

Abstract
Degeneration of nigrostriatal dopaminergic system is the principal lesion in Parkinson’s disease. Because glial cell line-derived neurotrophic factor (GDNF) promotes survival of dopamine neurons in vitro and in vivo, intracranial delivery of GDNF has been attempted for Parkinson’s disease treatment but with variable success. For improving GDNF-based therapies, knowledge on physiological role of endogenous GDNF at the sites of its expression is important. However, due to limitations of existing genetic model systems, such knowledge is scarce. Here, we report that prevention of transcription of Gdnf 3’UTR in Gdnf endogenous locus yields GDNF hypermorphic mice with increased, but spatially unchanged GDNF expression, enabling analysis of postnatal GDNF function. We found that increased level of GDNF in the central nervous system increases the number of adult dopamine neurons in the substantia nigra pars compacta and the number of dopaminergic terminals in the dorsal striatum. At the functional level, GDNF levels increased striatal tissue dopamine levels and augmented striatal dopamine release and re-uptake. In a proteasome inhibitor lactacystin-induced model of Parkinson’s disease GDNF hypermorphic mice were protected from the reduction in striatal dopamine and failure of dopaminergic system function. Importantly, adverse phenotypic effects associated with spatially unregulated GDNF applications were not observed. Enhanced GDNF levels up-regulated striatal dopamine transporter activity by at least five fold resulting in enhanced susceptibility to 6-OHDA, a toxin transported into dopamine neurons by DAT. Further, we report how GDNF levels regulate kidney development and identify microRNAs miR-9, miR-96, miR-133, and miR-146a as negative regulators of GDNF expression via interaction with Gdnf 3’UTR in vitro. Our results reveal the role of GDNF in nigrostriatal dopamine system postnatal development and adult function, and highlight the importance of correct spatial expression of GDNF. Furthermore, our results suggest that 3’UTR targeting may constitute a useful tool in analyzing gene function.
Author Summary
Intracranial delivery of GDNF has been attempted for Parkinson’s disease (PD) treatment but with variable success. For improving GDNF-based therapies, knowledge on physiological role of endogenous GDNF at the sites of its expression is important. However, due to limitations of existing genetic model systems, such knowledge is scarce. Here, we utilize an innovative genetic approach by targeting the 3’UTR regulation of Gdnf in mice. Such animals express elevated levels of Gdnf exclusively in natively Gdnf-expressing cells, enabling dissection of endogenous GDNF functions in vivo. We show that endogenous GDNF regulates dopamine system development and function and protects mice in a rodent PD model without side effects associated with ectopic GDNF applications. Further, we report how GDNF levels regulate kidney development and identify microRNAs which control GDNF expression. Our study highlights the importance of correct spatial expression of GDNF and opens a novel approach to study gene function in mice.
Introduction
Exogenously applied glial cell line-derived neurotrophic factor (GDNF) promotes the survival, function, and neurite growth of nigrostriatal dopamine (DA) neurons both in vitro and in vivo [1,2]. The classic motor deficit in Parkinson’s disease is characterized by a gradual loss of nigrostriatal DA neurons, leading to a reduction in striatal dopamine levels, resting tremor, rigidity, and an inability to initiate voluntary movement [3]. Intracranial delivery of GDNF has been tested in clinical trials for treating Parkinson’s disease (PD); however, both the efficacy and the side effects of this treatment vary widely [3–6]. Increasing the therapeutic efficacy of GDNF requires a better understanding of its physiological role; however, our knowledge regarding the postnatal role of GDNF is currently limited. Knockout mice that lack Gdnf or its receptors (Gfrα1 and Ret) die at birth due to a lack of kidneys, but with intact nigrostriatal DA system which undergoes developmental maturation during the first three post-natal weeks [7,8]. It has been reported that 50% reduction in GDNF levels in adult Gdnf conditional knock-out mice has profound consequences on midbrain dopamine neuron survival upon aging [9]. However, our recent study with Gdnf conditional knock-out mice utilizing three Cre systems including the repetition of the experiments performed in [9] did not reveal loss of DA neurons after GDNF deletion or reduction at any age [10]. Based on current evidence it is possible that GDNF either has no physiological role in the brain DA system, that GDNF reduction or deletion in the brain is compensated by another mechanism, or that GDNF regulates the DA system at the functional level, rather than at the level of supporting the survival of the DA cell bodies in the midbrain. Moreover, although GDNF is known to be essential for initiating kidney development [7], our understanding of the role of endogenous GDNF in kidney maturation has remained limited.
Here, we report generation and analysis of mice carrying Gdnf hypermorphic (Gdnf hyper) allele, generated by insertion of a cassette containing bovine growth hormone polyA signal after the stop-codon in Gdnf endogenous locus preventing transcription into Gdnf wild type 3’UTR. These mice have increased–but spatially unchanged–expression of the endogenous Gdnf gene. While Gdnf hyper/hyper mice die by postnatal day 18 (P18) due to kidney defects, Gdnf wt/hyper mice are healthy and only display mild occasional reduction in kidney size. Gdnf wt/hyper animals revealed that GDNF has an important role in the postnatal nigrostriatal system development and adult function and clarified which aspects of the nigrostriatal dopaminergic system structure and function are regulated by GDNF. They also enabled analysis of GDNF function in kidney maturation beyond the first steps in renal development.
Results
Generation of Gdnf hypermorphic allele and in vitro analysis
In the process of generating a conditional knockout (or “floxed”) Gdnf allele [10], we noted that the 3’UTR of Gdnf is relatively long and evolutionarily conserved (Fig 1A). Since Gdnf 3’UTR inhibits reporter gene expression in a cell line [11] we decided to analyze Gdnf 3’UTR function in vivo by insertion of an FRT-flanked puΔtk cassette [12] after the stop codon in the Gdnf locus in embryonic stem (ES) cells. The puΔtk cassette contains the bovine growth hormone polyadenylation (bGHpA) signal, which induces termination of transcription and is commonly used in gene-trap experiments in mice (Fig 1B). We used a luciferase-based reporter assay to confirm that the bGHpA signal prevents transcription into the Gdnf 3’UTR in our construct (S1A and S1B Fig) and yields correctly sized fusion mRNA (S1C Fig). Using a reporter gene assay, we found an 8-fold increase in luciferase expression from the construct containing Firefly-puΔtk proceeded by Gdnf 3’UTR (relative to Firefly-Gdnf 3’UTR) in a cell line derived from human embryonic kidney cells (HEK293) and a 2-fold increase in a cell line derived from human brain cells (U87) (S1D Fig). We also observed similar inhibitory effects on reporter gene expression, regardless of whether the Gdnf 3’UTR was cloned downstream of a sea pansy (Renilla reniformis) or Photinini firefly (Photinus pyralis) luciferase in both cell lines (S1E Fig), suggesting that the inhibition of expression by Gdnf 3’UTR is not limited to one cell type or dependent on the preceding gene. Blocking transcription with actinomycin D revealed that the Firefly-puΔtk yields a more stable gene product than Firefly-Gdnf 3’UTR, suggesting that negative regulation via Gdnf 3’UTR occurs at the post-transcriptional level (S1F Fig).
Fig 1. Gdnf 3’UTR and generation of GDNF hypermorphic allele.
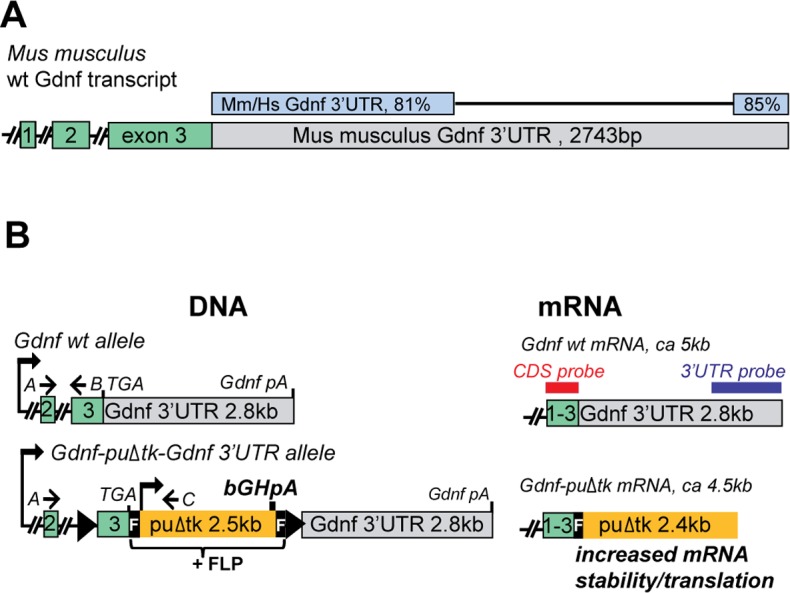
(A) The 3’UTR of the Gdnf mRNA is evolutionarily conserved. Exons and the 3’UTR are drawn to scale. Percent of identity between the human (Hs) and mouse (Mm) sequences is indicated. Source: Blast. (B) Schematic representation of the Gdnf hypermorphic allele. A and C designate the primers used for Gdnf mRNA sequence analysis in targeted mice (see S2F and S2G Fig); primers A and B were used for Gdnf QPCR. The bGHpA, puΔtk, loxP sites (black triangles), and F-FRT sites are indicated. The blue and red bars indicate the probes used for in situ hybridization.
Next, we generated mice carrying the Gdnf-FRT-puΔtk-FRT-Gdnf 3’UTR allele (S1G–S1I Fig). Based on the above experiments and a previously published study [11] the allele was hypothesized to result in elevated expression of endogenous Gdnf (Fig 1B, S1D–S1F Fig). Homozygous Gdnf puΔtk/puΔtk mice died before P18 due to extremely small, morphologically disorganized kidneys (see below). In contrast, kidney defects were mild (or absent) in heterozygous mice (S1 Table), which were born at the expected Mendelian frequency (S2 Table) and appeared otherwise phenotypically normal (see below).
Analysis of Gdnf mRNA expression levels and sites in peripheral tissues of Gdnf puΔtk mice
We first measured the location and levels of Gdnf expression in the developing kidney, testis and developing hind limb where the expression sites of Gdnf are well established [13,14], http://developingmouse.brain-map.org. Because antibodies that selectively and specifically bind endogenous GDNF protein in histology sections are not currently available, we used in situ hybridization to detect Gdnf mRNA. During embryonic kidney development, the expression of Gdnf is limited to a structure of metanephric mesenchyme called cap condensate, which surrounds a Gdnf-negative epithelial ureteric bud (Fig 2A). A probe spanning the Gdnf-coding sequence (comprising exons 1 through 3) revealed a Gdnf puΔtk allelic dose-dependent increase in Gdnf mRNA levels, but no difference in the site of expression between wild-type, heterozygous, and homozygous mice (Fig 2B, upper panel). Thus we designated the Gdnf puΔtk allele as Gdnf hypermorphic or Gdnf hyper allele. Further analysis revealed that a probe complementary to 525 bp of the 3’ end of the Gdnf 3’UTR revealed fewer Gdnf 3’UTR-containing transcripts in Gdnf wt/hyper mice compared to Gdnf w/wt mice. As expected, no signal was detected in Gdnf hyper/hyper mice using this probe (Fig 2B, lower panel), suggesting that transcription of Gdnf wt 3’UTR is prevented by the puΔtk cassette, as it did in cell line (S1B Fig). A similar increase in Gdnf expression was measured in Sertoli cells (in the testes) and in the developing hind limbs (S2A and S2B Fig). Quantification of Gdnf mRNA and GDNF protein levels in the developing kidneys and testes also revealed a Gdnf hyper allelic dose-dependent increase in both organs (Fig 2C–2E, S2C and S2D Fig), and removal of the puΔtk cassette by crossing with Deleter-FLP mice (Fig 1B; “Gdnf 3’UTR rest/rest mice”) restored GDNF expression to wild-type levels (Fig 2E, S2C and S2D Fig). Thus, we concluded that the Gdnf hyper allele indeed resulted in elevated expression of GDNF in peripheral tissues.
