzan et al., 2013). Briefly, on the conditioning day, each mouse was placed in the chamber and received two footshocks (0.5 mA intensity, 2 s duration, 2.5 min interval) starting after 2.5 min of free exploration. The contextual memory test was performed 7 d later by placing each mouse back into the same conditioning chamber for 5 min. Freezing behavior was assessed at 2.5 min intervals during conditioning, and every minute during contextual memory test.
Rotarod.
Mice were tested for balance and motor coordination on an accelerating rotarod (Ugo Basile), as described previously (Carter et al., 1999, 2001). Briefly, mice were trained to remain on a rotating rod set at the constant speed of 4 rpm. The speed of the rotating rod was then increased from 4 to 40 rpm over 5 min, and the time at which each animal fell from the rod was automatically recorded. The mice were given four consecutive trials with a minimum rest interval of 30 min.
Statistical analysis.
Group data are presented as the mean ± SEM. Electrophysiological and behavioral data were analyzed with either ANOVA or repeated-measures ANOVA (RM-ANOVA), using either time or trial as the within-subject factor, and genotype and treatment as the between-subject variable. Post hoc multiple-comparisons corrections were applied using Tukey's test.
Results
An increasing number of studies support the notion that oxidative stress may have a pathological role in several neurodevelopmental disorders including ASD (Chauhan and Chauhan, 2006; Kern and Jones, 2006). A recent investigation (Su et al., 2011) revealed altered mitochondria morphology and function in a mouse model of AS. Specifically, small and dense mitochondria were described in the hippocampus of AS model mice that were correlated with reduced complex III activity and oxidative phosphorylation defects in brain extracts. These findings are particularly interesting because mitochondria are considered to be a significant source of intracellular ROS that when not readily detoxified leads to oxidative stress (Boveris and Chance, 1973; Cadenas et al., 1977; Turrens, 2003). Thus, to determine a possible link between AS pathology and mitochondrial oxidative stress, we first asked whether there are increased levels of mitochondrial ROS in the brain of AS model mice. We stained hippocampal slices with MitoSOX Red, a fluorogenic dye targeted to the mitochondria that is rapidly oxidized by superoxide and becomes fluorescent (Fig. 1A,B). We observed a robust increase in the fluorescence intensity (FI) of oxidized MitoSOX Red in hippocampal CA1 pyramidal neurons from AS model mice compared with their wild-type littermates (Fig. 1D), which is consistent with increased levels of mitochondria-derived superoxide. Moreover, bath application of the mitochondria-targeted antioxidant MitoQ (Smith and Murphy, 2010; Ma et al., 2011) normalized the oxidized MitoSOX Red FI in hippocampal CA1 neurons from AS model mice to wild-type levels (Fig. 1A,B,D). These results indicate that levels of mitochondria-derived superoxide are elevated in the hippocampus of AS model mice and that the levels of superoxide can be restored by treatment with the antioxidant MitoQ.
Figure 1.

AS model mice exhibit enhanced levels of superoxide in area CA1 of hippocampal slices that are attenuated by MitoQ treatment. A, Schematic illustrating the MitoSOX Red staining procedure used to detect ROS in acute hippocampal slices (see also Materials and Methods). B, Hippocampal pyramidal neurons in area CA1 of AS model mice exhibit increased levels of superoxide, measured by MitoSOX Red FI, compared with WT littermates. Pretreatment with MitoQ (500 nm) for 30 min decreases MitoSOX Red FI. Scale bar, 50 μm. White bracketed rectangles indicate the portion of the images magnified in C. C, High-magnification image of the CA1 hippocampal pyramidal neurons shown in B. Scale bar, 5 μm. D, Purkinje cells in cerebellar slices of AS model mice exhibit levels of superoxide similar to those in WT littermates. E, Quantification of MitoSOX Red FI in relation to DAPI FI. All values are presented as a ratio to wild-type levels. The region of interest measured was over the CA1 pyramidal and Purkinje cell bodies. n = 15 slices and n = 5 mice in each group. ***p < 0.001, ANOVA, post hoc Tukey's multiple-comparison tests.
We performed a similar, but independent, set of experiments analyzing the levels of mitochondrial superoxide in the cerebellum, another brain region known to be affected in AS (Jiang et al., 1998). Surprisingly, we found similar levels of MitoSOX Red FI in cerebellar slices from AS model mice and their wild-type littermates (Fig. 1D,E). These results suggest that the oxidative stress selectively affects specific brain regions such as the hippocampus in AS model mice.
