

Abstract
The long-term (>18 months) protection of Ni surfaces against oxidation under atmospheric conditions is demonstrated by coverage with single-layer graphene, formed by chemical vapor deposition. In situ, depth-resolved X-ray photoelectron spectroscopy of various graphene-coated transition metals reveals that a strong graphene–metal interaction is of key importance in achieving this long-term protection. This strong interaction prevents the rapid intercalation of oxidizing species at the graphene–metal interface and thus suppresses oxidation of the substrate surface. Furthermore, the ability of the substrate to locally form a passivating oxide close to defects or damaged regions in the graphene overlayer is critical in plugging these defects and preventing oxidation from proceeding through the bulk of the substrate. We thus provide a clear rationale for understanding the extent to which two-dimensional materials can protect different substrates and highlight the key implications for applications of these materials as barrier layers to prevent oxidation.
Introduction
Graphene and other two-dimensional (2D) materials have been touted as promising ultrathin passivation coatings due to their extremely low permeability to gases1,2 and their ultimate thinness, offering the prospect of preserving the physical properties of surfaces with only a single atomic layer separating them from their surroundings. Standard exfoliation and transfer-based techniques for producing 2D materials are not well-suited to depositing such passivating layers, where a complete, conformal coating is typically desired to prevent ingress of oxidizing species. On the other hand, catalytic growth techniques such as chemical vapor deposition (CVD)3−5 offer direct routes for reducing a catalyst surface and forming a uniform, conformal 2D material layer that passivates the surface enabling simplified integration into devices.6 Indeed, CVD of graphene onto Ni electrodes has been shown to prevent their oxidation during week-long exposures to atmosphere, with functional tunneling spin valve devices successfully fabricated using these graphene-passivated electrodes.7−9 However, other studies investigating graphene grown on Cu foils, as well as graphene transferred onto Si surfaces, have highlighted that the expected passivation is not necessarily achieved and may not be maintained over the long term.10−12
Despite the huge progress in improving the quality and uniformity of CVD graphene,13,14 atomic defects still remain, both within graphene domains and at the boundaries where they join, which provide pathways for the permeation of oxidizing species.15 For single-layer graphene (SLG) on Cu, previous observations have revealed that, on atmospheric air exposure, oxidizing species are thereby able to intercalate between the SLG and Cu and thus access the whole Cu surface.12 Under these atmospheric conditions, where humidity is sufficient for condensed water vapor to serve as an electrolyte on the surface of the graphene–substrate couple, both dry oxidation and wet corrosion must be considered.16 Therefore, while the reduced permeation of oxidizing species through the SLG slows these processes over the short term, Cu oxidation still proceeds over time and is even observed to be electrochemically enhanced in the long term due to the galvanic couple formed by the SLG–Cu.10,11 Recent progress has been made in reducing the graphene permeability by stacking multiple layers17,18 or selectively blocking defects with post-treatments,19 yet these approaches yield barriers significantly thicker than SLG and the reported permeabilities are not sufficiently low to prevent surface oxidation over the long term (i.e., years),20 casting doubt on the level of passivation that can be achieved with atomically thin 2D materials.
Here we form continuous SLG coatings on various polycrystalline transition metal catalysts (Ni, Co, Fe, Pt) by CVD, and using in situ, depth-resolved X-ray photoelectron spectroscopy (XPS) we investigate the extent of oxidation following exposures to atmospheric (moist) air at room temperature for time frames ranging from several minutes up to many months. We thereby demonstrate that SLG can effectively passivate polycrystalline Ni surfaces and maintain a fully reduced Ni surface even following exposure to atmospheric conditions for more than 18 months. Our data reveal that of key importance to achieving this long-term passivation is a strong graphene–substrate interaction, which prevents the lateral diffusion of oxidizing species along the graphene–substrate interface. This enables the long-term passivation of the covered regions of a substrate that interacts strongly with graphene (e.g., Ni, Co), even in cases where the graphene does not form a complete, conformal coating. This is in stark contrast to metals that exhibit a weak interaction (e.g., Cu, Pt), where even short air exposures are sufficient to decouple the graphene from the catalyst by the intercalation of oxidizing species at edges or defects, opening a pathway by which oxidizing species can access the whole catalyst surface. We further show that the ability of the substrate to locally form a passivating oxide at defective or damaged regions in the graphene overlayer is critical in eliminating an alternative route by which oxidation of the substrate can proceed over the longer term. For catalysts that do not form such a passivating oxide (e.g., Fe in moist air), while a strong graphene–substrate interaction can suppress surface oxidation in the short term, for long-term exposures to atmospheric air, oxidation can proceed through the oxide layers that form close to graphene defects until eventually the metal becomes oxidized throughout.
These observations reveal that the interaction between a 2D material and the underlying substrate plays a significant role in determining its performance as a passivation layer and highlight that it is the combination of 2D material and substrate that is key to preventing oxidation. We thereby rationalize apparent inconsistencies in the literature regarding the extent to which graphene passivates different substrates and highlight the key advantages offered by direct integration techniques, such as CVD, that can inherently establish a strong graphene–substrate interaction.
Results and Discussion
Graphene is formed by CVD on polycrystalline transition metal foils (Co, Fe, Ni, Pt) and a Ni(111) single crystal or by vacuum annealing of a Pt(111) single crystal (see Methods), based on our extensive previous calibrations.3,21−24 Samples are then exposed to atmospheric (moist) air at room temperature from times ranging from several minutes to more than 18 months, where we note that condensed water vapor may serve as an electrolyte to facilitate wet corrosion.