Fig 2. Gdnf levels are a critical determinant of embryonic renal growth and morphogenesis.

(A) Schematic representation of Gdnf expression (blue) in the mouse kidney at E11.5. (B) Representative image of in situ hybridization of Gdnf mRNA (blue) in the urogenital tract of E11.5 mice. N = 4 mice/group. (C-E) Gdnf mRNA [E14.5 (C) and E18.5 (D)] and protein [E18.5; (E)] levels in the kidney measured using QPCR (C and D) and ELISA (E). N = 2–10 mice/group. (F) Representative image of kidneys obtained from P7.5 mice. (G) Representative image of hematoxylin-and-eosin‒stained sections from E18.5 kidneys. The renal cortex is indicated with a yellow bar, the medulla is indicated with a green bar, and collecting duct cysts are indicated with yellow arrowheads. (H) At the time of renal differentiation initiation (E11.75), a wild-type kidney (left) contains a typical UB branching pattern with an interim stalk (red arrowhead), elongated ureter stalk (white arrowhead), and locally enlarged UB tips (arrows). In contrast, a kidney from a Gdnf hyper/hyper embryo (right) contains one large UB that appears bumpy (arrows), lacks an interim stalk, and lacks normal elongation of the UB (white arrowhead). (I) Images of a wild-type (left) and Gdnf hyper/hyper (right) kidney at E13.5; the kidney from the Gdnf hyper/hyper embryo is smaller in size, has enlarged ureteric buds (arrows), and shortened stalks (arrow head). For F-I, N = 3–20 mice/group. Scale bars: B, 10 μm; F, 1 mm; G, 300 μm H, 50 μm; I, 100 μm. Abbreviations: E, embryonic day; MM, metanephric mesenchyme; P, postnatal day; UB, ureteric bud. In this and subsequent figures, all summary data are presented as the mean ± SEM; *P<0.05, **P<0.01, and ***P<0.001; Student’s t-test, unless noted otherwise.
To further analyze the Gdnf transcript in Gdnf hyper mice we applied Northern blotting to analyze Gdnf mRNA in testis. We observed a major band at the expected size of the predicted Gdnf-puΔtk fusion transcript (S2E Fig). We also sequenced the Gdnf transcripts from testis in Gdnf wt/wt, Gdnf hyper/hyper, and Gdnf 3’UTR rest/rest mice using RT-PCR. We used a forward primer spanning Gdnf exon 2 and a reverse primer spanning puΔtk (starting 428 bp downstream from the TGA stop codon of Gdnf) (S2F and S2G Fig); our analysis revealed a Gdnf-puΔtk fusion transcript with the predicted size and sequence (S2G Fig).
Kidney size and morphology are negatively regulated by excess GDNF levels
Ureteric bud outgrowth–the first step in kidney development–is induced by GDNF [7]. Thus, homozygous Gdnf-knockout mice lack kidneys, and 20–30% of heterozygous GDNF-knockout mice have only one kidney [15]. In vitro, exogenous GDNF promotes the growth of ectopic ureteric buds in embryonic tissue explants and expansion of endogenous ureteric buds tips [16]. However, how endogenous GDNF regulates subsequent steps in kidney development in vivo is poorly understood. In vitro GDNF protein application studies [16] and results from the Gdnf knockout mice [15] suggest that the overexpression of Gdnf in our hypermorphic mice might cause enlargement of the kidneys. In contrast, we found that overexpression of GDNF negatively regulates kidney size and morphological maturation (Fig 2F and 2G; S1 Table) in a concentration-dependent manner (Fig 2B–2E). An analysis of the early steps in kidney development (i.e., in E11.5 through E11.75) in Gdnf hyper/hyper mice revealed that excess GDNF (Fig 2B, upper panel) induces hypertrophic ureteric bud formation and impairs the development and elongation of the stalk (Fig 2H). At mid-gestation (i.e., E13.5), these enlarged ureteric buds and absent or shortened stalks are still observed, and the initial signs of reduced kidney size emerge (Fig 2I). During late embryogenesis, the kidneys in Gdnf hyper/hyper mice fail to reach normal size, resulting in severe renal hypodysplasia with disorganized medulla-cortex compartmentalization, reduced cortical and medullar areas, and cysts in the collecting ducts (Fig 2G). An analysis of kidney function by measuring serum electrolytes revealed that the kidneys of Gdnf hyper/hyper mice function poorly (S2H and S2I Fig). However, in Gdnf wt/hyper mice the kidney function remains relatively normal (S2H–S2K Fig). Finally, renal development was restored by crossing with Deleter-FLP mice (Gdnf 3’UTR rest/rest; S2L Fig). These data indicate that in kidney development, the correct GDNF levels are important as both lack of GDNF and excess GDNF result in failure of kidney development and function. Importantly, in Gdnf wt/hyper mouse the kidneys were functional and mice were healthy, providing us a unique animal model to study the postnatal function of GDNF in the CNS.
Analysis of Gdnf mRNA expression levels and sites in the CNS of Gdnf hyper mice
Next, we set to analyze the site of Gdnf expression in the CNS of GDNF hypermorphic mice. We utilized in situ hybridization analysis of Gdnf mRNA in thalamic nuclei and spinal cord and observed identical spatial expression patterns between genotypes (Fig 3A and 3B). In the striatum, where Gdnf mRNA levels peak at P12.5 [17,18], the spatial pattern of Gdnf expression was limited to a discrete set of sparsely distributed cells, interspaced with Gdnf nonexpressing cells, consistent with previous reports [17,19] and similar between Gdnf wt/hyper and Gdnf wt/wt mice (Fig 3C). In the striatum, Gdnf is expressed primarily by parvalbumin (Pvalb)—expressing inhibitory neurons [17]. We therefore used RNAscope, a novel high-sensitivity in situ hybridization method for double-staining Pvalb and Gdnf mRNAs in histological sections [20]. First, we verified the specificity of RNAscope probes for Pvalb and Gdnf mRNA in the adult cerebellum and E14.5 kidney (S2M Fig), where the expression of Pvalb and Gdnf, respectively, has been well characterized. Pvalb/Gdnf double-staining of the striatum of adult mice confirmed that the expression of Gdnf in Gdnf wt/hyper mice is largely retained to Pvalb-expressing cells, similar to wild-type controls (Fig 3D and 3E).
Fig 3. Gdnf expression is increased in cells that normally express Gdnf.

(A) In situ hybridization showing Gdnf mRNA expression in thalamic nuclei at P7.5 (white arrowheads). (B) In situ hybridization showing Gdnf mRNA expression in Clarke’s column in the thoracic part of the spinal cord at P12.5 (white arrowheads). (C) Gdnf mRNA-positive cells (black dots) in the whole striatum (upper panel); a magnified view of Gdnf mRNA-positive cells (arrowheads) is shown in the lower panel. (D) Representative images of Pvalb (PV, blue) and Gdnf (red) mRNA in the striatum of 3-month-old mice detected using RNAscope. (E) Summary of PV-positive only, Gdnf-positive only, and double-positive cells in striatal slices obtained from Gdnf wt/wt and Gdnf wt/hyper mice; N = 5 animals/group. (F) QPCR analysis of Gdnf mRNA levels in the indicated brain regions in P7.5 mice; N = 6–8 mice/group. (G) QPCR analysis of Gdnf mRNA levels in the indicated brain regions of adult mice; N = 4–8 mice/group. (H) ELISA analysis of GDNF protein levels in the dorsal striatum of adult mice; N = 6–8 mice/group. Scale bars: A, 150 μm; B, 200 μm; C (upper panels), 500 μm; C (lower panels), 50 μm; D, 10 μm. Abbreviations: dSTR, dorsal striatum; OB, olfactory bulb; Hyp, hypothalamus; PFC, prefrontal cortex; vSTR, ventral striatum; Hip, hippocampus; vMB, ventral midbrain; CB, cerebellum; RN, dorsal raphe nucleus; SC, spinal cord; SN, substantia nigra; VTA, ventral tegmental area; PV, parvalbumin; m, months; P, postnatal day.
To assess the level of Gdnf mRNA in several CNS regions including dorsal striatum, olfactory bulb, hypothalamus, ventral striatum, ventral midbrain and cerebellum in P7.5 Gdnf wt/hyper and Gdnf hyper/hyper animals and in 2.5–3 month old adult Gdnf wt/hyper mice we utilized quantitative RT-PCR. We observed an increase of Gdnf mRNA in Gdnf wt/hyper and Gdnf hyper/hyper mice in comparison to wild-type littermate controls (Fig 3F and 3G). To examine whether the increase in Gdnf mRNA level is reflected in the GDNF protein levels we used ELISA assay and noted that GDNF protein levels were increased by approximately two fold in the dorsal striatum of adult Gdnf wt/hyper mice (Fig 3H). Notably, we observed no elevation in Gdnf mRNA levels in the substantia nigra of adult Gdnf wt/hyper mice (Fig 3G). We conclude that elevation in Gdnf mRNA expression in Gdnf hyper mice occurs in most brain structures which naturally express Gdnf in the brain, and that Gdnf mRNA and protein levels in the dorsal striatum of adult Gdnf wt/hyper mice are two fold increased.
Increased GDNF levels increase the nigrostriatal dopaminergic system during development
In the first three postnatal weeks, DA neurons experience several major developmental changes, including maturation of striatal target innervation and programmed cell death in the substantia nigra pars compacta [7,8]. The postnatal function of endogenous GDNF in the brain’s dopaminergic system is poorly understood [10]. To assess whether increased GDNF levels in the GDNF hypermorphic mice result in changes in nigrostriatal DA system during development, we first analyzed the striatal levels of phosphorylated extracellular signal-regulated kinase (Erk) a known target in GDNF signaling [7] using western blotting. We found that the levels of phosphorylated ERK2 were increased in the striatum of Gdnf wt/hyper and Gdnf hyper/hyper mice at P7.5 (Fig 4A), indicating increased GDNF signaling. Next, we analyzed rostral brain DA levels and the number of DA neurons in the substantia nigra pars compacta. We found that at P7.5, DA levels in the rostral brain were increased by 25% to a similar extent in both Gdnf wt/hyper and Gdnf hyper/hyper mice, and normalizing Gdnf levels by crossing the mice with Deleter-FLP animals restored DA to wild-type levels (Fig 4B). Compared to wt mice, the number of DA neurons in the substantia nigra pars compacta revealed a similar 15% increase in Gdnf wt/hyper and Gdnf hyper/hyper animals at P7.5 (Fig 4C). Finally, the levels of DA metabolites in the rostral brain were similar between genotypes at P7.5 (S3A and S3B Fig). The development of the DA system was not linked with kidney development, as kidney function was severely impaired in the Gdnf hyper/hyper mice, but not in the Gdnf wt/hyper mice while the DA system parameters were comparable between the Gdnf wt/hyper and Gdnf hyper/hyper mice. This finding was further supported by the lack of correlation between serum urea and brain DA levels in individual heterozygous animals at P7.5 (S3 Table). We conclude that increased GDNF levels increase striatal DA levels and DA cell number in the substantia nigra pars compacta at P7.5.
Fig 4. Increased endogenous GDNF expression affects the development and function of the nigrostriatal dopaminergic system.