To eliminate the possibility that the acute slice preparation contributed to the increased levels of ROS in the hippocampus of the AS model mice, we examined ROS levels in vivo with MitoQ treatment (Fig. 2). We used intraperitoneal injections of DHE, an ROS-sensitive dye that crosses the blood–brain barrier and has been previously used to study the levels of superoxide in brain tissues (Hu et al., 2006). We also used a MitoQ treatment paradigm (5 mg/kg i.p.; see Materials and Methods; Fig. 4A,C) that results in brain levels of MitoQ sufficient to induce antioxidant effects in mice (Smith et al., 2003; Chandran et al., 2009; Ghosh et al., 2010; Smith and Murphy, 2010). Consistent with the MitoSox experiments, we observed increased ROS levels in the hippocampus of the AS mice (Fig. 2A,B,D), which were normalized to wild-type levels following MitoQ treatment (Fig. 2A,B,D). Moreover, we did not detect statistical differences in ROS levels in the cerebellum between wild-type and AS model mice (Fig. 2C,D). Finally, we also examined neurons in layer 5 of the cortex, and we observed no difference between either genotypes or treatments (DHE/DAPI FI: WT + veh = 1.00 ± 0.06; WT + MitoQ = 0.99 ± 0.08; AS + veh = 0.98 ± 0.06; AS + MitoQ = 1 ± 0.08). These results confirm that ROS levels are elevated in the hippocampus of AS model mice in vivo, which can be restored by treatment with the mitochondrial antioxidant MitoQ.
Figure 2.
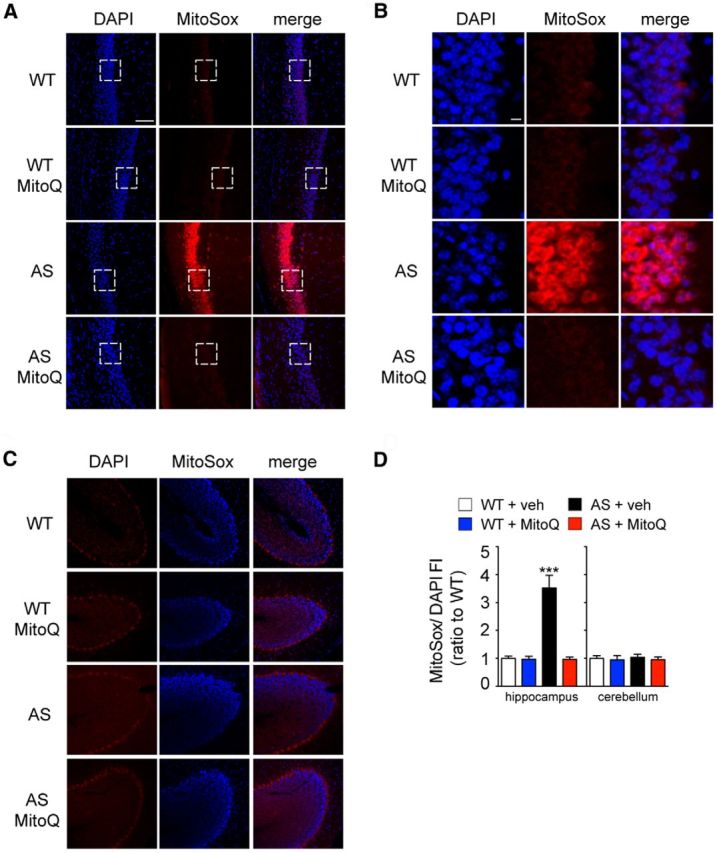
AS model mice exhibit enhanced levels of superoxide in the hippocampus in vivo that are normalized by MitoQ treatment. A, Hippocampal pyramidal neurons in area CA1 of AS model mice exhibit increased levels of superoxide, measured by DHE FI, compared with WT littermates. Treatment of the AS mice with MitoQ (5 mg/kg; see Materials and Methods; Fig. 4A, C) decreases DHE FI. Scale bar, 50 μm. White bracketed rectangles indicate the portion of the images magnified in B. B, High-magnification image of the CA1 pyramidal neurons shown in A. Scale bar, 5 μm. C, Purkinje cells in the cerebellum of AS model mice exhibit levels of superoxide similar to those in WT littermates. D, Quantification of DHE FI in relation to DAPI FI. All values are presented as a ratio to wild-type levels. The region of interest measured was over the CA1 pyramidal and Purkinje cell bodies. n = 25 slices and n = 5 mice in each group. ***p < 0.001, ANOVA, post hoc Tukey's multiple-comparison tests.