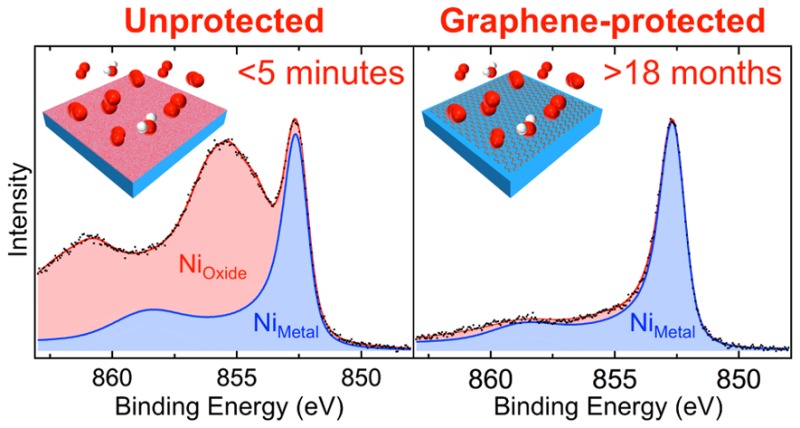
Figure 1 compares the depth-resolved XP Ni 2p3/2 core level spectra of polycrystalline Ni foils with different extents of graphene coverage and lengths of exposure to atmospheric conditions. Depth resolution is achieved by varying the incident photon energy, Ephoton, which in turn varies the kinetic energy of the photoelectrons and thus their mean escape depth, λescape. The spectra for a bare Ni (25 μm) foil that has been annealed [600 °C, H2 (10–1 mbar) for 15 min] and kept under vacuum conditions (Figure 1a) correspond to metallic Ni with a dominant peak at 852.6 eV (NiM), confirming that the foil is fully reduced across the depths probed.25 Following exposure to atmosphere for just 5 min (Figure 1b), strong Ni oxide and hydroxide peaks26 (NiOx) appear in the most surface-sensitive spectrum (λescape ≈ 7 Å), indicating that the Ni surface has become heavily oxidized. Probing deeper into the sample (λescape ≈ 9–11 Å) reveals a lower extent of oxidation, as seen from the decrease in the intensities of the NiOx peaks relative to the NiM peaks, consistent with the rapid formation of an oxide layer on atmospheric exposure, that passivates the surface and limits oxidation from proceeding throughout the Ni bulk.27,28 Conversely, for a Ni (25 μm) foil covered with a complete SLG film (Figure 1c), the XP Ni 2p3/2 core level spectra remain very similar to those measured for the fully reduced Ni foil (Figure 1a), even following 18 months in air. This confirms the SLG-covered Ni surface remains reduced during extended exposure to atmospheric air and, therefore, that long-term passivation can be achieved with SLG on Ni surfaces. Figure 1d shows that, even for a Ni (250 μm) foil covered with noncontinuous SLG islands, the surface remains largely reduced with only small NiOx contributions visible following 18 months of air exposure. The extent of the oxidation correlates with the area of uncovered Ni, as determined by scanning electron microscopy (Figure S1), indicating that only these regions are oxidized while the areas beneath the SLG remain reduced. It should be noted that the absolute intensity of the Ni 2p3/2 signals is weakened by graphene coverage, as apparent from the increased signal-to-noise ratio in Figure 1c,d.
Figure 1.

Depth-resolved XP Ni 2p3/2 core level spectra for polycrystalline Ni (25 μm) in situ immediately following annealing [600 °C, H2 (10–1 mbar) for 15 min] (a) and after subsequent exposure to atmosphere for <5 min (b), as well as for Ni (25 μm) covered with a complete SLG layer (c), and Ni (250 μm) covered with noncontinuous SLG islands (d) following exposure to atmosphere for >18 months. The SLG was grown by CVD [600 °C, C6H6 (10–5 mbar) for 15 min]. Spectra are collected at photon energies, Ephoton, of 1010, 1150, 1300, and 1450 eV [from upper to lower spectra, respectively, λescape ≈ 7, 9, 10, and 11 Å] and are normalized to have the same maximum intensity. Fitted components for metallic Ni (NiM) and Ni oxide/hydroxide (NiOx) are shaded blue and red, respectively.
This long-term passivation behavior of graphene on Ni is in contrast to that reported for graphene on Cu, where long-term passivation under atmospheric conditions is not achieved, and the presence of graphene is even found to electrochemically enhance the oxidation of Cu by the formation of a galvanic couple.10−12 It has been proposed that such disparities in graphene passivation behavior may relate to differences in the defect densities of the graphene formed on these different metals.29 However, comparison of SLG grown on Ni using the conditions herein and SLG grown on Cu that subsequently oxidizes in air reveals very similar defect densities as determined by Raman spectroscopy (following transfer to SiO2 (300 nm)/Si substrates).3,12 A significant difference between these two systems, however, does lie in the strength of the metal–graphene interaction. We therefore draw a distinction between strongly interacting metals, such as Ni,22,30−33 Co,34,35 Fe,33,36 Ru,37−39 Rh,40 and Pd,41 where the hybridization between the graphene π and metal d valence band states destroys the characteristic linear band dispersion of graphene at the K point, and weakly interacting metals, such as Cu,42−44 Ag,42 Ir,45 Pt,31,46,47 and Au,32,42,48 where this linear dispersion is preserved but charge transfer between the metal and graphene (i.e., doping) typically shifts the Fermi level position of the graphene.31,33,47,49 Dahal and Batzill quantify this distinction in terms of the energy of the metal d band center with respect to the Fermi level, with the transition between weakly and strongly interacting metal suggested to occur at ∼2 eV.47 We note that this refers to graphene on idealized low-index, single-crystal surfaces and that the situation for polycrystalline surfaces is more complex, with possible variations in the graphene–substrate interaction for different crystal facets.
Figure 2 compares the effect of atmospheric exposures on SLG grown on prototypical strongly interacting Ni(111)32 and more weakly interacting Pt(111). The spectra are consistently fitted (see Methods) based on our previous XPS investigations of graphene on Ni21−24 and other catalysts.12,50 The spectral resolution of ∼0.3 eV allows the relatively small shifts in binding energy associated with changes in graphene–catalyst interaction to be readily resolved. The CVD condition used on Ni(111) [400 °C, C2H4 (10–6 mbar) for 2 h] results in epitaxial SLG formation by the transformation of an intermediate Ni2C surface carbide.21,22Figure 2a shows the resulting XP C 1s spectrum following growth, which has a majority component at ∼284.8 eV corresponding to epitaxial SLG, whose higher binding energy compared to isolated graphene (∼284.4 eV) relates to the alteration of the band structure by the strong interaction with Ni(111).21,47 Very minor contributions are also observed at ∼284.4 and ∼283.2 eV, which correspond to small amounts of rotated SLG and residual Ni2C, respectively.21 We note that, under such low-temperature growth conditions, numerous defects are included within the graphene lattice as previously confirmed by scanning tunneling microscopy.21Figure 2a also shows the C 1s spectrum for epitaxial SLG on Ni(111) following exposure to atmosphere for 5 days. Significantly, the dominant component remains at ∼284.8 eV, with the absence of a shift in the peak position during air exposure confirming that the strong SLG–Ni(111) coupling is maintained, in spite of the presence of defects in the SLG. A shift toward the binding energy of isolated graphene (∼284.4 eV) would be expected if the graphene–catalyst interaction is weakened, as seen for rotated SLG,21,22 additional graphene layers,22 or for epitaxial SLG on Ni(111) intercalated with Au.32,51 Following air exposure there is some broadening of the fitted components, consistent with the accumulation of atmospheric contaminants (e.g., hydrocarbons, oxygen).52 The CA component also disappears, indicating that residual Ni2C is unstable under atmospheric conditions and likely oxidizes. Furthermore, given that no bulk carbidic phases are found to be stabilized under these growth conditions on Ni,23,24 the passivation we observe is attributed to the presence of SLG rather than any surface or bulk carbidic phase.
Figure 2.