(A) Levels of phosphorylated ERK2 at P7.5 in the striatum of Gdnf wt/wt, Gdnf wt/hyper and Gdnf hyper/hyper mice. N = 5 mice/group; ERK was used for normalization. (B) HPLC analysis of DA levels in the rostral brain; N = 5–8 mice/group (F = 7.44, P = 0.016). (C) Quantification of tyrosine hydroxylase (TH)-positive (a marker of DA neurons) cells in the SNpc; N = 6–8 mice/group (F = 7.44, P = 0.0048). (D) HPLC analysis of DA levels in the dSTR; N = 11 for Gdnf wt/wt, 8 for Gdnf wt/hyper mice/group (P = 0.000164). HPLC analysis of DA levels in the dorsal striatum of Gdnf 3’UTR wt/wt and Gdnf wt/KO mice; N = 6 mice/group. (E-F) The number of TH-positive (E; N = 8 Gdnf wt/wt, N = 7 Gdnf wt/hyper; P = 0.025) and VMAT2-positive neurons (F; N = 7 Gdnf wt/wt, N = 7 Gdnf wt/hyper; P = 0.016) in the SNpc. (G) The number of DAT+ varicosities (N = 9 Gdnf wt/wt, N = 7 Gdnf wt/hyper; P = 0.042) in the dSTR. (H-K) Cyclic voltammetry analysis of acute striatal slices (see also S3J Fig); N = 5–7 mice/group with 1–3 slices per mouse. (H) DA release in response to electrical stimulation [two-way repeated measures ANOVA, F (1,29) = 5.866]; (I) Averaged traces of DA events. (J) Short-term depression of striatal DA release after prior DA exocytosis, shown as percent of the first DA release. (K) The ratio of DA release after a single stimulus and after a 5 pulse burst at 20Hz. (L) In vivo amperometry following intrastriatal DA injection reveals that dopamine transporter (DAT) activity in Gdnf wt/hyper mice is dependent on the concentration of DA; N = 4 mice/group (F = 47.931). (M) Locomotor activity after an injection of amphetamine (1 mg/kg, i.p.); N = 9–10 mice/group (F = 4.386, P = 0.04). (N) In vivo microdialysis analysis of extracellular striatal DA levels; amphetamine was applied as indicated by the horizontal bar; N = 9 mice/group. (O) Cyclic voltammetry analysis shows that amphetamine (5 μM) depletes stimulated DA release faster in the striata of Gdnf wt/hyper mice compared to Gdnf wt/wt mice; two-way repeated-measures ANOVA reveals an effect of time (P<0.0001) and genotype (P = 0.031), as well as an interaction between time and genotype (P = 0.049); N = 6 mice/group with 1–3 slices per mouse. (P-Q) Analysis of a 6-OHDA induced PD model. (P) Quantification of DA in the dSTR 2 weeks after striatal 6-OHDA injection, relative to the intact side (N = 12 Gdnf wt/wt; N = 10 Gdnf wt/hyper), (F = 40.62, P = 0.00549, Students t-test). The intact and lesioned side differed significantly (P = 2.71×10−15). (Q) Quantification of TH-positive neurons in the SNpc 2 weeks after striatal 6-OHDA injection, relative to the intact side, (F = 7.04, P = 0.0143, Students t-test). The intact and lesioned side differed significantly (P = 3.00×10−11). (R-T) Analysis of a lactacystin-induced PD model. (R) The percentage of sugar pellet retrievals from the contralateral side in the corridor test; N = 5–7 mice/group (F = 6.087, P = 0.033). (S) Quantification of DA, DOPAC, and HVA in the dSTR 5 weeks after supranigral lactacystin injection, relative to the intact side; N = 5 Gdnf wt/wt, N = 7 Gdnf wt/hyper; P = 0.046 for DA, P = 0.015 for DOPAC, P = 0.011 for HVA. The intact and lesioned side differed significantly; P = 0.00016 for DA, P = 0.015 for DOPAC, P = 0.010 for HVA. (T) Quantification of TH-positive neurons in the SNpc 5 weeks after lactacystin injection, relative to the intact side; N = 4 Gdnf wt/wt, N = 7 Gdnf wt/hyper; P = 0.236. The intact and lesioned side differed significantly (P = 0.00029). (U-W) Evaluation of side effects associated with intracranial ectopic GDNF expression. (U) Spontaneous locomotor activity in an open field; N = 31–34 mice/group. (V) Food intake by adult mice during a 72-hour period; N = 10 mice/group. (W) Body weight of adult mice; N = 9–34 mice/group. Abbreviations: DA, dopamine; DOPAC, 3,4-dihydroxyphenylacetic acid; HVA, homovanillic acid; dSTR, dorsal striatum; SNpc, substantia nigra pars compacta.
Increased GDNF levels increase the nigrostriatal dopaminergic system in adult mice
To assess whether DA system was changed in adult animals, we studied Gdnfwt/hyper mice at 2.5–4 months of age and noted that the DA levels in the striatum of Gdnf wt/hyper mice were increased by 25% compared to wild-type littermates (Fig 4D). Analysis of striatal DA metabolites (S3C and S3D Fig) revealed a 35–40% increase in 3,4-dihydroxyphenylacetic acid (DOPAC) levels (S3C Fig), suggesting that also DA release in Gdnf wt/hyper mice may be increased. To assess the effect of the absence of one Gdnf allele in the comparable genetic background, we also measured DA levels in heterozygous GDNF-knockout mice (Gdnf wt/KO) [10]. Consistent with previous studies [21] the levels of DA in Gdnf wt/KO mice were similar to wild-type levels (Fig 4D). Restoring wild-type expression levels of GDNF by crossing Gdnf wt/hyper mice with Deleter-FLP mice also restored DA levels (S3E Fig). In the substantia nigra pars compacta the number of DA neurons in the Gdnf wt/hyper mice was 15% increased relative to the controls (Fig 4E and 4F), indicating that the moderate 15% increase in the number of substantia nigra pars compacta DA neurons noted at P7.5 is retained in adulthood. Similarly, we found that the number of dopaminergic terminals in the dorsal striatum is increased by 15% in the Gdnf wt/hyper mice (Fig 4G, S3F Fig). Immunohistochemical examination of nigrostriatal DA system revealed no gross anatomical differences between the Gdnf wt/hyper and Gdnf wt/wt mice (S3G and S3H Fig) and the size of the striatum appeared unaffected (S3I Fig).
Collectively, these data indicate that an increase in GDNF expression levels increases the adult number of DA cells in the substantia nigra pars compacta and the number of dorsal striatal DA terminals by 15%. In addition, increase in GDNF expression increases striatal tissue DA and its metabolite DOPAC levels by 25% and 35–40%, respectively, suggesting that DA release in Gdnf wt/hyper mice may be enhanced.
Increased GDNF levels increase specific DA system functions in adult mice
To determine whether increasing the levels of endogenous GDNF affects the function of the nigrostriatal DA system in adult mice, we performed fast-scan cyclic voltammetry measurements in acute striatal slices at 3–4 months of age, and measured the clearance rate of extracellular exogenous DA in the striatum using in vivo amperometry at the same age. To study various functional aspects of the DA system, we used a range of stimulation patterns for the voltammetry measurements (S3J Fig). We observed that stimulations in the striatal tissue of Gdnf wt/hyper mice released about 35–45% more DA (Fig 4H and 4I) with a steeper rising slope (S3K Fig) when compared to Gdnf wt/wt mice. No differences were found between the genotypes in the rise time and decay parameters of the DA events (S3L–S3N Fig). We observed no difference in paired stimulus depression of DA release (Fig 4J), and we found no difference in DA release probability (Fig 4K).
Since more DA is released but DA reuptake parameters are comparable between the genotypes, increased DA uptake in the striatum of Gdnf wt/hyper mice can result at least in part, from the observed 15% increase in the number of dopaminergic terminals in the striatum. However, increased DA transporter (DAT) levels and/or increased DAT activity could also contribute to the observed increase in DA re-uptake. To test these possibilities, we measured the clearance rate of extracellularly applied DA in the striatum using in vivo amperometry. We found that compared to Gdnf wt/wt mice, DAT activity in the Gdnf wt/hyper mice was increased at least up to five-fold in a DA concentration-dependent manner (Fig 4L). Next, we examined whether this difference is due to differences in the levels of DAT between genotypes. We measured total and surface levels of DAT protein in the striata at P7.5 and total DAT levels in the striata of adult mice; we found no differences between the genotypes (S4A–S4C Fig). Taken together, our findings suggest that increasing endogenous GDNF levels increases striatal tissue DA content and striatal DA release and re-uptake without affecting the DA release probability. The observed increase in striatal DA re-uptake in Gdnf wt/hyper mice is most likely explained by the combined effects of increased DAT activity and 15% increase in the number of striatal dopaminergic terminals.
Gdnf wt/hyper mice have an increased response to amphetamine
In order to investigate whether increased GDNF levels result in functional consequences in the DA system in behaving animals we administered amphetamine, a dopaminergic stimulant. Amphetamine is taken up by the DAT into the presynaptic terminal, where it is then loaded into synaptic vesicles. Amphetamine releases DA from these vesicles into the cytoplasm and reverses its active transport across the presynaptic membrane; the result is increased DA concentration in the synaptic cleft and increased locomotor activity in treated animals [22]. Compared to Gdnf wt/wt littermates, Gdnf wt/hyper mice had increased amphetamine-induced locomotor activity (Fig 4M), suggesting that amphetamine has a stronger effect in terms of driving higher extracellular DA levels in the striatum of Gdnf wt/hyper mice. To test this hypothesis, we measured extracellular dopamine levels in the striatum following local amphetamine delivery via in vivo microdialysis. Consistent with our hypothesis, we observed an increase in amphetamine-induced extracellular DA levels in the striata of Gdnf wt/hyper mice (Fig 4N). We also measured the effect of amphetamine in striatal slices using cyclic voltammetry. Because amphetamine depletes DA from nerve terminals, cyclic voltammetry can detect the gradual decrease in stimulated DA release following amphetamine application. Consistent with increased DAT function in Gdnf wt/hyper mice, we found that amphetamine depletes the synaptic DA storages with a faster time course in Gdnf wt/hyper mice compared to Gdnf wt/wt littermate controls (Fig 4O).
Based on these results, the increased amphetamine-induced locomotor activity in Gdnf wt/hyper mice is likely due to a combination of three effects. First, the increased DAT activity in Gdnf wt/hyper mice likely causes amphetamine to accumulate in the nerve terminals more rapidly. Second, the 15% increase in the number of striatal dopaminergic terminals increases the number of striatal DA release sites. And third, more DA is released by the terminals. Collectively, these DA system features explain the increased amphetamine-induced locomotor activity in Gdnf wt/hyper mice.
Increasing GDNF levels protects the dopaminergic system in a lactacystin-induced model of Parkinson’s disease
Currently, no mouse model is available that phenocopies the slow disease progression of patients with PD [23]. Thus, the most widely used animal models for studying PD are based on the toxins MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) and 6-hydroxydopamine (6-OHDA), both of which are taken up specifically into DA neurons via the DAT [23]. Given that we found at least up to five-fold increase in DAT activity in Gdnf wt/hyper mice, we expect Gdnf wt/hyper mice to be sensitized to DAT-based toxins, unless the trophic effect of increased endogenous GDNF expression dampens or reverses the phenotype. We found that relative to the controls, Gdnf wt/hyper mice are five-fold more sensitive to striatal 6-OHDA injection. More specifically, following striatal 6-OHDA injection we found five-fold aggravated decrease in striatal DA levels and two-fold aggravated decrease in substantia nigra pars compacta DA neuron numbers in Gdnf wt/hyper mice relative to the littermate controls (Fig 4P and 4Q). To overcome the confounding effect of enhanced DAT activity in Gdnf wt/hyper mice in DAT based PD models, we looked for alternative models of PD. Abnormal aggregation of proteins is a generally accepted pathological process common to most neurodegenerative disorders, including PD. Consistent with this notion, intracranial application of proteasome inhibitors such as lactacystin can induce a PD-like phenotype in both rodents and fish [24,25]. We found that unilateral lactacystin injection just above the substantia nigra induced significant side bias (measured using the corridor test) in Gdnf wt/wt mice; however, lactacystin-injected Gdnf wt/hyper mice did not develop this bias (Fig 4R). In addition, DA and its metabolite levels were better preserved in the striatum of lactacystin-injected Gdnf wt/hyper mice compared to lactacystin-injected wild-type mice (Fig 4S). However, the number of DA cells in the substantia nigra pars compacta was comparably reduced by lactacystin injection in both genotypes (Fig 4T), suggesting that the protective effect from lactacystin-induced PD in Gdnf wt/hyper mice occurs at the functional level in the striatum.