Previous studies have shown that excessive mitochondrial ROS impairs LTP. For example, it was shown that amyloid-β application induces an increase in hippocampal mitochondrial superoxide, which results in impaired LTP that is rescued by bath application of MitoQ (Ma et al., 2011). To determine whether the increased levels of mitochondrial ROS in the hippocampus play a role in LTP impairments displayed by AS model mice, we delivered HFS to Schaffer collateral inputs in hippocampal slices in the presence of either MitoQ or veh. Consistent with previous results (Jiang et al., 1998; van Woerden et al., 2007; Kaphzan et al., 2012, 2013), AS mice exhibited impaired LTP, which was normalized to wild-type levels of potentiation by the application of MitoQ (Fig. 3B,C; p < 0.01, RM-ANOVA, post hoc Tukey's multiple-comparison test). Moreover, MitoQ did not change baseline synaptic transmission in either wild-type or AS mice (Fig. 3B). Finally, MitoQ treatment neither impacted LTP in the wild-type mice nor caused any further enhancement of LTP over the veh treatment (Fig. 3B,C).
Figure 3.

Impaired hippocampal LTP of AS model mice is rescued by pretreatment with MitoQ. A, Representative fEPSPs in hippocampal slices from WT and AS model mice treated with either vehicle (veh) or MitoQ (500 nm) before (black traces corresponding to 1 of B) and after (red traces corresponding to 2 of B) stimulation with 2 trains of 100 Hz high-frequency stimulation (2XHFS). B, Hippocampal slices were treated with veh or with MitoQ (500 nm) for 30 min before stimulation with 2XHFS (indicated by the arrows). n = 8 slices and n = 4 mice for each group. ***p < 0.001 RM-ANOVA, post hoc Tukey's multiple-comparison tests. C, Comparison of the level of potentiation 80 min after 2XHFS. *p < 0.05, ANOVA, post hoc Tukey's multiple-comparison tests. D, Paired pulse facilitation of hippocampal slices from AS model mice treated with MitoQ (500 nm), before and after 2XHFS, showing no change after potentiation was induced.
To examine whether normalization of LTP induced by MitoQ in the AS mice occurred via a presynaptic or postsynaptic mechanism, we also examined paired pulse facilitation (PPF) before and after HFS. We found no difference in PPF before and after HFS, suggesting that the altered potentiation in the AS model mice likely arises from a postsynaptic mechanism (Fig. 3D). Overall, these results indicate that increased levels of mitochondria-derived ROS contribute to the LTP deficits displayed by AS model mice.
Previous studies (Jiang et al., 1998; van Woerden et al., 2007; Kaphzan et al., 2012) have shown that contextual fear memory, which is hippocampus dependent, is impaired in AS model mice. Given the ability of MitoQ to reduce mitochondria-derived superoxide and normalize LTP, we determined whether elevated ROS also contribute to hippocampus-dependent memory impairments displayed by AS model mice. Thus, we delivered intraperitoneal injections of either veh or MitoQ (5 mg/kg; Fig. 4A) to AS model mice and their wild-type littermates before and during the acquisition and consolidation phases of the fear conditioning. Similar to previous results, we found that the AS model mice displayed a decrease in freezing time 7 d after contextual fear conditioning. Treatment with MitoQ normalized the impaired contextual fear memory displayed by the AS model mice, restoring it to wild-type levels (p < 0.01, RM-ANOVA, post hoc Tukey's multiple-comparison test; Fig. 4E,F). MitoQ treatment did not alter contextual fear memory in the wild-type mice (Fig. 4E,F). Moreover, the acquisition curves of all groups of mice were similar, suggesting that the MitoQ treatment did not affect the acquisition of contextual fear memory (Fig. 4D). It should be noted that cued fear memory was not impaired in AS model mice (data not shown), which is consistent with the findings of previous studies (Jiang et al., 1998). Overall, these results indicate that elevated mitochondrial ROS contributes to the deficits in contextual fear memory exhibited by AS model mice.