(a) XP C 1s core level lines of Ni(111) covered with SLG grown by CVD [400 °C, C2H4 (10–6 mbar) for 2 h] in situ immediately following growth (lower) and after exposure to atmosphere for 5 days (upper). (b) XP C 1s core level lines of Pt(111) covered with SLG grown by vacuum annealing [10–8 mbar, 1000 °C, for 2 h] in situ immediately following growth (lower) and after exposure to atmosphere for 5 min (middle) and 2 days (upper). All spectra are collected at photon energy, Ephoton, of 425 eV (λescape ≈ 7 Å) and are normalized to have the same maximum intensity. (c) SEM micrographs of SLG island on polycrystalline Pt (25 μm) after exposure to atmosphere for 5 min (lower) and 1 day (upper), with inset schematic indicating the coupled (purple) and decoupled (blue) regions.
Figure 2b shows XP C 1s spectra measured for SLG grown on a Pt(111) surface by the diffusion of adventitious carbon from within the bulk of the crystal during vacuum annealing [10–8 mbar, 1000 °C, for 2 h]. The dominant component immediately following growth is at ∼284.0 eV, which corresponds to SLG coupled to the Pt surface, with the peak position shifted to lower binding energy than isolated graphene due to the p-type charge transfer doping by the higher work function Pt(111).31,40,46,47,49 On exposure to atmosphere for only short times (5 min), additional peaks begin to appear with the strongest at ∼284.4 eV, which grows in intensity at the expense of the ∼284.0 eV peak for longer exposures (2 days). This shift in peak position toward that of free-standing, undoped graphene50,52 is attributed to the weakening of the SLG–Pt coupling by the gradual intercalation of oxidizing species at the SLG–Pt interface, leading to the decoupling of the SLG and loss of the charge transfer, i.e., loss of p-type doping. The other more minor peaks are tentatively attributed to carbidic species (∼283.6 eV)53 and reconstruction of platinum regions beneath the SLG (∼285 eV).54
This decoupling behavior is also apparent in scanning electron microscopy (SEM) images (secondary electron, in-lens detector) of an isolated SLG island on polycrystalline Pt (Figure 2c), where the brighter decoupled region near the perimeter (lower image, 5 min air exposure) proceeds inward with continuing air exposure until the whole SLG island is decoupled (upper image, 1 day air exposure). The reduction in secondary electron yield for the coupled SLG region is attributed to the higher work function of the SLG when it is p-doped.55 The shorter time scale for complete decoupling of this isolated SLG island is ascribed to the shorter lateral distances over which intercalants must diffuse in comparison to the continuous SLG film of Figure 2b. We note that a similar decoupling behavior on air exposure is observed for graphene on other weakly interacting catalysts such as Cu,12 albeit occurring over even shorter time scales, and with coupled regions showing brighter secondary electron yield than decoupled regions.12,50 This increased secondary electron yield can again be explained by alteration of the coupled graphene’s work function, which in this case is expected to be lowered due to n-type charge transfer doping by Cu.47
Having established that with Ni a strong graphene–metal interaction protects against rapid surface oxidation by preventing intercalation of oxidizing species at the graphene–metal interface, we now consider the performance of graphene in protecting other transition metals that interact strongly with graphene, namely, Co (Figure 3) and Fe (Figure 4). Figure 3a shows Co 2p3/2 core level spectra for bare Co (25 μm) after annealing [H2 (1 mbar) at 600 °C for 15 min], which indicate that the Co is fully reduced with a dominant metallic peak at ∼778.2 eV (CoM), for both of the depths probed (λescape ≈ 8 and 11 Å). Following air exposure of the bare Co surface for ∼5 min (Figure 3b), peaks related to Co oxides/hydroxides with binding energies above 780 eV (CoOx) dominate the most surface-sensitive Co 2p3/2 spectrum (λescape ≈ 8 Å). They are also readily apparent in the more bulk-sensitive spectra (λescape ≈ 11 Å), but the CoM peak remains the most intense component, indicating a lower extent of oxidation. This closely parallels the oxide formation on Ni and is again consistent with previous results showing the rapid formation of an oxide layer on exposure to atmospheric air that passivates the surface and slows further oxidation of the metal bulk.56Figure 3c reveals that, for SLG-covered Co (25 μm) exposed to air for >6 months, the Co 2p3/2 spectra are very similar to those of reduced Co (Figure 3a). Importantly, the absence of any significant CoOx peaks confirms that oxidation of the Co surface is very limited and that the SLG-covered Co is maintained in a reduced state. We note that no significant carbidic phases are apparent in the Co 2p3/2 spectra or corresponding C 1s spectra (not shown) and the passivation is thus attributed to the presence of SLG. This behavior observed for Co is qualitatively very similar to that for Ni, with almost no oxidation of the SLG-covered Co evident after >6 months, while the bare Co forms a passivating oxide layer across its surface.
Figure 3.

Depth-resolved XP Co 2p3/2 core level spectra for polycrystalline Co (25 μm) in situ immediately following annealing [600 °C, H2 (10–1 mbar) for 15 min] (a) and after subsequent exposure to atmosphere for <5 min (b); and for Co (25 μm) covered with a complete SLG layer grown by CVD [700 °C, C2H2 (∼10–6 mbar) for 15 min followed by C2H2 (∼10–5 mbar) for 5 min] following exposure to atmosphere for >6 months. Spectra are collected at photon energies, Ephoton, of 1020 (upper) and 1400 eV (lower) [respectively, λescape ≈ 8 and 11 Å] and are normalized to have the same maximum intensity.
Figure 4.

Depth-resolved XP Fe 2p3/2 core level spectra for polycrystalline Fe (100 μm) in situ immediately following annealing [1000 °C, H2 (10–1 mbar) for 15 min] (a) and after subsequent exposure to atmosphere for 1 h (b); and for Fe (100 μm) covered with a complete SLG layer grown by CVD [650 °C, C2H2 (∼10–4 mbar) for 30 min] following exposure to atmosphere for 1 week (c) and >6 months (d). Spectra are collected at photon energies, Ephoton, of 920 (upper) and 1150 eV (lower) [respectively, λescape ≈ 8 and 10 Å] and are normalized to have the same maximum intensity.