GDNF derepression does not induce side effects associated with ectopic GDNF application
Despite its dopaminotrophic benefits both in PD models and in clinical trials, the delivery of GDNF to the nigrostriatal DA system also induces adverse side effects, including hyperactivity [5,26–29], reduced levels of striatal tyrosine hydroxylase (TH)–the rate-limiting enzyme in DA synthesis [5,30], reduced food intake, and loss of body weight [6,26]. Importantly, none of these effects were observed in our Gdnf wt/hyper mice as assessed by open field test, measurements of food intake in physiological cage, bodyweight measurements of adult mice at 3–4 months of age (Fig 4U–4W) and by western blotting and immunohistochemical measurements of striatal TH levels at P7.5 and in adult mice at 3–4 months of age (S4D–S4G Fig).
miR-9, miR-96, miR-133 and miR-146a are novel regulators of GDNF
To get insight on the mechanism how Gdnf 3’UTR negatively regulates gene expression (S1D–S1F Fig) we looked for trans-acting factors that regulate transcripts containing the Gdnf 3’UTR. Sequence analysis revealed three putative binding sites for RNA-binding proteins (RBPs) and conserved binding sites for several miRNAs in the Gdnf 3’UTR (Fig 5A). We found that the RBPs tristetraprolin (TTP, a negative regulator of brain-derived neurotrophic factor BDNF [31]); embryonic lethal abnormal vision-like protein 1 (ELAVL1); and the AU-rich element-binding protein AUF1 had little or no effect on the expression of a reporter construct containing the Gdnf 3’UTR (S4H Fig), consistent with a previous report [11].
Fig 5. Identification of Gdnf-regulating miRNAs.

(A) Putative miR binding sites cluster within the conserved areas of the Gdnf 3’UTR. The miRNAs underlined with red bars were co-immunoprecipitated with Gdnf mRNA in a genome-wide screen of a mouse brain tissue [34]. Predicted putative RNA-binding protein (RBP) sites are indicated with triangles. Source: Blast, TragetScan, AREsite, and http://servers.binf.ku.dk/antar/; Hs, human; Mm, mouse. (B) Luciferase expression from a Ren-Gdnf 3’UTR construct after co-transfection with the indicated pre-miRNAs in HEK293 cells. scr1 and scr2 are scrambled pre-miRNA controls; N = 3 experiments/miRNA with 3–5 biological repeats/miRNA/experiment. (C) Predicted binding sites (ddG<-3) for miR-9 (green), miR-96 (blue), miR-133a/b (orange), and miR-146a (red) based on “predict microRNA targets” analysis (http://genie.weizmann.ac.il/). Boxes indicate the mutated miRNA-binding sites in each mutant. Note that miR-9/96/133m contains overlapping sites for miR-9, miR-96, and miR-133, all of which were mutated in this construct. (D) Luciferase assay of miR-9, miR-96, miR-133a, and miR-146a mutants (mutated sequences indicated with boxes in panel C; scr1 is a scrambled pre-miRNA control; N = 2 experiments/miRNA with 3 biological repeats/miRNA/experiment. (E-F) Expression of endogenous GDNF mRNA (E) and GDNF protein (F) is inhibited in U87 cells by co-transfection with the indicated pre-miRNAs; N = 3–4 experiments with 2–3 biological replicates/experiment. (G) Adenoviral transduction of Cre—but not GFP—in homozygous “floxed” Dicer-1F/F primary cortical neurons increases endogenous GDNF protein levels; N = 2 experiments with 3–4 mice/group. (H) AAV-based constructs encoding shRNAs against miR-9, miR-96, and miR-146a, as well as siRNA against Dicer, increase endogenous GDNF expression in HEK293 cells; N = 5 experiments/construct (except for the shRNA against miR-9, where N = 2 experiments/construct) with 2 biological repeats per experiment. Ad, adenovirus; m, mutant.
Next, we investigated the effect of miRNAs on regulating Gdnf expression. We examined the miRNAs miR-133a, miR-133b, miR-125a-5p, miR-125b-5p, miR-30a, miR-30b, miR-96, miR-9, and miR-146a, which were selected based on their co-expression with Gdnf in several brain areas [17,19,32,33]; see also www.microrna.org and presence of conserved binding sites within Gdnf 3’UTR (Fig 5A; www.targetscan.org). Our analysis of miRNA expression revealed that miR-9, miR-133a, miR-133b, miR-125a-5p, miR-125b-5p, miR-30a, miR-30b, and miR-146a are all expressed in the developing forebrain, adult dorsal striatum and in the developing kidney (S4 Table). Next, we transfected HEK293 cells with the above-mentioned putative Gdnf-regulating pre-miRNAs. Compared to the control miRNAs, the specific miRNAs negatively regulated the expression of a reporter construct containing the Gdnf 3’UTR by 30–50% (Fig 5B). Next, we examined whether the in silico (genie.weizmann.ac.il, www.targetscan.org) predicted conserved miRNA sites mediate the interaction between these miRNAs and the Gdnf 3’UTR. We paid particular interest to miR-9 and miR-96, as previous data obtained from two genome-wide screens suggested that these miRNAs interact with Gdnf mRNA in the mouse brain; and overexpressing them in human cell line suppresses the expression of GDNF (summarized in [34]). Mutating some of the predicted miRNA seed sites (Fig 5C; see S1 Materials and Methods for details) in the Gdnf 3’UTR either reduced or abolished the ability of miR-9, miR-96, miR-133a, and miR-146a to inhibit expression (Fig 5D), suggesting a direct interaction between these miRNAs and some of the predicted sites in the Gdnf 3’UTR. Based on in silico analysis, puΔtk contains approximately half the number of potential miRNA-binding sites and about 10% of the conserved miRNA sites present in the Gdnf 3’UTR (S4I and S4J Fig). Consistent with this prediction, we found that miRNAs had no effect on expression of a reporter construct containing the puΔtk-Gdnf 3’UTR cassette (S4K Fig). To analyze the effect of miR-9, miR-96, miR-133b, and miR-146a on endogenous GDNF expression, we transiently overexpressed these miRNAs in U87 cells (a human glioblastoma cell line that expresses endogenous GDNF at detectable levels). We found that compared to the control miRNAs, transient co-expression of these four miRNAs reduced the Gdnf mRNA level (Fig 5E) and GDNF protein level (Fig 5F) without affecting cell survival (S4L Fig).
Dicer is required for the maturation of miRNAs. To gain further evidence that the GDNF expression is regulated by miRNAs, we next examined the effect of deleting Dicer on endogenous GDNF expression in primary Dicerflox/flox cortical neurons [35] using adenovirus-mediated delivery of Cre. Deleting Dicer resulted in up-regulation of endogenous GDNF expression in cortical neurons (Fig 5G). Finally, to gain insight into how endogenous miR-9, miR-96 and miR-146a impact endogenous GDNF mRNA levels in human cells we tested six different shRNA constructs targeted to each miRNA for their ability to derepress endogenous GDNF expression in HEK293 cells (S4M Fig). Based on this analysis, we identified the three shRNA constructs targeting miR-9, miR-96 and miR-146a with the highest potency for derepressing endogenous GDNF mRNA expression (Fig 5H). An siRNA against Dicer was used as a positive control (Fig 5H). We conclude that miR-9, miR-96, miR-133, and miR-146a interact directly with their binding sites in the Gdnf 3’UTR; moreover, miR-9, miR-96, and miR-146a regulate the expression of GDNF in vitro.
Discussion
Due to the limitations associated with existing genetic tools, the function of endogenous GDNF has remained poorly understood [7,10]. Despite multiple attempts by several research groups, transgenic animals in which GDNF expression is restricted to cells that normally express GDNF are not available. The dramatic consequences of ectopic GDNF expression—for example, on the development of the urogenital tract [36]—make it difficult to draw conclusions regarding the function of endogenous GDNF based solely on ectopic GDNF expression in the brain.
Analysis of GDNF hypermorphic mice
GDNF hypermorphic mice provided us with an opportunity to study the function of endogenous GDNF with the focus on the postnatal nigrostriatal DA system and in renal development.
We found that increasing the endogenous levels of GDNF increases the number of DA neurons in the substantia nigra pars compacta during the development, and that this increase in DA neuron numbers is retained in adulthood. Highlighting the importance of the correct expression site, GDNF has no such effects when overexpressed in the mouse brain under the promoter not specific to GDNF-expressing neurons [8].
In the dorsal striatum, increase in GDNF levels resulted in increased number of dopaminergic terminals, increased levels of DA and enhanced DA release and reuptake, explaining the enhanced amphetamine-induced locomotor activity in GDNF hypermorphic mice. Since the nigrostriatal DA system is well known for its capacity to compensate for changes [37], the observed increase in DA transporter activity in GDNF hypermorphic mice likely reflects a measure to counterbalance elevated striatal DA content and release to maintain normal extracellular DA levels. We observed no change in striatal DA transporter levels, suggesting that DAT activity in GDNF hypermorphic mice is regulated by other mechanisms, such as post-translational modifications or protein-protein interactions. Since currently methods allowing temporal analysis of those endpoints in the mouse striatum are not available, the question of how GDNF regulates DAT activity remains to be resolved.
Notably, we found that increased GDNF levels have a protective effect in supranigrally delivered lactacystin model of Parkinson’s disease; this effect is not mediated by protecting DA cell bodies in the substantia nigra pars compacta, but by enhancing the dopaminergic function in the striatum. These findings are consistent with a previous study reporting the lack of effect of selective GDNF deletion or reduction on the survival of DA cell bodies in the substantia nigra pars compacta upon aging [10] and with a study reporting that deletion of GDNF receptor RET does not modulate MPTP toxicity on the dopaminergic system but is required for regeneration of striatal dopaminergic axon terminals [38]. Collectively, these results suggest that in adult mice GDNF acts as a local trophic factor for DA axons in the striatum.
Furthermore, our Gdnf wt/hyper mice do not develop any of the adverse side effects usually associated with ectopic GDNF expression, which can include hyperactivity, loss of body weight, and a decrease in the levels of tyrosine hydroxylase—the rate-limiting enzyme in DA synthesis. Because GDNF is not applied ectopically in our model, the common feature in both experimental animals and human studies—specifically, the massive sprouting of DA fibers towards the site of GDNF delivery, with unknown consequences with respect to side effects, treatment efficacy and behavior—was not observed in Gdnf wt/hyper mice. Together, these results imply that measures that promote elevation in endogenous GDNF levels in the striatum may have clinical potential in the treatment of Parkinson’s disease. However, because we found increased DAT activity in GDNF hypermorphic mice, the simultaneous use of GDNF with DAT-based toxins or drugs should be carefully considered and calls for further investigation.
To date, data on GDNF function is mainly gathered using various ectopic GDNF application methods in rodent and primate models and using constitutive or conditional GDNF knock-out mice. Our results bring an important new dimension since we analyze the effect of the elevation of endogenous GDNF. In Table 1 we illustrate the qualitative difference between ectopic and endogenous GDNF sources.
Table 1. Summary of GDNF functions identified using GDNF hypermorphic mice.