Because motor dysfunction is a clinically relevant component of AS symptomatology (Lossie et al., 2001; Williams et al., 2006), we also determined the role of mitochondrial ROS in motor coordination and learning. Thus, we tested the AS mice using the rotarod to test motor coordination and learning, which is in part dependent on the cerebellum. Consistent with previous studies (van Woerden et al., 2007), we found that the AS model mice were impaired on the rotarod test (Fig. 4G). Moreover, we saw no improvement in the performance of the AS model mice on the task after either short MitoQ treatment (MitoQS; Fig. 4B,G) or long MitoQ treatment (MitoQL; Fig. 4C,G). These results are in agreement with the data demonstrating that mitochondrial ROS levels were not altered in the cerebellum of the AS model mice (Figs. 1D,E, 2C,D) and suggest that the contribution of elevated levels of mitochondrial ROS in AS model mice are specific to hippocampal pathophysiology. Alternatively, it is possible that motor deficits of adult AS mice are resistant to treatment, which is consistent with recent studies (Silva-Santos et al., 2015) demonstrating that reinstating Ube3A in adult mice that develop in the absence of Ube3A does not improve rotarod performance.
Discussion
In this study, we observed that AS model mice exhibited a robust increase in mitochondrial superoxide levels in the hippocampus (Figs. 1, 2). In addition, impairments in hippocampal LTP and contextual fear conditioning exhibited by AS model mice were normalized by the antioxidant MitoQ (Figs. 3, 4), suggesting that the increased levels of mitochondrial superoxide are detrimental for hippocampal function in AS model mice. Oxidative stress occurs in other neurological disorders, including fragile X syndrome and autism, where it has been linked to cognitive deficits (Chauhan and Chauhan, 2006; Kern and Jones, 2006). In addition, it was found previously that increased mitochondrial superoxide plays a role in impaired hippocampal synaptic plasticity and memory deficits in Alzheimer's disease model mice that can be rescued with either treatment with MitoQ or genetic overexpression of SOD-2, the mitochondrial superoxide dismutase (Massaad et al., 2009; Ma et al., 2011; McManus et al., 2011). These results suggest that high levels of oxidative stress via increased levels of mitochondrial superoxide could be a common theme among many brain diseases associated with cognitive deficits.
Although we provide evidence for the involvement of excessive mitochondrial superoxide levels in the hippocampal synaptic and behavioral deficits displayed by AS mice, the link between the lack of expression of the E3 ligase E6-AP and the increased expression of superoxide is unknown. One possible mechanism is an altered ETC, which commonly leads to oxidative stress. Consistent with this idea, a previous study (Su et al., 2011) reported morphological alterations in the mitochondria of AS model mice that were correlated with a reduction in the activity of complex III of the ETC. It is possible that E6-AP may control the expression of either components or modifiers of the ETC, and therefore the loss of E6-AP could cause dysregulation that results in oxidative stress. Further investigation is required to determine how E6-AP regulates the ETC.
The cerebellum is a brain region known to be affected in AS (Jiang et al., 1998; Dindot et al., 2008), but we found no difference in the levels of mitochondrial superoxide in the cerebellum of the AS model mice compared with wild-type littermates (Figs. 1, 2). This was correlated with no improvement in the motor performance on the rotarod test, which is in part dependent on the cerebellum, with MitoQ treatment (Fig. 4G). Consistent with the widespread genomic imprinting of Ube3a, both the hippocampus and cerebellum lack E6-AP (Gustin et al., 2010). Thus, the differences in the levels of mitochondrial superoxide that we discovered in these brain regions of the AS model mice may result from different mechanisms of detoxification of ROS. In agreement with this idea, a marked difference in the expression of enzymes that reduce oxidative stress in the hippocampus and cerebellum of postmortem AD patients has been reported where mRNA for peroxide detoxifying enzymes were elevated in the hippocampus, but not in the cerebellum (Aksenov et al., 1998). Alternatively, the lack of E6-AP may differentially impact the generation of mitochondrial ROS in the hippocampus compared with the cerebellum.
In summary, our results suggest that hippocampal pathophysiology exhibited by AS model mice are related to increased oxidative stress that can be reversed by therapeutically targeting mitocondrial ROS. Notably, previous studies have successfully administered antioxidant treatments to AS patients, though only to treat specific symptoms. For example, melatonin has been efficacious in treating AS-related insomnia (Zhdanova et al., 1999; Braam et al., 2008), and an ω-3 fatty acid-enriched diet decreased epileptic activity (Schlanger et al., 2002). Although other symptoms, such as overall cognitive improvement, were not tested, these studies provide a promising basis for the use of antioxidants for the treatment of AS.