Parts a and b of Figure 4 show Fe 2p3/2 spectra for an annealed [H2 (1 mbar) at 900 °C for 15 min] Fe foil, before and after exposure to atmosphere for ∼1 h, respectively. Initially (Figure 4a) a dominant peak at ∼706.7 eV (FeM) is apparent for both excitation energies used (corresponding to λescape ≈ 8 and 10 Å), consistent with the Fe being fully reduced. Following the short atmospheric exposures, components related to Fe oxides/oxyhydroxides57,58 appear around 711 eV (FeOx), which are dominant across the depths probed, with only a very weak FeM component remaining. This significant oxidation of the Fe that is observed even in the more depth-sensitive spectrum is in contrast to the behavior of the bare Co and Ni films where the initial rapid formation of a thin oxide layer passivates the surface, limiting further oxidation of the metal bulk. This is, however, consistent with the well-established and often experienced behavior of Fe in moist air where a hydrated oxide forms as a loose deposit that provides little or no passivation of the Fe surface and allows oxidation to proceed throughout the bulk,16 in contrast to cases of purely dry or purely wet Fe oxidation.58,59
Figure 4c shows that, for SLG-covered Fe exposed to air for 5 days, while some weak FeOx contributions are detectable in the most surface-sensitive spectrum (λescape ≈ 8 Å), the spectra remain largely similar to those of the reduced Fe (Figure 4a), with FeM remaining by far the dominant component. However, following a longer, >6 month air exposure (Figure 4d), the Fe 2p3/2 spectra now more closely resemble the air-exposed bare Fe foil (Figure 4b), showing only FeOx components with no detectable FeM contribution. This highlights that, while the SLG coverage can protect the Fe from oxidation for air exposures of a few weeks, for longer-term exposure the underlying Fe foil gradually oxidizes and eventually no reduced Fe remains close to the surface. This same general behavior is confirmed for Fe covered with thicker few-layer graphene (FLG) films; however, a small FeM peak is still detectible in the XP spectra even after >18 months (see Supporting Information, Figure S3). This reveals that the Fe oxidation occurs more slowly, with some Fe still preserved in a metallic state, presumably as a result of the reduced permeation of oxidizing species through the thicker FLG film. No significant carbidic phases are observed in the Fe 2p3/2 spectra or corresponding C 1s spectra (not shown) of the samples measured, and thus their involvement in the observed oxidation behavior can be largely excluded.
Figure 5 illustrates the general model for 2D material passivation that is developed herein by investigating graphene passivation on different substrates during atmospheric air exposures. First the presence of the 2D material provides a low-permeability barrier that limits the access of oxidizing species to the substrate below, with thicker layers having even lower permeability. However, this low permeabilty alone does not typically afford long-term passivation, as intrinsic defects such as grain boundaries and atomic vacancies in the graphene still allow oxidizing species to reach the substrate close to these defects. For weakly interacting metals (e.g., Cu, Pt), the graphene is rapidly decoupled from the surface on air exposure by the intercalation of oxidizing species, allowing ready access to the whole metal surface and thus its rapid oxidation. For strongly interacting metals (e.g., Ni, Co, Fe) however, the graphene remains coupled on air exposure and the oxidizing species are thus only able to access the metal surface close to graphene defects, suppressing oxidation of the surface over the short term. For metals that form a passivating oxide (e.g., Ni, Co), these exposed regions near to defects are quickly “plugged” by oxide formation, and the majority of the metal surface is thereby protected from oxidation over the long term. In the case of metals whose oxide is not passivating (e.g., Fe in moist air), while the strong graphene–metal interaction provides short-term protection of the surface, over the longer term, oxidation can proceed through the oxide layers initially formed close to graphene defects until eventually the metal becomes oxidized throughout.
Figure 5.

Schematic illustrating the passivation behavior of different graphene-covered metals. Graphene is easily decoupled from the surface of weakly interacting metals (e.g., Cu, Pt) on air exposure, providing a pathway for the intercalation of oxidizing species at the graphene–catalyst interface and ready access for these oxidizing species to the whole metal surface. For strongly interacting metals (e.g., Ni, Co, Fe), graphene is not decoupled on air exposure, and the oxidizing species are thus only able to access the metal surface close to graphene defects. For metals that form a passivating oxide (e.g., Ni, Co), these exposed regions near to defects are quickly “plugged” by oxide formation, protecting the substrate from oxidation over the long term. For metals whose oxide is not passivating (e.g., Fe), oxidation is initially slowed by the already formed oxide, and thus the graphene coverage provides short-term passivation. However, oxidation can proceed through the already formed oxide, eventually allowing the metal to become oxidized throughout for long-term air exposures.
We emphasize that, although a passivating oxide is key to the observed long-term passivation of graphene-covered strongly interacting metals, a passivating oxide alone does not afford equivalent protection. The surfaces of bare Ni and Co are heavily oxidized within minutes of atmospheric exposure, whereas with graphene present negligible oxidation of the surface occurs for atmospheric exposures of several months or even years, time scales of >5 orders of magnitude longer. This suppression of surface oxidation is important for various applications where even limited surface oxidation of metals can severely undermine performance, including ferromagnetic spin injectors,7−9 bipolar plates for polymer electrolyte membrane fuel cells,60,61 and non-noble plasmonic materials.62
Our model is consistent with various reports in literature of the effective passivation of metals that strongly interact with graphene such as Ni and Ru in atmospheric air7−9,63,64 and Fe in dry O2.65 More weakly interacting catalysts covered with graphene, most typically Cu10,12,66 but also Ir,67,68 are reported to show oxygen intercalation even for relatively modest air exposures, and in the case of Cu, surface oxidation is observed within hours of exposure to atmosphere.10,12 More direct comparisons of the gaseous oxidation of strongly and weakly interacting metals covered with 2D materials formed by CVD are scarcely reported in the literature; however, the behavior observed during electrochemical corrosion studies of different graphene-covered metals in aqueous solutions appears consistent with the model developed herein.69 Anodic reactions are found to be strongly suppressed for graphene-covered Ni, which has a strong graphene–catalyst interaction and forms a passivating oxide. In contrast only a very minor reduction in anodic reaction rate is observed for graphene-covered Cu, whose weak graphene–catalyst interaction and lack of passivating oxide offer little protection. While our focus herein has been on passivation with 2D materials at room temperature in atmospheric air, we note that, under the more extreme conditions of full immersion in a liquid environment, the formation of a stable passivating oxide is expected to be particularly important in suppressing wet (i.e., electrochemical) corrosion.
Under more aggressive chemical environments or at elevated temperatures, further factors may need to be taken into account when applying the graphene passivation framework that we have developed. For example, the oxidizing species may be more readily able to intercalate beneath 2D materials even when a strong interaction with the substrate exists39,63 and/or passivating oxides formed at room temperature may no longer be stable.56 Furthermore, depending on the nature of the substrate, it can serve as a catalyst to accelerate the breakdown of the 2D material layer under such conditions. Indeed the etching of h-BN on Cu in the presence of oxygen has been observed at temperatures well below those at which isolated h-BN starts to degrade.70 Therefore, careful consideration of the compatibility of the 2D material and substrate under the specific operating conditions is required, and further experimental verification of the passivation that can be achieved under such conditions may be needed.
A number of previous publications have compared the passivation performance of graphene grown directly on a substrate with that of graphene transferred onto a substrate, with the latter typically giving much poorer results.10,29,71 This can be understood in the context of the model developed herein, given that a strong interaction with the substrate is not expected to result from typical 2D material-transfer techniques. This also applies to passivation barriers based on percolation of liquid-phase-exfoliated platelets,72 and thus in both of these cases any potential strong interaction with the substrate that could improve the barrier’s performance may need to be activated by, for example, postdeposition thermal annealing.73 This highlights a key advantage of catalytic deposition techniques such as CVD, in that the establishment of an interaction between the catalyst and 2D material is integral to the growth process,22 allowing the direct integration of 2D materials into device structures to provide long-term passivation.6−9 We further note that, while the permeability of 2D material passivation layers can be improved by decorating defects using atomic layer deposition (ALD),19 this does not strengthen the graphene–substrate interaction and thus does not benefit from the synergy between the substrate and the passivation layer.