The main findings of this study are reported in comparison with published reports from ectopic GDNF applications and GDNF knock-out mice. The forth column indicates whether the observations made in GDNF hypermorphic mice are consistent with function predicted from ectopic GDNF applications and from Gdnf gene deletion studies, expand the previous knowledge or provide novel information.
| Endogenous GDNF elevation (this study) | Reported ectopic GDNF expression or gene knock-out approaches | Novelty of the finding | |
|---|---|---|---|
| Observed phenotype | Observed phenotype | Approach | |
| Increased motor activity upon amphetamine injection | Increased motor activity upon amphetamine injection | Nigrostriatal recombinant GDNF injection and viral gene delivery [5,26] | Consistent with ectopic/KO approaches |
| GDNF controls DA cell number at P7.5 | GDNF controls DA cell number at P7.5 | 2–3 fold transgenic over-expression of GDNF in the forebrain using CaMKIIα promoter [8] | Consistent with ectopic/KO approaches |
| Striatal DA levels are maintained upon supra-nigral lactacystin injection | Striatal DA levels are maintained upon supra-nigral lactacystin injection | Striatal viral gene delivery [39] | Consistent with ectopic/KO approaches |
| Increased stimulated and amphetamine -induced DA release | Increased stimulated and amphetamine -induced DA release | Intranigral GDNF protein injection [27,28] | Consistent with ectopic/KO approaches |
| GDNF levels regulate kidney size and morphological maturation | GDNF is required for the induction of the ureteric bud development during early embryogenesis | GDNF gene deletion [15] | Consistent and expanding ectopic/KO approaches |
| Elevated GDNF increases the number of DA neurons and enhances DA system functionduring development and in adulthood | GDNF deletion or reduction has no effect on the number of DA neurons in adulthood and upon aging; no gross effect on animal behavior | Conditional GDNF deletion or reduction using three Cre lines [10] | Novel |
| Elevated GDNF increases the number of DA neurons during development which is retained in adulthood. | Elevated GDNF does not increase the number of DA neurons after two weeks of age | 2–3 fold transgenic over-expression of GDNF in the forebrain using CaMKIIα promoter [8] | Novel |
| Elevated GDNF increases striatal DA release and re-uptake | Elevated GDNF has no effect on DA release and re-uptake | 2–3 fold transgenic over-expression of GDNF in the forebrain using CaMKIIα promoter [8] | Novel |
| No effect on food intake and bodyweight at least until 3–4 months of age | Reduction in food intake and weight loss within 1–2 weeks after GDNF overexpression | Nigrostriatal recombinant GDNF injection and viral gene delivery [6,26] | Novel |
| No effect on striatal tyrosine hydroxylase levels at least until 3 months of age | Downregulation of striatal tyrosine hydroxylase levels after 6 weeks | Nigrostriatal viral gene delivery [5,30] | Novel |
| No effect on spontaneous motor activity | Spontaneous motor hyperactivity | Nigrostriatal recombinant GDNF injection [26–29] | Novel |
| Elevation in total striatal DA levels | ; No effect on total striatal DA levels; elevation in DA turnover | Nigrostriatal recombinant GDNF injection and viral gene delivery [5,26,28] | Novel |
| No aberrant sprouting of DA fibers | Sprouting of DA fibers towards GDNF injection site | Nigrostriatal recombinant GDNF injection and viral gene delivery [4,26,40] | Novel |
| Elevated GDNF augments DA concentration -dependent increase in DAT activity | Not addressed previously | Novel | |
With respect to the role of GDNF in renal development, we found that excess GDNF levels negatively regulate kidney growth and morphogenesis. This result is contrary to expectations based on the majority of in vitro studies, where the effect of ectopic GDNF on embryonic urogenital block can be followed for the few days, and likely arose from an overstimulation of ureteric bud growth, a key process in renal development.
A comparison between our Gdnf wt/hyper mice and MEN2B mice, which express a constitutively active form of the GDNF receptor RET [41], revealed several common features and key differences between these two models. For example, striatal DA levels, TH-positive cell numbers, and striatal DAT activity are increased in both mutants. In contrast, only MEN2B mice develop increased levels of DAT and TH, and reduced spontaneous locomotion [42–44]. Thus, increasing endogenous GDNF levels is fundamentally different than constitutively activating RET; this finding may have broad-reaching implications with respect to drug design and studies of receptor-ligand biology.
miRNAs and regulation of GDNF expression via the 3’UTR
Previously published genome-wide screens suggested that overexpressing miR-9 and miR-96 reduce the levels of GDNF mRNA in a human cell line and that these miRNAs interact with the Gdnf mRNA in the mouse brain [34]. We identified binding sites for miR-9 and miR-96 in the 3’UTR of Gdnf; in addition, we identified binding sites for miR-133 and miR-146a. We also found that reducing the levels of Dicer, an enzyme required for the maturation of miRNAs, derepresses endogenous GDNF expression both in human cells and in mouse primary neurons. Moreover, overexpressing miR-9, miR-96, miR-133b, and miR-146a represses the expression of endogenous GDNF mRNA and protein in a human cell line. Finally, we found that shRNAs against miR-9, miR-96, and miR-146a derepress endogenous GDNF mRNA levels in human cell line. Taken together, these results confirm the miRNA target in vitro [45]. However, every miRNA can have several hundred mRNA targets; thus, assigning specific observations to the direct effect of a given miRNA acting on one target in vivo is currently not possible. Future work with a conditional targeting of Gdnf 3’UTR is necessary to overcome this limitation. Once the effects of adult-onset GDNF elevation in the striatum are characterized, we can then utilize various anti-miRNA strategies to evaluate a given miRNA’s role in GDNF-induced changes in the DA system. Our data also suggest that miR-125a-5p, miR-125b-5p, miR-30a, and miR-30b are possible regulators of GDNF expression. Whether the effect of these miRNAs on GDNF expression is direct or indirect warrants further research.
Interestingly, we found that GDNF derepression in GDNF hypermorphic mice was stronger in the kidneys than in the brain and in the brain areas in which Gdnf mRNA levels are higher. The reasons underlying this finding are currently unknown; however, it may be related to the relative ratio between the mRNA and miRNAs; in addition, miRNAs can have different effects on their target mRNAs depending on the tissue context [46].
Conclusions
Using GDNF hypermorphic mice we found that endogenous GDNF regulates postnatal development and function of nigrostriatal dopamine system. Moreover, some of the identified GDNF functions overlapped with results from earlier studies with ectopic GDNF, whereas others were novel, highlighting the importance of correct spatial expression of GDNF. We also found that about two-fold elevation in endogenous GDNF levels protects mice in lactacystin-based model of Parkinson’s disease without side effects associated with ectopic GDNF applications. Whether increasing endogenous GDNF levels is a viable strategy for developing new therapeutic approaches for treating Parkinson’s disease and other diseases is an important question. Finally, since negative regulation via 3’UTR is shared by many genes, our data pinpoints that 3’UTR-s could provide an important target for genetic studies in vivo. More specifically, prevention of transcription of negatively regulated 3’UTR-s could provide a measure to elevate endogenous gene expression while avoiding mis-expression commonly associated with transgenesis.
Materials and Methods
Detailed descriptions of all materials and methods are provided in S1 Materials and Methods.
Ethics statement
The animal experiments were performed according to the EU legislation harmonized with Finnish legislation and have been approved by the National Animal Experiment Board of Finland (permit no. ESAVI/3770/04.10.03/2012).
Cell culture and molecular biology assays
Cell culture and molecular biology assays were performed using routine methods in the field; please see S1 Materials and Methods for details.
Generation of GDNF hypermorphic animals, tissue dissection and analysis
Briefly, 5667bp 5’ homologous arm spanning the second intron of the Gdnf gene, 6055bp 3’ homologous arm and GDNF protein coding part of Gdnf exon 3 including the stop codon were amplified with PCR from Gdnf-containing PAC (RP21-583-K20, CHORI) and cloned into pFlexible [12,47], to generate Gdnf targeted allele. ES clones were screened with standard Southern blotting. Mice were maintained in 129Ola/ICR/C57bl6 mixed genetic background.
Lactacystin model of Parkinson’s disease
4 μg of lactacystin (AG Scientific) in 4 μl of PBS was injected just above the SN at: antero-posterior (AP) -3.3 mm; medio-lateral (ML) -1.2 mm and dorso-ventral (DV) -4.6 mm. The animals were subjected to corridor test 5 weeks after the injection, and sacrificed for tissue isolation and IHC analysis.
In situ hybridization
In situ hybridization was performed using a probe spanning Gdnf exons or 525bp in the 3’ end of Gdnf 3’UTR. RNAscope [20] probes detecting Gdnf (red) and Parvalbumin (PV, blue) mRNA were custom made by Advanced Cell Diagnostics.
Fast-scan cyclic voltammetry
Dopamine release was evoked on acute striatal slices with electrical stimulations and measured with a carbon fiber electrode calibrated with known dopamine concentrations. The signal was amplified with Axopatch 200B amplifier (Molecular Devices), digitized (ITC-18 board; InstruTech) and analyzed with a computer routine in IGOR Pro (WaveMetrics).
In vivo chronoamperometry
In anesthetized mice (urethane 1.7–1.9 g/kg, i.p.; Sigma) the electrode was mounted in parallel with a micropipette used for application of dopamine. Recordings were performed at two rostrocaudal striatal tracks in each hemisphere, at AP +0.3 or +1.0 mm; ML ±1.8 mm, using Fast Analytical Sensing Technology (FAST-16) system (Quanteon). At each recording site, data was collected from three depths below the dura: at -2.0, -2.5, and -3.0 mm.
In vivo microdialysis
A microdialysis guide cannula (MAB 4.1, AgnTho’s AB) was inserted into the dorsal striatum (AP +0.6 mm; ML +1.8 mm and DV -2.2 mm) of mice under isoflurane anaesthesia. After obtaining a stable baseline, the Ringer solution was switched into 100 μM of D-amphetamine for 60 minutes. The dialysis flow rate was 2 μl/min. Concentration of dopamine was analyzed using HPLC.
Measurements of GDNF protein levels from tissues
Striatal tissues lysates were prepared and analyzed immediately after sacrificing with GDNF Emax ImmunoAssay System (Promega).
Behavioral analyses
Open field test was performed in three independent cohorts of comparable size (N = 10–12 male mice per genotype with littermate controls). Metabolic monitoring was performed using Comprehensive Lab Animal Monitoring System (CLAMS).
Statistical analysis
Statistical analysis for pairwise comparisons was performed using Student’s t-test with two tailed distribution using the unequal variance option. Data from amperometry was analyzed by one-way ANOVA followed by Bonferroni post hoc test. Behavioral data were analyzed using factorial ANOVA design with genotype and cohort as between-subject factors, where appropriate. Post hoc analysis after significant ANOVA was carried out using Student-Newman-Keuls test. Data from CV was analyzed by two-way repeated measures ANOVA, which in the amphetamine analysis was followed by multiple comparisons (Sidak´s). All numerical results are reported as mean ± standard error of the mean. SPSS (IBM Corp., Armonk NY, USA) or STATISTICA 11 (StatSoft Inc., Tulsa) were used for analysis.
Supporting Information
(DOCX)
Gdnf KO animals were obtained by crossing Gdnf wt/hyper animals to Deleter-Cre mice, which results in deletion of GDNF protein coding exon 3 (Fig 1B) [10].
(DOCX)
(DOCX)
Correlational analyses of rostral brain dopamine and serum urea levels in individual animals in 10 Gdnf wt/wt and 12 Gdnf wt/hyper animals at P7.5 using the Correlation function in Microsoft Excel.
(DOCX)
Levels of mature miRs expressed as fold difference relative to sno202 in adult mouse brain, developing mouse kidney and in HEK293 cells. N = 2 experiments. dSTR, dorsal striatum, E, embryonic day; Hs, Homo sapiens; Mm, Mus musculus; P, postnatal day; w, weeks.