One important factor when considering therapies to reduce oxidative stress is that a baseline level of ROS is required for normal synaptic plasticity. For example, it was shown previously that exogenous application of SOD-1 and similar compounds to reduce ROS levels also prevented LTP (Klann et al., 1998; Klann, 1998), and young mice that overexpress SOD-1 and extracellular SOD have impaired LTP and deficits in contextual fear memory (Thiels et al., 2000; Hu et al., 2006). Thus, it is important to note that the addition of treatment with MitoQ had no effect on the basal levels of mitochondrial superoxide, hippocampal LTP, and contextual fear memory in wild-type mice (Figs. 1, 2, 3, 4), which is consistent with the findings of a previous study (Hu et al., 2007) demonstrating that overexpression of SOD-2, the mitochondrial SOD, had no impact on hippocampal LTP and memory. Because MitoQ targets mitochondrial superoxide, and not other sources of oxidative stress, it could be beneficial for the treatment of AS because it would minimally affect the normal role of ROS in synaptic plasticity and memory. MitoQ has been tested therapeutically for the treatment of Parkinson's disease, and, although it was not efficacious for treating the disorder, it was found to be safe when administered to patients for a prolonged period of time (Snow et al., 2010). Another clinical trial targeting liver inflammation in hepatitis C patients was successful in phase II trials (Smith and Murphy, 2011). Therefore, targeting mitochondrial superoxide with MitoQ may be a promising therapy for individuals with AS.
Footnotes
This work was supported by National Institutes of Health Grants NS087112 (E.S.) and NS078718 (E.K.).
M.P.M. holds stock in Antipodean Pharmaceuticals, which is commercializing MitoQ as a pharmaceutical. The authors declare no other competing financial interests.
References
- Aksenov MY, Tucker HM, Nair P, Aksenova MV, Butterfield DA, Estus S, Markesbery WR. The expression of key oxidative stress-handling genes in different brain regions in Alzheimer's disease. J Mol Neurosci. 1998;11:151–164. doi: 10.1385/JMN:11:2:151. [DOI] [PubMed] [Google Scholar]
- Anitha A, Nakamura K, Thanseem I, Matsuzaki H, Miyachi T, Tsujii M, Iwata Y, Suzuki K, Sugiyama T, Mori N. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol. 2013;23:294–302. doi: 10.1111/bpa.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braam W, Didden R, Smits MG, Curfs LM. Melatonin for chronic insomnia in Angelman syndrome: a randomized placebo-controlled trial. J Child Neurol. 2008;23:649–654. doi: 10.1177/0883073808314153. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Boveris A, Ragan CI, Stoppani AO. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys. 1977;180:248–257. doi: 10.1016/0003-9861(77)90035-2. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, Morton J, Dunnett SB. Motor coordination and balance in rodents. Curr Protoc Neurosci Chapter. 2001;8 doi: 10.1002/0471142301.ns0812s15. Unit 8.12. [DOI] [PubMed] [Google Scholar]
- Chamberlain SJ, Lalande M. Angelman syndrome, a genomic imprinting disorder of the brain. J Neurosci. 2010;30:9958–9963. doi: 10.1523/JNEUROSCI.1728-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, Antholine WE, Zielonka J, Srinivasan S, Avadhani NG, Kalyanaraman B. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: cardioprotection by Mito-Q. Biophys J. 2009;96:1388–1398. doi: 10.1016/j.bpj.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17:111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Chandran K, Kalivendi SV, Joseph J, Antholine WE, Hillard CJ, Kanthasamy A, Kanthasamy A, Kalyanaraman B. Neuroprotection by a mitochondria-targeted drug in a Parkinson's disease model. Free Radic Biol Med. 2010;49:1674–1684. doi: 10.1016/j.freeradbiomed.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu F, Chauhan V, Kaur K, Brown WT, LaFauci G, Wegiel J, Chauhan A. Alterations in mitochondrial DNA copy number and the activities of electron transport chain complexes and pyruvate dehydrogenase in the frontal cortex from subjects with autism. Transl Psychiatry. 2013;3:e299. doi: 10.1038/tp.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin RM, Bichell TJ, Bubser M, Daily J, Filonova I, Mrelashvili D, Deutch AY, Colbran RJ, Weeber EJ, Haas KF. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis. 2010;39:283–291. doi: 10.1016/j.nbd.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Serrano F, Oury TD, Klann E. Aging-dependent alterations in synaptic plasticity and memory in mice that overexpress extracellular superoxide dismutase. J Neurosci. 