We have so far only considered graphene-covered elemental substrates, but we note that, for strongly interacting metals that do not form the required passivating oxide, alloying of the metal may offer a route to achieving long-term passivation. For example, by exchanging Fe for stainless steel, a strong graphene–metal interaction is still expected while the alloy can form a stable passivating oxide that can plug defects in the graphene, allowing improved corrosion resistance.74 Similarly, where long-term passivation of a substrate with a 2D material is desired but no strong interaction exists, it may be possible to intercalate a thin layer of strongly interacting atoms at the interface to provide the required protection,75 although this will of course further alter the surface properties.
The ability for metal atoms to intercalate beneath graphene indicates that defects are typically present in as-grown graphene or are readily formed on annealing. Nevertheless, our results highlight that the protection against oxidation achieved on metals that interact strongly with graphene and form a passivating oxide is maintained even if the graphene coating is relatively defective (Figure 2a) or not completely continuous (Figure 1d). This makes such protection promising even for applications where some in-service damage or wear might be expected. This raises the question of what quality of 2D material is actually required to achieve passivation of the substrate surface. The strong graphene−metal interaction that is key to preventing oxidation arises from the hybridization between the graphene π and metal d valence band states. Thus, if this hybridization is not maintained due to significant changes in the electronic structure of the graphene, the protection against surface oxidation is also expected to be lost. The formation of a passivating oxide is also critical in preventing oxidation from proceeding through the metal bulk over the longer term. Therefore, as the distance between defects approaches the thickness of the metal’s passivating oxide, the oxidation behavior will approach that of the bare metal surface. Thus, for Ni and Co substrates, with typical passivating oxide thicknesses of the order of nanometers,27,56 reasonable passivation can still be expected from relatively defective and even nanocrystalline graphene, but this is likely to be lost on moving further along the amorphization trajectory toward tetrahedral amorphous carbon.4 A similar line of argument can be applied to understanding whether the suppression of intercalation on certain metals arises due to the anchoring of the graphene at edges/defects or the interaction between the graphene basal plane and the metal surface. While differences in the anchoring of graphene layers on different metals might be expected, the thicknesses of the passivating oxides on Ni and Co, i.e., the distance over which oxidizing species can penetrate, would easily bypass the anchoring of the atom-thick graphene, and thus the strong interaction between the graphene basal plane and the metal surface is chiefly implicated.
Conclusions
In summary, we have shown that SLG can effectively protect Ni and Co surfaces from oxidation over extended periods, even when the SLG does not form a continuous film. We find that crucial to achieving this long-term passivation is a strong interaction between the 2D material and the underlying substrate, preventing the intercalation of oxidizing species along their interface, which otherwise allows the rapid oxidation of the whole substrate surface. This reveals a route to the long-term protection of metal surfaces, based on the synergy between the substrate and the passivation layer, rather than just the passivation layer’s standalone permeability. Furthermore, we highlight that the ability of the substrate to form a passivating oxide is critical in preventing oxidation from instead proceeding through the substrate bulk, fed through defects or damaged regions in the 2D material overlayer. We are thus able to provide a consistent explanation for apparent disparities in literature regarding the ability for graphene to provide long-term passivation, which, as we highlight, depends critically on the properties of the underlying substrate. These insights are highly relevant to the application of 2D materials as effective passivation barriers, where they offer the prospect of preserving the physical properties of surfaces over the long term.
Methods
We investigate commercially available polycrystalline foils of Ni (25 or 250 μm thick), Co (25 μm thick), Fe (100 μm thick), and Pt (25 μm thick), as well as ∼1 mm thick Ni(111) and Pt(111) single crystals. The polycrystalline foils and Ni(111) are annealed [600–900 °C, H2 (1 mbar), 15 min, heated at ∼100 °C min–1], exposed to hydrocarbons [400–700 °C, C2H4 (10–6–10–4 mbar)], and then cooled [under vacuum (∼10–7 mbar) at ∼100 °C/min] in custom-built cold-wall reactors unless otherwise stated. For growth on Pt(111), the sample is first annealed [1000 °C, O2 (10–4 mbar), 30 min] to leave a carbon-free surface (as confirmed by in situ XP C 1s spectra) and, following removal of O2, graphene growth proceeds during vacuum annealing [1000 °C, 10–8 mbar], presumably supplied by carbon dissolved with the sample’s bulk. Samples are exposed to atmospheric (moist) air at room temperature for between 5 min and 18 months and are stored in polystyrene sample boxes during this time to minimize the buildup of dust that may otherwise alter their oxidation behavior.
In situ XPS measurements were performed at the BESSY II synchrotron at the ISISS end station of the FHI-MPG. The high-pressure setup consists of a reaction cell (base pressure ≈ 10–8 mbar) attached to a set of three differentially pumped electrostatic lenses and a differentially pumped analyzer (Phoibos 150, SPECS GmbH), as described elsewhere.76 All spectra are collected in normal emission geometry, with a spot size of 80 μm × 150 μm and a spectral resolution of ∼0.3 eV. Ephoton is varied to achieve depth resolution, by changing the kinetic energy of the emitted photoelectrons and thus their inelastic mean free paths, λescape. All spectra are background-corrected (Shirley) and analyzed by performing a nonlinear mean square fit of the data, using Doniach-Šùnjić functions convoluted with Gaussian profiles. All binding energies are referenced to contemperaneously measured Fermi edges. The extent of S-/FLG growth is confirmed ex situ on as-grown samples using scanning electron microscopy (SEM, Zeiss SigmaVP, 1–2 kV, in-lens detector) or after transfer of the S-/FLG films to SiO2 (300 nm)/Si substrates using optical microscopy and Raman spectroscopy (Renishaw Raman InVia Microscope, 532 nm excitation).
Acknowledgments
R.S.W. acknowledges a Research Fellowship from St. John’s College, Cambridge, and a Marie Skłodowska-Curie Individual Fellowship (Global) under Grant ARTIST (no. 656870) from the European Union’s Horizon 2020 research and innovation programme. L.D. and S.C. acknowledge EPSRC Doctoral Training Awards, and A.C.-V. acknowledges a Conacyt Cambridge Scholarship and the Roberto Rocca Fellowship. S.H. acknowledges funding from ERC Grant InsituNANO (no. 279342). This research was partially supported by the EUFP7 Work Programme under Grant GRAFOL (project reference 285275) and EPSRC under Grant GRAPHTED (project reference EP/K016636/1). We acknowledge the Helmholtz-Zentrum-Berlin Electron storage ring BESSY II for provision of synchrotron radiation at the ISISS beamline, and we thank the BESSY staff for ongoing support of our experiments. We are grateful to Bruno Dlubak for fruitful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b08729.