(DOCX)
(A) Schematic representation of the reporter constructs and the derived mRNAs used in this study. Red and blue bar indicate probes used in the Northern blot for firefly luciferase CDS and Gdnf 3’UTR, respectively. (B) The puΔtk cassette blocks transcription to the Gdnf 3’UTR, measured by Northern blot analysis of expression from a construct harboring the Firefly (FF) coding sequence upstream of the puΔtk-Gdnf 3’UTR cassette in HEK293 cells; loading control: 28S ribosomal RNA. (C) Northern blot analysis of expression from construct harboring FF coding sequence proceeded with puΔtk-Gdnf 3’UTR cassette (S1 Fig A) in HEK293 cells (S1 Fig D); loading control: 28S ribosomal RNA. (D) Expression from equimolar amounts of the FF-Gdnf 3’UTR, FF-puΔtk-Gdnf 3’UTR and FF-SV40 pA constructs in U87 and HEK293 cells; FF-firefly luciferase; N = 3 experiments/construct with 3 replicates/experiment. (E) Gdnf 3’UTR reduces expression of both Renilla luciferase (Ren) and FF in HEK293 and U87 cells; N = 3 experiments/construct with 3 replicates/experiment (F) FF expression from constructs containing SV40 pA, puΔtk-Gdnf 3’UTR cassette and Gdnf 3’UTR in HEK293 cells after treatment with actinomycin D. Note that prevention of transcription of the native Gdnf 3’UTR by preceding puΔtk alleviates the post-transcriptional inhibition of the reporter. Renilla luciferase expression from a separate plasmid was used for normalization. N = 3 experiments/construct with 3 replicates/experiment. (G) Targeting strategy of the Gdnf locus. 5667 bp 5’ (green bar) and 6055 bp 3’ (blue bar) homologous arms, and Gdnf exon 3 until and including the stop codon were amplified with PCR from Gdnf-containing PAC and cloned into PmeI, NotI and HindIII sites in pFlexible [31], respectively, to generate Gdnf targeted allele. Sequence lengths are not drawn to scale. Arrows indicate primers used for routine genotyping; black bar indicates probe used for Southern blotting using BlpI restriction enzyme shown on (H). (I) Representative image of routine genotyping of the mice using primers indicated on G. Abbreviations: puΔtk, a cassette with PGK promoter encoding for fusion protein between puromycin N-acetyltransferase and a truncated version of herpes simplex virus type 1 thymidine kinase (TK) with 3’ bovine growth hormone polyadenylation signal (pA); FF-firefly luciferase; CDS-coding sequence.
(TIF)
(A-B) Representative images of in situ hybridization of Gdnf mRNA using a probe against the CDS (stained blue, indicated with white arrow heads) in mice in whole-mount preparations of E11.5 hindlimb (A) and in paraffin sections from P7.5 testis (B). Note that Gdnf expression sites are comparable between genotypes, whereas the signal appears stronger in Gdnf hyper/hyper mice. N = 4 mice/group in A and N = 2 in B. (C-D) Gdnf mRNA (C) and protein (D) expression in the testis at E18.5 measured with QPCR and ELISA, respectively, shows allele dose-dependent increase in GDNF levels in GDNF hypermorphic mice. Note that GDNF protein levels in Gdnf 3’UTR rest/rest mice are normal (D). N = 2–5 mice/group in 2–3 experiments with 2–3 replicates/experiment (E) Representative image of Northern blot analysis of Gdnf mRNA in E18.5 testis using Gdnf CDS probe. Note that the size of the band is ca 500 bp shorter in Gdnf hyper/hyper mice compared to the Gdnf wt/wt mice, consistent with the size of transcripts derived from Gdnf-puΔtk fusion transcript (S1A–S1C Fig; and see S2 Fig F, G). N = 2 mice/group. (F) Schematic representation of the puΔtk-Gdnf 3’UTR (Gdnf hyper) allele for Gdnf, A and B designate primers used in QPCR analysis of Gdnf mRNA levels (C); primers A and C depict primers used for Gdnf transcript sequence analysis in (G). (G) Representative image of RT-PCR analysis from E18.5 testis using primers A and C as depicted in (F). The observed PCR product in Gdnf hyper/hyper mice was of expected length (977 bp) and it was sequenced for validation; N = 3 mice; beta-actin was used to verify cDNA quality. (H-I) Kidneys of P7.5 Gdnf hyper/hyper mice function poorly, as indicated by serum creatinine (H) and serum urea (I) levels. H, N = 3–12; I, N = 7–26 mice/group. (J-K) The blood serum of adult Gdnf wt/hyper mice contains slightly higher levels of urea (J), but not creatinine (K) compared to wild type mice. Note that two-fold variation in serum urea levels is considered normal in humans. N = 16–19 mice/group. (L) Representative image of kidneys in wild type and Gdnf 3’UTRrest/rest mice. In total, 20 mice were analyzed from each genotype. (M) Verification of the specificity of RNAscope probes against parvalbumin (left panel; blue) and Gdnf (right panel; red). Note that the signal matches known parvalbumin mRNA expression in Purkinje cells (black triangles) and in molecular layer interneurons (open triangles) at three months of age. Similarly, Gdnf mRNA can be detected in the metanephric mesenchyme (MM) and not in the ureteric bud (UB) in the developing kidney at E14.5. Scale bars: A, 1.5 mm; B, 5 μm; M, 1 mm; L, 70 μm (left) and 100 μm (right). Abbreviations: bGHpA, bovine growth hormone polyadenylation signal; bp, base pairs; CDS, coding sequence; Cp, quantification cycle; dSTR, dorsal striatum; E, embryonic day; F, FRT sites for Flp recombinase; IHC, immunohistochemistry; kb, kilo-base pairs; m, months; MM, metanephric mesenchyme; P, postnatal day; PV, parvalbumin; puΔtk, a cassette with PGK promoter encoding for fusion protein between puromycin N-acetyltransferase and a truncated version of herpes simplex virus type 1 thymidine kinase (TK) with 3’ bovine growth hormone polyadenylation signal; UB, ureteric bud. *P< 0.05, **P< 0.01, and ***P< 0.001; error bars indicate SEM.
(TIF)
(A-B) HPLC analysis of DA metabolites DOPAC (A) and HVA (B) in P7.5 rostral brain. N = 5–8 mice/group. (C-D) HPLC analysis of DA metabolites DOPAC (C) and HVA (D) in the dSTR at 2.5–3 months of age. N = 11 for Gdnf wt/wt, 8 for Gdnf wt/hyper; P = 0.00521 for DOPAC and P = 0.0569 for HVA. (E) HPLC analysis of DA levels in the dSTR of Gdnf 3’UTR rest/rest and Gdnf wt/wt mice; N = 6 mice/group. (F) Representative images of DAT immunostaining in the dSTR, N = 7–9 mice/group. (G-H) Representative images of TH immunostaining in the dSTR (G) and midbrain (H). In total, 8 mice were analyzed from each genotype. (I) Average striatal area size measured from DAT+ sections. (N = 9 for Gdnf wt/wt, 7 for Gdnf wt/hyper (J) Representative recording trace of DA events stimulated by single electrical pulses at 2 min intervals, by paired stimulations at shown intervals, and by a burst of 5 stimuli at 20 Hz. (K-O) Rise and decay parameters of the first five DA events evoked by single-pulses. N = 5–7 mice/group with 1–3 striatal slices/mouse analyzed. (K) DA rise slope is steeper in the striata of Gdnf wt/hyper mice [two-way repeated measures ANOVA, F (1,29) = 11.47, P = 0.0021], indicating that more dopamine is released from the terminals during the rise phase. (L) T ½ times, i.e., the width of the event at half of the maximum. (M) Rise time, i.e. the time for DA events to rise from baseline to the maximum. (N) The fall time of the DA events. (O) The exponential decay time constant of the DA transients. Abbreviations: DA, dopamine; DOPAC, 3,4-dihydroxyphenylacetic acid; dSTR, dorsal striatum; HPLC, high performance liquid chromatography; HVA, homovanillic acid; IHC, immunohistochemistry; m, months; P, postnatal day. Scale bars: F, 10 μm; G, 75 μm; in inset 1 mm; H, 0.5 mm; in inset 30 μm.
(TIF)
(A-B). Total DAT protein levels (A) and plasma membrane associated DAT levels (B) at P7.5, measured by western blotting. N = 4 mice/group. (C) DAT protein levels in the dSTR and SNpc at 2.5-3m, measured by western blotting. Gapdh or α-tubulin was used to normalize loading, as indicated in the figure. N = 5 mice/group. (D) OD of striatal TH+ fibers at P7.5, reflecting striatal TH levels and the density of striatal dopaminergic innervation at the macroscopic level. N = 7 mice/group. (E) TH protein levels in the dSTR at P7.5, measured by western blotting. Gapdh was used to normalize loading. N = 4 mice/group. (F) OD of striatal TH+ fibers at 2.5–3 months of age. N = 10–11 mice/group. (G) TH protein levels in the dSTR at 2.5–3 months of age, measured by western blotting. Gapdh was used to normalize loading. N = 5 mice/group. (H) Luciferase expression from Firefly-Gdnf 3’UTR construct in HEK293 cells after co-transfection with constructs encoding RBPs. Renilla luciferase expression from a separate plasmid was used for normalization and GFP was used as a negative control. N = 3 experiments with 3 biological repeats each. (I) The number of predicted miR binding sites (ddG<0) in Gdnf 3’UTR, puΔtk and SV40 late pA signal, analyzed with PITA software [4]. (J) The number of predicted strong binding sites for Gdnf-specific miRs (ddG<-10) in Gdnf 3’UTR, puΔtk and SV40 late pA signal, analyzed with PITA software [4]. Selection of Gdnf-specific miRs was based on TargetScan analysis using the most stringent conditions. (K) Luciferase expression from Firefly-puΔtk-Gdnf 3’UTR construct in HEK293 cells after co-transfection with Gdnf-regulating pre-miRs. Note that miRs that inhibit luciferase expression via the native Gdnf 3’UTR (Fig 5B) do not suppress the expression from reporter construct containing the Firefly-puΔtk-Gdnf 3’UTR cassette. N = 3 experiments with 3–5 biological repeats. (L) Luciferase activity in an ATP-based survival assay in U87 cells after co-transfection with pre-miRs. Note that the viability of U87 cells is not affected by the overexpression of pre-miRs, compared to scrambled control pre-miRs. N = 3 experiments with 3 replicates/experiment. (M) Gdnf mRNA levels in HEK293 cells after transfection with pAAV constructs encoding shRNAs against miR-9, miR-96 and miR-146. A number of shRNA-coding plasmids from two different producers, Signagen (left panel) and Vector Biosystems (right panel), were tested. N = 5 experiments with 2 repeats (all constructs from Signagen) and N = 1–2 experiments (constructs from Vector Biosystems). $ indicates that the constructs were tested 2 times with 2 repeats. The rest of the constructs from Vector Biosystems were tested once with 2 repeats. siRNA against Dicer was included as a positive control. N = 5 experiments with 2 repeats. Abbreviations: AAV, adeno-associated virus; RBP, RNA-binding protein; GFP, green fluorescent protein; AUF1, AU-rich element binding protein 1; ELAVL1, embryonic lethal, abnormal vision-like protein 1; TTP, tristetraprolin; puΔtk, a cassette with PGK promoter encoding for fusion protein between puromycin N-acetyltransferase and a truncated version of herpes simplex virus type 1 thymidine kinase (TK) with 3’ bovine growth hormone polyadenylation signal; scr, scrambled control; pA, polyadenylation signal. *P<0.05, **P<0.01, and ***P<0.001; error bars indicate SEM.