2006;26:3933–3941. doi: 10.1523/JNEUROSCI.5566-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Cao P, Thiels E, Chu CT, Wu GY, Oury TD, Klann E. Hippocampal long-term potentiation, memory, and longevity in mice that overexpress mitochondrial superoxide dismutase. Neurobiol Learn Mem. 2007;87:372–384. doi: 10.1016/j.nlm.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/S0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- Kaphzan H, Hernandez P, Jung JI, Cowansage KK, Deinhardt K, Chao MV, Abel T, Klann E. Reversal of impaired hippocampal long-term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitors. Biol Psychiatry. 2012;72:182–190. doi: 10.1016/j.biopsych.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphzan H, Buffington SA, Ramaraj AB, Lingrel JB, Rasband MN, Santini E, Klann E. Genetic reduction of the α1 subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Rep. 2013;4:405–412. doi: 10.1016/j.celrep.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern JK, Jones AM. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J Toxicol Environ Health B Crit Rev. 2006;9:485–499. doi: 10.1080/10937400600882079. [DOI] [PubMed] [Google Scholar]
- Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J Neurophysiol. 1998;80:452–457. doi: 10.1152/jn.1998.80.1.452. [DOI] [PubMed] [Google Scholar]
- Klann E, Roberson ED, Knapp LT, Sweatt JD. A role for superoxide in protein kinase C activation and induction of long-term potentiation. J Biol Chem. 1998;273:4516–4522. doi: 10.1074/jbc.273.8.4516. [DOI] [PubMed] [Google Scholar]
- Knoll JH, Nicholls RD, Magenis RE, Graham JM, Jr, Lalande M, Latt SA. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. Am J Med Genet. 1989;32:285–290. doi: 10.1002/ajmg.1320320235. [DOI] [PubMed] [Google Scholar]
- Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, Hutson A, Nicholls RD, Zori RT, Williams CA, Driscoll DJ. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. 2001;38:834–845. doi: 10.1136/jmg.38.12.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Hoeffer CA, Wong H, Massaad CA, Zhou P, Iadecola C, Murphy MP, Pautler RG, Klann E. Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci. 2011;31:5589–5595. doi: 10.1523/JNEUROSCI.6566-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaad CA, Pautler RG, Klann E. Mitochondrial superoxide: a key player in Alzheimer's disease. Aging (Albany NY) 2009;1:758–761. doi: 10.18632/aging.100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, Rommens JM, Beaudet AL. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2011;31:15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlanger S, Shinitzky M, Yam D. Diet enriched with omega-3 fatty acids alleviates convulsion symptoms in epilepsy patients. Epilepsia. 2002;43:103–104. doi: 10.1046/j.1528-1157.2002.13601.x. [DOI] [PubMed] [Google Scholar]
- Silva-Santos S, van Woerden GM, Bruinsma CF, Mientjes E, Jolfaei MA, Distel B, Kushner SA, Elgersma Y. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Invest. 2015;125:2069–2076. doi: 10.1172/JCI80554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RAJ, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- Smith RA, Murphy MP. Mitochondria-targeted antioxidants as therapies. Discov Med. 2011;11:106–114. [PubMed] [Google Scholar]
- Smith RA, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- Su H, Fan W, Coskun PE, Vesa J, Gold JA, Jiang YH, Potluri P, Procaccio V, Acab A, Weiss JH, Wallace DC, Kimonis VE. Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci Lett. 2011;487:129–133. doi: 10.1016/j.neulet.2009.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JS, Jiang YH, Galijaard RJ, Matsuura T, Fang P, Kubota T, Christian SL, Bressler J, Cattanach B, Ledbetter DH, Beaudet AL. The E6-Ap ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 1997;7:368–377. doi: 10.1101/gr.7.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiels E, Urban NN, Gonzalez-Burgos GR, Kanterewicz BI, Barrionuevo G, Chu CT, Oury TD, Klann E. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. J Neurosci. 2000;20:7631–7639. doi: 10.1523/JNEUROSCI.20-20-07631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, Jiang YH, Elgersma Y, Weeber EJ. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci. 2007;10:280–282. doi: 10.1038/nn1845. [DOI] [PubMed] [Google Scholar]
- Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet. 2006;140:413–418. doi: 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- Zhdanova IV, Wurtman RJ, Wagstaff J. Effects of a low dose of melatonin on sleep in children with Angelman syndrome. J Pediatr Endocrinol Metab. 1999;12:57–67. doi: 10.1515/jpem.1999.12.1.57. [DOI] [PubMed] [Google Scholar]