SE micrographs showing extents of graphene coverage for samples corresponding to Figures 1, 3, and 4 and XP spectra for FLG-covered Fe following >18 months of air exposure (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bunch J. S.; Verbridge S. S.; Alden J. S.; van der Zande A. M.; Parpia J. M.; Craighead H. G.; McEuen P. L. Nano Lett. 2008, 8, 2458–2462 10.1021/nl801457b. [DOI] [PubMed] [Google Scholar]
- Bohm S. Nat. Nanotechnol. 2014, 9, 741–742 10.1038/nnano.2014.220. [DOI] [PubMed] [Google Scholar]
- Weatherup R. S.; Dlubak B.; Hofmann S. ACS Nano 2012, 6, 9996–10003 10.1021/nn303674g. [DOI] [PubMed] [Google Scholar]
- Weatherup R. S.; Baehtz C.; Dlubak B.; Bayer B. C.; Kidambi P. R.; Blume R.; Schloegl R.; Hofmann S. Nano Lett. 2013, 13, 4624–4631 10.1021/nl401601x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caneva S.; Weatherup R. S.; Bayer B.; Brennan B.; Spencer S. J.; Mingard K.; Cabrero-Vilatela A.; Baehtz C.; Pollard A. J.; Hofmann S. Nano Lett. 2015, 15, 1867–1875 10.1021/nl5046632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann S.; Braeuninger-Weimer P.; Weatherup R. S. J. Phys. Chem. Lett. 2015, 6, 2714–2721 10.1021/acs.jpclett.5b01052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M.-B.; Dlubak B.; Weatherup R. S.; Piquemal-Banci M.; Yang H.; Blume R.; Schloegl R.; Collin S.; Petroff F.; Hofmann S.; Robertson J.; Anane A.; Fert A.; Seneor P. Appl. Phys. Lett. 2015, 107, 012408. 10.1063/1.4923401. [DOI] [Google Scholar]
- Dlubak B.; Martin M.-B.; Weatherup R. S.; Yang H.; Deranlot C.; Blume R.; Schloegl R.; Fert A.; Anane A.; Hofmann S.; Seneor P.; Robertson J. ACS Nano 2012, 6, 10930–10934 10.1021/nn304424x. [DOI] [PubMed] [Google Scholar]
- Martin M.-B.; Dlubak B.; Weatherup R. S.; Yang H.; Deranlot C.; Bouzehouane K.; Petroff F.; Anane A.; Hofmann S.; Robertson J.; Fert A.; Seneor P. ACS Nano 2014, 8, 7890–7895 10.1021/nn5017549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriver M.; Regan W.; Gannett W. J.; Zaniewski A. M.; Crommie M. F.; Zettl A. ACS Nano 2013, 7, 5763–5768 10.1021/nn4014356. [DOI] [PubMed] [Google Scholar]
- Zhou F.; Li Z.; Shenoy G. J.; Li L.; Liu H. ACS Nano 2013, 7, 6939–6947 10.1021/nn402150t. [DOI] [PubMed] [Google Scholar]
- Kidambi P. R.; Bayer B. C.; Blume R.; Wang Z.-J.; Baehtz C.; Weatherup R. S.; Willinger M.-G.; Schloegl R.; Hofmann S. Nano Lett. 2013, 13, 4769–4778 10.1021/nl4023572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y.; Bharathi M.; Wang L.; Liu Y.; Chen H.; Nie S.; Wang X.; Chou H.; Tan C.; Fallahazad B.; Ramanarayan H.; Magnuson C. W.; Tutuc E.; Yakobson B. I.; McCarty K. F.; Zhang Y.-W.; Kim P.; Hone J.; Colombo L.; Ruoff R. S. Science 2013, 342, 720–723 10.1126/science.1243879. [DOI] [PubMed] [Google Scholar]
- Petrone N.; Dean C. R.; Meric I.; van der Zande A. M.; Huang P. Y.; Wang L.; Muller D.; Shepard K. L.; Hone J. Nano Lett. 2012, 12, 2751–2756 10.1021/nl204481s. [DOI] [PubMed] [Google Scholar]
- Walker M. I.; Weatherup R. S.; Bell N. A. W.; Hofmann S.; Keyser U. F. Appl. Phys. Lett. 2015, 106, 023119. 10.1063/1.4906236. [DOI] [Google Scholar]
- Ashby M. F.; Jones D. R. H. Engineering Materials I: An Introduction to Properties, Applications, and Design 2012, 385–400 10.1016/B978-0-08-096665-6.00026-X. [DOI] [Google Scholar]
- Choi K.; Nam S.; Lee Y.; Lee M.; Jang J.; Kim S. J.; Jeong Y. J.; Kim H.; Bae S.; Yoo J.-B.; Cho S. M.; Choi J.-B.; Chung H. K.; Ahn J.-H.; Park C. E.; Hong B. H. ACS Nano 2015, 9, 5818–5824 10.1021/acsnano.5b01161. [DOI] [PubMed] [Google Scholar]
- Wirtz C.; Berner N. C.; Duesberg G. S. Adv. Mater. Interfaces 2015, 2, n/a. 10.1002/admi.201500082. [DOI] [Google Scholar]
- Hsieh Y.; Hofmann M.; Chang K.; Jhu J.; Li Y.-Y.; Chen K. Y.; Yang C. C.; Chang W.-S.; Chen L.-C. ACS Nano 2014, 8, 443–448 10.1021/nn404756q. [DOI] [PubMed] [Google Scholar]
- Burrows P. E.; Graff G. L.; Gross M. E.; Martin P. M.; Hall M.; Mast E.; Bonham C. C.; Bennett W. D.; Michalski L. A.; Weaver M. S.; Brown J. J.; Fogarty D.; Sapochak L. S. Proc. SPIE 2000, 4105, 75–83 10.1117/12.416878. [DOI] [Google Scholar]
- Patera L. L.; Africh C.; Weatherup R. S.; Blume R.; Bhardwaj S.; Castellarin-Cudia C.; Knop-Gericke A.; Schloegl R.; Comelli G.; Hofmann S.; Cepek C. ACS Nano 2013, 7, 7901–7912 10.1021/nn402927q. [DOI] [PubMed] [Google Scholar]
- Weatherup R. S.; Amara H.; Blume R.; Dlubak B.; Bayer B. C.; Diarra M.; Bahri M.; Cabrero-vilatela A.; Caneva S.; Kidambi P. R.; Martin M.; Deranlot C.; Seneor P.; Schloegl R.; Bichara C.; Hofmann S.; Ducastelle F.; Bichara C.; Hofmann S. J. Am. Chem. Soc. 2014, 136, 13698–13708 10.1021/ja505454v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherup R. S.; Bayer B. C.; Blume R.; Ducati C.; Baehtz C.; Schlögl R.; Hofmann S. Nano Lett. 2011, 11, 4154–4160 10.1021/nl202036y. [DOI] [PubMed] [Google Scholar]