(TIF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
MS was supported by grants from the Sigrid Jusélius Foundation, the H. Lundbeck Foundation, the Michael J. Fox Foundation for Parkinson’s Research, the Academy of Finland (grant 11186236), the NIH (NS 0708259), and the University of Helsinki. JOA was supported by the Academy of Finland grants 136591, 140983, and 263700 and by the Institute of Biotechnology. TPP was supported by Päivikki and Sakari Sohlberg Foundation. AK and LP were supported by grants from Integrative Life Science doctoral program, and KV was supported by doctoral program Brain and Mind. JK was supported by Research Foundation of the University of Helsinki and The Finnish Cultural Foundation. KL was supported by an institutional investigation grant (IUT20-41) from the Estonian Research Council. AP was supported by the Jane and Aatos Erkko Foundation. The Mouse Behavioral Unit (VV) was supported by the Biocenter Finland. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F (1993) GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260: 1130–1132. [DOI] [PubMed] [Google Scholar]
- 2. Hoffer BJ, Hoffman A, Bowenkamp K, Huettl P, Hudson J, et al. (1994) Glial cell line-derived neurotrophic factor reverses toxin-induced injury to midbrain dopaminergic neurons in vivo. Neurosci Lett 182: 107–111. [DOI] [PubMed] [Google Scholar]
- 3. Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, et al. (2011) Priorities in Parkinson's disease research. Nat Rev Drug Discov 10: 377–393. 10.1038/nrd3430 [DOI] [PubMed] [Google Scholar]
- 4. Georgievska B, Kirik D, Bjorklund A (2002) Aberrant sprouting and downregulation of tyrosine hydroxylase in lesioned nigrostriatal dopamine neurons induced by long-lasting overexpression of glial cell line derived neurotrophic factor in the striatum by lentiviral gene transfer. Exp Neurol 177: 461–474. [DOI] [PubMed] [Google Scholar]
- 5. Georgievska B, Kirik D, Bjorklund A (2004) Overexpression of glial cell line-derived neurotrophic factor using a lentiviral vector induces time- and dose-dependent downregulation of tyrosine hydroxylase in the intact nigrostriatal dopamine system. J Neurosci 24: 6437–6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Manfredsson FP, Tumer N, Erdos B, Landa T, Broxson CS, et al. (2009) Nigrostriatal rAAV-mediated GDNF overexpression induces robust weight loss in a rat model of age-related obesity. Mol Ther 17: 980–991. 10.1038/mt.2009.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Airaksinen MS, Saarma M (2002) The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3: 383–394. [DOI] [PubMed] [Google Scholar]
- 8. Kholodilov N, Yarygina O, Oo TF, Zhang H, Sulzer D, et al. (2004) Regulation of the development of mesencephalic dopaminergic systems by the selective expression of glial cell line-derived neurotrophic factor in their targets. J Neurosci 24: 3136–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pascual A, Hidalgo-Figueroa M, Piruat JI, Pintado CO, Gomez-Diaz R, et al. (2008) Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat Neurosci. [DOI] [PubMed] [Google Scholar]
- 10. Kopra J, Vilenius C, Grealish S, Harma MA, Varendi K, et al. (2015) GDNF is not required for catecholaminergic neuron survival in vivo. Nat Neurosci 18: 319–322. 10.1038/nn.3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oh-Hashi K, Hirata Y, Kiuchi K (2012) Characterization of 3'-untranslated region of the mouse GDNF gene. BMC Mol Biol 13: 2 10.1186/1471-2199-13-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen YT, Bradley A (2000) A new positive/negative selectable marker, puDeltatk, for use in embryonic stem cells. Genesis 28: 31–35. [DOI] [PubMed] [Google Scholar]
- 13. Kramer ER, Knott L, Su F, Dessaud E, Krull CE, et al. (2006) Cooperation between GDNF/Ret and ephrinA/EphA4 signals for motor-axon pathway selection in the limb. Neuron 50: 35–47. [DOI] [PubMed] [Google Scholar]
- 14. Meng X, Lindahl M, Hyvonen ME, Parvinen M, de Rooij DG, et al. (2000) Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science 287: 1489–1493. [DOI] [PubMed] [Google Scholar]
- 15. Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, et al. (1996) Defects in enteric innervation and kidney development in mice lacking GDNF. Nature 382: 73–76. [DOI] [PubMed] [Google Scholar]
- 16. Sainio K, Suvanto P, Davies J, Wartiovaara J, Wartiovaara K, et al. (1997) Glial-cell-line-derived neurotrophic factor is required for bud initiation from ureteric epithelium. Development 124: 4077–4087. [DOI] [PubMed] [Google Scholar]
- 17. Hidalgo-Figueroa M, Bonilla S, Gutierrez F, Pascual A, Lopez-Barneo J (2012) GDNF Is Predominantly Expressed in the PV+ Neostriatal Interneuronal Ensemble in Normal Mouse and after Injury of the Nigrostriatal Pathway. J Neurosci 32: 864–872. 10.1523/JNEUROSCI.2693-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blum M, Weickert CS (1995) GDNF mRNA expression in normal postnatal development, aging, and in Weaver mutant mice. Neurobiol Aging 16: 925–929. [DOI] [PubMed] [Google Scholar]
- 19. Trupp M, Belluardo N, Funakoshi H, Ibanez CF (1997) Complementary and overlapping expression of glial cell line-derived neurotrophic factor (GDNF), c-ret proto-oncogene, and GDNF receptor-alpha indicates multiple mechanisms of trophic actions in the adult rat CNS. J Neurosci 17: 3554–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang F, Flanagan J, Su N, Wang LC, Bui S, et al. (2012) RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 14: 22–29. 10.1016/j.jmoldx.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Airavaara M, Mijatovic J, Vihavainen T, Piepponen TP, Saarma M, et al. (2006) In heterozygous GDNF knockout mice the response of striatal dopaminergic system to acute morphine is altered. Synapse 59: 321–329. [DOI] [PubMed] [Google Scholar]
- 22. Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, et al. (1995) Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci 15: 4102–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Westerlund M, Hoffer B, Olson L (2010) Parkinson's disease: Exit toxins, enter genetics. Prog Neurobiol 90: 146–156. 10.1016/j.pneurobio.2009.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bentea E, Van der Perren A, Van Liefferinge J, El Arfani A, Albertini G, et al. (2015) Nigral proteasome inhibition in mice leads to motor and non-motor deficits and increased expression of Ser129 phosphorylated alpha-synuclein. Front Behav Neurosci 9: 68 10.3389/fnbeh.2015.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matsui H, Ito H, Taniguchi Y, Inoue H, Takeda S, et al. (2010) Proteasome inhibition in medaka brain induces the features of Parkinson's disease. J Neurochem 115: 178–187. 10.1111/j.1471-4159.2010.06918.x [DOI] [PubMed] [Google Scholar]
- 26. Hudson J, Granholm AC, Gerhardt GA, Henry MA, Hoffman A, et al. (1995) Glial cell line-derived neurotrophic factor augments midbrain dopaminergic circuits in vivo. Brain Res Bull 36: 425–432. [DOI] [PubMed] [Google Scholar]
- 27. Hebert MA, Gerhardt GA (1997) Behavioral and neurochemical effects of intranigral administration of glial cell line-derived neurotrophic factor on aged Fischer 344 rats. J Pharmacol Exp Ther 282: 760–768. [PubMed] [Google Scholar]
- 28. Hebert MA, Van Horne CG, Hoffer BJ, Gerhardt GA (1996) Functional effects of GDNF in normal rat striatum: presynaptic studies using in vivo electrochemistry and microdialysis. J Pharmacol Exp Ther 279: 1181–1190. [PubMed] [Google Scholar]
- 29. Emerich DF, Plone M, Francis J, Frydel BR, Winn SR, et al. (1996) Alleviation of behavioral deficits in aged rodents following implantation of encapsulated GDNF-producing fibroblasts. Brain Res 736: 99–110. [PubMed] [Google Scholar]
- 30. Rosenblad C, Georgievska B, Kirik D (2003) Long-term striatal overexpression of GDNF selectively downregulates tyrosine hydroxylase in the intact nigrostriatal dopamine system. Eur J Neurosci 17: 260–270. [DOI] [PubMed] [Google Scholar]
- 31. Kumar A, Varendi K, Peranen J, Andressoo JO (2014) Tristetraprolin is a novel regulator of BDNF. Springerplus 3: 502 10.1186/2193-1801-3-502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, et al. (2007) A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129: 1401–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. John B, Enright AJ, Aravin A, Tuschl T, Sander C, et al. (2004) Human MicroRNA targets. PLoS Biol 2: e363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wen J, Parker BJ, Jacobsen A, Krogh A (2011) MicroRNA transfection and AGO-bound CLIP-seq data sets reveal distinct determinants of miRNA action. RNA 17: 820–834. 10.1261/rna.2387911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ (2005) The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci U S A 102: 10898–10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shakya R, Jho EH, Kotka P, Wu Z, Kholodilov N, et al. (2005) The role of GDNF in patterning the excretory system. Dev Biol 283: 70–84. [DOI] [PubMed] [Google Scholar]
- 37. Parish CL, Finkelstein DI, Drago J, Borrelli E, Horne MK (2001) The role of dopamine receptors in regulating the size of axonal arbors. J Neurosci 21: 5147–5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kowsky S, Poppelmeyer C, Kramer ER, Falkenburger BH, Kruse A, et al. (2007) RET signaling does not modulate MPTP toxicity but is required for regeneration of dopaminergic axon terminals. Proc Natl Acad Sci U S A 104: 20049–20054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Du Y, Zhang X, Tao Q, Chen S, Le W (2013) Adeno-associated virus type 2 vector-mediated glial cell line-derived neurotrophic factor gene transfer induces neuroprotection and neuroregeneration in a ubiquitin-proteasome system impairment animal model of Parkinson's disease. Neurodegener Dis 11: 113–128. 10.1159/000334527 [DOI] [PubMed] [Google Scholar]
- 40. Love S, Plaha P, Patel NK, Hotton GR, Brooks DJ, et al. (2005) Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat Med 11: 703–704. [DOI] [PubMed] [Google Scholar]
- 41. Smith-Hicks CL, Sizer KC, Powers JF, Tischler AS, Costantini F (2000) C-cell hyperplasia, pheochromocytoma and sympathoadrenal malformation in a mouse model of multiple endocrine neoplasia type 2B. Embo J 19: 612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mijatovic J, Piltonen M, Alberton P, Mannisto PT, Saarma M, et al. (2009) Constitutive Ret signaling is protective for dopaminergic cell bodies but not for axonal terminals. Neurobiol Aging. [DOI] [PubMed] [Google Scholar]
- 43. Mijatovic J, Airavaara M, Planken A, Auvinen P, Raasmaja A, et al. (2007) Constitutive Ret activity in knock-in multiple endocrine neoplasia type B mice induces profound elevation of brain dopamine concentration via enhanced synthesis and increases the number of TH-positive cells in the substantia nigra. J Neurosci 27: 4799–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mijatovic J, Patrikainen O, Yavich L, Airavaara M, Ahtee L, et al. (2008) Characterization of the striatal dopaminergic neurotransmission in MEN2B mice with elevated cerebral tissue dopamine. J Neurochem 105: 1716–1725. 10.1111/j.1471-4159.2008.05265.x [DOI] [PubMed] [Google Scholar]
- 45. Varendi K, Matlik K, Andressoo JO (2015) From microRNA target validation to therapy: lessons learned from studies on BDNF. Cell Mol Life Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vasudevan S (2012) Posttranscriptional upregulation by microRNAs. Wiley Interdiscip Rev RNA 3: 311–330. 10.1002/wrna.121 [DOI] [PubMed] [Google Scholar]
- 47. van der Weyden L, Adams DJ, Harris LW, Tannahill D, Arends MJ, et al. (2005) Null and conditional Semaphorin 3B alleles using a flexible puro Delta tk LoxP/FRT vector. Genesis 41: 171–178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
Gdnf KO animals were obtained by crossing Gdnf wt/hyper animals to Deleter-Cre mice, which results in deletion of GDNF protein coding exon 3 (Fig 1B) [10].
(DOCX)
(DOCX)
Correlational analyses of rostral brain dopamine and serum urea levels in individual animals in 10 Gdnf wt/wt and 12 Gdnf wt/hyper animals at P7.5 using the Correlation function in Microsoft Excel.