- Weatherup R. S.; Bayer B. C.; Blume R.; Baehtz C.; Kidambi P. R.; Fouquet M.; Wirth C. T.; Schlögl R.; Hofmann S. ChemPhysChem 2012, 13, 2544–2549 10.1002/cphc.201101020. [DOI] [PubMed] [Google Scholar]
- Grosvenor A. P.; Biesinger M. C.; Smart R. S. C.; McIntyre N. S. Surf. Sci. 2006, 600, 1771–1779 10.1016/j.susc.2006.01.041. [DOI] [Google Scholar]
- Biesinger M. C.; Payne B. P.; Lau L. W. M.; Gerson A.; Smart R. S. C. Surf. Interface Anal. 2009, 41, 324–332 10.1002/sia.3026. [DOI] [Google Scholar]
- Holloway P. H. J. Vac. Sci. Technol. 1981, 18, 653–659 10.1116/1.570847. [DOI] [Google Scholar]
- Lambers E. S.; Dykstal C. N.; Seo J. M.; Rowe J. E.; Holloway P. H. Oxid. Met. 1996, 45, 301–321 10.1007/BF01046987. [DOI] [Google Scholar]
- Prasai D.; Tuberquia J. C.; Harl R. R.; Jennings G. K.; Bolotin K. I. ACS Nano 2012, 6, 1102–1108 10.1021/nn203507y. [DOI] [PubMed] [Google Scholar]
- Nagashima A.; Tejima N.; Oshima C. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50, 17487–17495 10.1103/PhysRevB.50.17487. [DOI] [PubMed] [Google Scholar]
- Oshima C.; Nagashima A. J. Phys.: Condens. Matter 1997, 9, 1–20 10.1088/0953-8984/9/1/004. [DOI] [Google Scholar]
- Varykhalov A.; Sánchez-Barriga J.; Shikin A.; Biswas C.; Vescovo E.; Rybkin A.; Marchenko D.; Rader O. Phys. Rev. Lett. 2008, 101, 157601. 10.1103/PhysRevLett.101.157601. [DOI] [PubMed] [Google Scholar]
- Voloshina E.; Dedkov Y. Phys. Chem. Chem. Phys. 2012, 14, 13502–13514 10.1039/c2cp42171b. [DOI] [PubMed] [Google Scholar]
- Eom D.; Prezzi D.; Rim K. T.; Zhou H.; Lefenfeld M.; Xiao S.; Nuckolls C.; Hybertsen M. S.; Heinz T. F.; Flynn G. W. Nano Lett. 2009, 9, 2844–2848 10.1021/nl900927f. [DOI] [PubMed] [Google Scholar]
- Varykhalov A.; Rader O. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 80, 035437. 10.1103/PhysRevB.80.035437. [DOI] [Google Scholar]
- Vinogradov N. A.; Zakharov A. A.; Kocevski V.; Rusz J.; Simonov K. A.; Eriksson O.; Mikkelsen A.; Lundgren E.; Vinogradov A. S.; Mårtensson N.; Preobrajenski A. B. Phys. Rev. Lett. 2012, 109, 026101. 10.1103/PhysRevLett.109.026101. [DOI] [PubMed] [Google Scholar]
- Brugger T.; Günther S.; Wang B.; Dil J. H.; Bocquet M. L.; Osterwalder J.; Wintterlin J.; Greber T. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 79, 045407. 10.1103/PhysRevB.79.045407. [DOI] [Google Scholar]
- Enderlein C.; Kim Y. S.; Bostwick A.; Rotenberg E.; Horn K. New J. Phys. 2010, 12, 033014. 10.1088/1367-2630/12/3/033014. [DOI] [Google Scholar]
- Sutter P.; Sadowski J. T.; Sutter E. A. J. Am. Chem. Soc. 2010, 132, 8175–8179 10.1021/ja102398n. [DOI] [PubMed] [Google Scholar]
- Preobrajenski A.; Ng M.; Vinogradov A.; Mårtensson N. Phys. Rev. B: Condens. Matter Mater. Phys. 2008, 78, 073401. 10.1103/PhysRevB.78.073401. [DOI] [Google Scholar]
- Kwon S. Y.; Ciobanu C. V.; Petrova V.; Shenoy V. B.; Bareño J.; Gambin V.; Petrov I.; Kodambaka S. Nano Lett. 2009, 9, 3985–3990 10.1021/nl902140j. [DOI] [PubMed] [Google Scholar]
- Shikin A. M.; Adamchuk V. K.; Rieder K.-H. Phys. Solid State 2009, 51, 2390–2400 10.1134/S1063783409110316. [DOI] [Google Scholar]
- Walter A. L.; Nie S.; Bostwick A.; Kim K. S.; Moreschini L.; Chang Y. J.; Innocenti D.; Horn K.; McCarty K. F.; Rotenberg E. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 84, 195443. 10.1103/PhysRevB.84.195443. [DOI] [Google Scholar]
- Wilson N. R.; Marsden A. J.; Saghir M.; Bromley C. J.; Schaub R.; Costantini G.; White T. W.; Partridge C.; Barinov A.; Dudin P.; Sanchez A. M.; Mudd J. J.; Walker M.; Bell G. R. Nano Res. 2013, 6, 99–112 10.1007/s12274-013-0285-y. [DOI] [Google Scholar]
- Nie S.; Walter A. L.; Bartelt N. C.; Starodub E.; Bostwick A.; Rotenberg E.; McCarty K. F. ACS Nano 2011, 5, 2298–2306 10.1021/nn103582g. [DOI] [PubMed] [Google Scholar]
- Sutter P.; Sadowski J. T.; Sutter E. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 80, 245411. 10.1103/PhysRevB.80.245411. [DOI] [Google Scholar]
- Dahal A.; Batzill M. Nanoscale 2014, 6, 2548–2562 10.1039/c3nr05279f. [DOI] [PubMed] [Google Scholar]
- Shikin A.; Prudnikova G.; Adamchuk V.; Moresco F.; Rieder K.-H. Phys. Rev. B: Condens. Matter Mater. Phys. 2000, 62, 13202–13208 10.1103/PhysRevB.62.13202. [DOI] [Google Scholar]
- D’Arsié L.; Esconjauregui S.; Weatherup R.; Guo Y.; Bhardwaj S.; Centeno A.; Zurutuza A.; Cepek C.; Robertson J. Appl. Phys. Lett. 2014, 105, 103103. 10.1063/1.4895025. [DOI] [Google Scholar]