(DOCX)
Levels of mature miRs expressed as fold difference relative to sno202 in adult mouse brain, developing mouse kidney and in HEK293 cells. N = 2 experiments. dSTR, dorsal striatum, E, embryonic day; Hs, Homo sapiens; Mm, Mus musculus; P, postnatal day; w, weeks.
(DOCX)
(A) Schematic representation of the reporter constructs and the derived mRNAs used in this study. Red and blue bar indicate probes used in the Northern blot for firefly luciferase CDS and Gdnf 3’UTR, respectively. (B) The puΔtk cassette blocks transcription to the Gdnf 3’UTR, measured by Northern blot analysis of expression from a construct harboring the Firefly (FF) coding sequence upstream of the puΔtk-Gdnf 3’UTR cassette in HEK293 cells; loading control: 28S ribosomal RNA. (C) Northern blot analysis of expression from construct harboring FF coding sequence proceeded with puΔtk-Gdnf 3’UTR cassette (S1 Fig A) in HEK293 cells (S1 Fig D); loading control: 28S ribosomal RNA. (D) Expression from equimolar amounts of the FF-Gdnf 3’UTR, FF-puΔtk-Gdnf 3’UTR and FF-SV40 pA constructs in U87 and HEK293 cells; FF-firefly luciferase; N = 3 experiments/construct with 3 replicates/experiment. (E) Gdnf 3’UTR reduces expression of both Renilla luciferase (Ren) and FF in HEK293 and U87 cells; N = 3 experiments/construct with 3 replicates/experiment (F) FF expression from constructs containing SV40 pA, puΔtk-Gdnf 3’UTR cassette and Gdnf 3’UTR in HEK293 cells after treatment with actinomycin D. Note that prevention of transcription of the native Gdnf 3’UTR by preceding puΔtk alleviates the post-transcriptional inhibition of the reporter. Renilla luciferase expression from a separate plasmid was used for normalization. N = 3 experiments/construct with 3 replicates/experiment. (G) Targeting strategy of the Gdnf locus. 5667 bp 5’ (green bar) and 6055 bp 3’ (blue bar) homologous arms, and Gdnf exon 3 until and including the stop codon were amplified with PCR from Gdnf-containing PAC and cloned into PmeI, NotI and HindIII sites in pFlexible [31], respectively, to generate Gdnf targeted allele. Sequence lengths are not drawn to scale. Arrows indicate primers used for routine genotyping; black bar indicates probe used for Southern blotting using BlpI restriction enzyme shown on (H). (I) Representative image of routine genotyping of the mice using primers indicated on G. Abbreviations: puΔtk, a cassette with PGK promoter encoding for fusion protein between puromycin N-acetyltransferase and a truncated version of herpes simplex virus type 1 thymidine kinase (TK) with 3’ bovine growth hormone polyadenylation signal (pA); FF-firefly luciferase; CDS-coding sequence.
(TIF)
(A-B) Representative images of in situ hybridization of Gdnf mRNA using a probe against the CDS (stained blue, indicated with white arrow heads) in mice in whole-mount preparations of E11.5 hindlimb (A) and in paraffin sections from P7.5 testis (B). Note that Gdnf expression sites are comparable between genotypes, whereas the signal appears stronger in Gdnf hyper/hyper mice. N = 4 mice/group in A and N = 2 in B. (C-D) Gdnf mRNA (C) and protein (D) expression in the testis at E18.5 measured with QPCR and ELISA, respectively, shows allele dose-dependent increase in GDNF levels in GDNF hypermorphic mice. Note that GDNF protein levels in Gdnf 3’UTR rest/rest mice are normal (D). N = 2–5 mice/group in 2–3 experiments with 2–3 replicates/experiment (E) Representative image of Northern blot analysis of Gdnf mRNA in E18.5 testis using Gdnf CDS probe. Note that the size of the band is ca 500 bp shorter in Gdnf hyper/hyper mice compared to the Gdnf wt/wt mice, consistent with the size of transcripts derived from Gdnf-puΔtk fusion transcript (S1A–S1C Fig; and see S2 Fig F, G). N = 2 mice/group. (F) Schematic representation of the puΔtk-Gdnf 3’UTR (Gdnf hyper) allele for Gdnf, A and B designate primers used in QPCR analysis of Gdnf mRNA levels (C); primers A and C depict primers used for Gdnf transcript sequence analysis in (G). (G) Representative image of RT-PCR analysis from E18.5 testis using primers A and C as depicted in (F). The observed PCR product in Gdnf hyper/hyper mice was of expected length (977 bp) and it was sequenced for validation; N = 3 mice; beta-actin was used to verify cDNA quality. (H-I) Kidneys of P7.5 Gdnf hyper/hyper mice function poorly, as indicated by serum creatinine (H) and serum urea (I) levels. H, N = 3–12; I, N = 7–26 mice/group. (J-K) The blood serum of adult Gdnf wt/hyper mice contains slightly higher levels of urea (J), but not creatinine (K) compared to wild type mice. Note that two-fold variation in serum urea levels is considered normal in humans. N = 16–19 mice/group. (L) Representative image of kidneys in wild type and Gdnf 3’UTRrest/rest mice. In total, 20 mice were analyzed from each genotype. (M) Verification of the specificity of RNAscope probes against parvalbumin (left panel; blue) and Gdnf (right panel; red). Note that the signal matches known parvalbumin mRNA expression in Purkinje cells (black triangles) and in molecular layer interneurons (open triangles) at three months of age. Similarly, Gdnf mRNA can be detected in the metanephric mesenchyme (MM) and not in the ureteric bud (UB) in the developing kidney at E14.5. Scale bars: A, 1.5 mm; B, 5 μm; M, 1 mm; L, 70 μm (left) and 100 μm (right). Abbreviations: bGHpA, bovine growth hormone polyadenylation signal; bp, base pairs; CDS, coding sequence; Cp, quantification cycle; dSTR, dorsal striatum; E, embryonic day; F, FRT sites for Flp recombinase; IHC, immunohistochemistry; kb, kilo-base pairs; m, months; MM, metanephric mesenchyme; P, postnatal day; PV, parvalbumin; puΔtk, a cassette with PGK promoter encoding for fusion protein between puromycin N-acetyltransferase and a truncated version of herpes simplex virus type 1 thymidine kinase (TK) with 3’ bovine growth hormone polyadenylation signal; UB, ureteric bud. *P< 0.05, **P< 0.01, and ***P< 0.001; error bars indicate SEM.
(TIF)
(A-B) HPLC analysis of DA metabolites DOPAC (A) and HVA (B) in P7.5 rostral brain. N = 5–8 mice/group. (C-D) HPLC analysis of DA metabolites DOPAC (C) and HVA (D) in the dSTR at 2.5–3 months of age. N = 11 for Gdnf wt/wt, 8 for Gdnf wt/hyper; P = 0.00521 for DOPAC and P = 0.0569 for HVA. (E) HPLC analysis of DA levels in the dSTR of Gdnf 3’UTR rest/rest and Gdnf wt/wt mice; N = 6 mice/group. (F) Representative images of DAT immunostaining in the dSTR, N = 7–9 mice/group. (G-H) Representative images of TH immunostaining in the dSTR (G) and midbrain (H). In total, 8 mice were analyzed from each genotype. (I) Average striatal area size measured from DAT+ sections. (N = 9 for Gdnf wt/wt, 7 for Gdnf wt/hyper (J) Representative recording trace of DA events stimulated by single electrical pulses at 2 min intervals, by paired stimulations at shown intervals, and by a burst of 5 stimuli at 20 Hz. (K-O) Rise and decay parameters of the first five DA events evoked by single-pulses. N = 5–7 mice/group with 1–3 striatal slices/mouse analyzed. (K) DA rise slope is steeper in the striata of Gdnf wt/hyper mice [two-way repeated measures ANOVA, F (1,29) = 11.47, P = 0.0021], indicating that more dopamine is released from the terminals during the rise phase. (L) T ½ times, i.e., the width of the event at half of the maximum. (M) Rise time, i.e. the time for DA events to rise from baseline to the maximum. (N) The fall time of the DA events. (O) The exponential decay time constant of the DA transients. Abbreviations: DA, dopamine; DOPAC, 3,4-dihydroxyphenylacetic acid; dSTR, dorsal striatum; HPLC, high performance liquid chromatography; HVA, homovanillic acid; IHC, immunohistochemistry; m, months; P, postnatal day. Scale bars: F, 10 μm; G, 75 μm; in inset 1 mm; H, 0.5 mm; in inset 30 μm.
(TIF)
(A-B). Total DAT protein levels (A) and plasma membrane associated DAT levels (B) at P7.5, measured by western blotting. N = 4 mice/group. (C) DAT protein levels in the dSTR and SNpc at 2.5-3m, measured by western blotting. Gapdh or α-tubulin was used to normalize loading, as indicated in the figure. N = 5 mice/group. (D) OD of striatal TH+ fibers at P7.5, reflecting striatal TH levels and the density of striatal dopaminergic innervation at the macroscopic level. N = 7 mice/group. (E) TH protein levels in the dSTR at P7.5, measured by western blotting. Gapdh was used to normalize loading. N = 4 mice/group. (F) OD of striatal TH+ fibers at 2.5–3 months of age. N = 10–11 mice/group. (G) TH protein levels in the dSTR at 2.5–3 months of age, measured by western blotting. Gapdh was used to normalize loading. N = 5 mice/group. (H) Luciferase expression from Firefly-Gdnf 3’UTR construct in HEK293 cells after co-transfection with constructs encoding RBPs. Renilla luciferase expression from a separate plasmid was used for normalization and GFP was used as a negative control. N = 3 experiments with 3 biological repeats each. (I) The number of predicted miR binding sites (ddG<0) in Gdnf 3’UTR, puΔtk and SV40 late pA signal, analyzed with PITA software [4]. (J) The number of predicted strong binding sites for Gdnf-specific miRs (ddG<-10) in Gdnf 3’UTR, puΔtk and SV40 late pA signal, analyzed with PITA software [4]. Selection of Gdnf-specific miRs was based on TargetScan analysis using the most stringent conditions. (K) Luciferase expression from Firefly-puΔtk-Gdnf 3’UTR construct in HEK293 cells after co-transfection with Gdnf-regulating pre-miRs. Note that miRs that inhibit luciferase expression via the native Gdnf 3’UTR (Fig 5B) do not suppress the expression from reporter construct containing the Firefly-puΔtk-Gdnf 3’UTR cassette. N = 3 experiments with 3–5 biological repeats. (L) Luciferase activity in an ATP-based survival assay in U87 cells after co-transfection with pre-miRs. Note that the viability of U87 cells is not affected by the overexpression of pre-miRs, compared to scrambled control pre-miRs. N = 3 experiments with 3 replicates/experiment. (M) Gdnf mRNA levels in HEK293 cells after transfection with pAAV constructs encoding shRNAs against miR-9, miR-96 and miR-146. A number of shRNA-coding plasmids from two different producers, Signagen (left panel) and Vector Biosystems (right panel), were tested. N = 5 experiments with 2 repeats (all constructs from Signagen) and N = 1–2 experiments (constructs from Vector Biosystems). $ indicates that the constructs were tested 2 times with 2 repeats. The rest of the constructs from Vector Biosystems were tested once with 2 repeats. siRNA against Dicer was included as a positive control. N = 5 experiments with 2 repeats. Abbreviations: AAV, adeno-associated virus; RBP, RNA-binding protein; GFP, green fluorescent protein; AUF1, AU-rich element binding protein 1; ELAVL1, embryonic lethal, abnormal vision-like protein 1; TTP, tristetraprolin; puΔtk, a cassette with PGK promoter encoding for fusion protein between puromycin N-acetyltransferase and a truncated version of herpes simplex virus type 1 thymidine kinase (TK) with 3’ bovine growth hormone polyadenylation signal; scr, scrambled control; pA, polyadenylation signal. *P<0.05, **P<0.01, and ***P<0.001; error bars indicate SEM.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.