- Blume R.; Kidambi P. R.; Bayer B. C.; Weatherup R. S.; Wang Z.-J.; Weinberg G.; Willinger M.-G.; Greiner M.; Hofmann S.; Knop-Gericke A.; Schlögl R. Phys. Chem. Chem. Phys. 2014, 16, 25989–26003 10.1039/C4CP04025B. [DOI] [PubMed] [Google Scholar]
- Koch R. J.; Weser M.; Zhao W.; Viñes F.; Gotterbarm K.; Kozlov S. M.; Höfert O.; Ostler M.; Papp C.; Gebhardt J.; Steinrück H. P.; Görling A.; Seyller T. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 86, 075401. 10.1103/PhysRevB.86.075401. [DOI] [Google Scholar]
- Blume R.; Rosenthal D.; Tessonnier J.-P.; Li H.; Knop-Gericke A.; Schlögl R. ChemCatChem 2015, 7, 2871–2881 10.1002/cctc.201500344. [DOI] [Google Scholar]
- Viñes F.; Neyman K.; Görling A. J. Phys. Chem. A 2009, 113, 11963–11973 10.1021/jp903653z. [DOI] [PubMed] [Google Scholar]
- Parkinson C. R.; Walker M.; McConville C. F. Surf. Sci. 2003, 545, 19–33 10.1016/j.susc.2003.08.029. [DOI] [Google Scholar]
- Cazaux J. Appl. Surf. Sci. 2010, 257, 1002–1009 10.1016/j.apsusc.2010.08.007. [DOI] [Google Scholar]
- Tompkins H. G.; Augis J. a. Oxid. Met. 1981, 16, 355–369 10.1007/BF00611349. [DOI] [Google Scholar]
- Welsh I. D.; Sherwood P. M. a. Phys. Rev. B: Condens. Matter Mater. Phys. 1989, 40, 6386–6392 10.1103/PhysRevB.40.6386. [DOI] [PubMed] [Google Scholar]
- Grosvenor A. P.; Kobe B. A.; McIntyre N. S. Surf. Sci. 2004, 572, 217–227 10.1016/j.susc.2004.08.035. [DOI] [Google Scholar]
- Grosvenor A. P.; Kobe B. A.; McIntyre N. S. Surf. Sci. 2004, 565, 151–162 10.1016/j.susc.2004.06.210. [DOI] [Google Scholar]
- Pu N.-W.; Shi G.-N.; Liu Y.-M.; Sun X.; Chang J.-K.; Sun C.-L.; Ger M.-D.; Chen C.-Y.; Wang P.-C.; Peng Y.-Y.; Wu C.-H.; Lawes S. J. Power Sources 2015, 282, 248–256 10.1016/j.jpowsour.2015.02.055. [DOI] [Google Scholar]
- Stoot A. C.; Camilli L.; Spiegelhauer S.-A.; Yu F.; Bøggild P. J. Power Sources 2015, 293, 846–851 10.1016/j.jpowsour.2015.06.009. [DOI] [Google Scholar]
- Kravets V. G.; Jalil R.; Kim Y.-J.; Ansell D.; Aznakayeva D. E.; Thackray B.; Britnell L.; Belle B. D.; Withers F.; Radko I. P.; Han Z.; Bozhevolnyi S. I.; Novoselov K. S.; Geim A. K.; Grigorenko A. N. Sci. Rep. 2014, 4, 5517. 10.1038/srep05517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter E.; Albrecht P.; Camino F. E.; Sutter P. Carbon 2010, 48, 4414–4420 10.1016/j.carbon.2010.07.058. [DOI] [Google Scholar]
- Nayak P. K.; Hsu C. J.; Wang S. C.; Sung J. C.; Huang J. L. Thin Solid Films 2013, 529, 312–316 10.1016/j.tsf.2012.03.067. [DOI] [Google Scholar]
- Dedkov Y. S.; Fonin M.; Rüdiger U.; Laubschat C. Appl. Phys. Lett. 2008, 93, 022509. 10.1063/1.2953972. [DOI] [PubMed] [Google Scholar]
- Chen S.; Brown L.; Levendorf M.; Cai W.; Ju S.-Y.; Edgeworth J.; Li X.; Magnuson C. W.; Velamakanni A.; Piner R. D.; Kang J.; Park J.; Ruoff R. S. ACS Nano 2011, 5, 1321–1327 10.1021/nn103028d. [DOI] [PubMed] [Google Scholar]
- Grånäs E.; Knudsen J.; Schröder U. a; Gerber T.; Busse C.; Arman M. a; Schulte K.; Andersen J. N.; Michely T. ACS Nano 2012, 6, 9951–9963 10.1021/nn303548z. [DOI] [PubMed] [Google Scholar]
- Larciprete R.; Ulstrup S.; Lacovig P.; Dalmiglio M.; Bianchi M.; Mazzola F.; Hornekær L.; Orlando F.; Baraldi A.; Hofmann P.; Lizzit S. ACS Nano 2012, 6, 9551–9558 10.1021/nn302729j. [DOI] [PubMed] [Google Scholar]
- Kirkland N. T.; Schiller T.; Medhekar N.; Birbilis N. Corros. Sci. 2012, 56, 1–4 10.1016/j.corsci.2011.12.003. [DOI] [Google Scholar]
- Kidambi P. R.; Blume R.; Kling J.; Wagner J. B.; Baehtz C.; Weatherup R. S.; Schloegl R.; Bayer B. C.; Hofmann S. Chem. Mater. 2014, 26, 6380–6392 10.1021/cm502603n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mišković-Stanković V.; Jevremović I.; Jung I.; Rhee K. Carbon 2014, 75, 335–344 10.1016/j.carbon.2014.04.012. [DOI] [Google Scholar]
- Su Y.; Kravets V. G.; Wong S. L.; Waters J.; Geim A. K.; Nair R. R. Nat. Commun. 2014, 5, 4843. 10.1038/ncomms5843. [DOI] [PubMed] [Google Scholar]
- Kang D.; Kwon J. Y.; Cho H.; Sim J.; Hwang H. S.; Kim C. S.; Kim Y. J.; Ruoff R. S.; Shin H. S. ACS Nano 2012, 6, 7763–7769 10.1021/nn3017316. [DOI] [PubMed] [Google Scholar]
- Dumée L. F.; He L.; Wang Z.; Sheath P.; Xiong J.; Feng C.; Tan M. Y.; She F.; Duke M.; Gray S.; Pacheco A.; Hodgson P.; Majumder M.; Kong L. Carbon 2015, 87, 395–408 10.1016/j.carbon.2015.02.042. [DOI] [Google Scholar]
- Coraux J.; N’Diaye A. T.; Rougemaille N.; Vo-Van C.; Kimouche A.; Yang H. X.; Chshiev M.; Bendiab N.; Fruchart O.; Schmid A. K. J. Phys. Chem. Lett. 2012, 3, 2059–2063 10.1021/jz3007222. [DOI] [Google Scholar]
- Bluhm H.; Hävecker M.; Knop-Gericke A.; Kiskinova M.; Schlögl R.; Salmeron M. MRS Bull. 2007, 32, 1022–1030 10.1557/mrs2007.211. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.