

Background
Kidney transplantation corrects or improves many complications of chronic kidney disease, but its impact on disordered mineral metabolism is incompletely understood.
Methods
We performed a multicenter, prospective, observational cohort study of 246 kidney transplant recipients in the United States to investigate the evolution of mineral metabolism from pretransplant through the first year after transplantation. Participants were enrolled into 2 strata defined by their pretransplant levels of parathyroid hormone (PTH), low PTH (>65 to ≤300 pg/mL; n = 112), and high PTH (>300 pg/mL; n = 134) and underwent repeated, longitudinal testing for mineral metabolites.
Results
The prevalence of posttransplant, persistent hyperparathyroidism (PTH >65 pg/mL) was 89.5%, 86.8%, 83.1%, and 86.2%, at months 3, 6, 9, and 12, respectively, among participants who remained untreated with cinacalcet, vitamin D sterols, or parathyroidectomy. The results did not differ across the low and high PTH strata, and rates of persistent hyperparathyroidism remained higher than 40% when defined using a higher PTH threshold greater than 130 pg/mL. Rates of hypercalcemia peaked at 48% at week 8 in the high PTH stratum and then steadily decreased through month 12. Rates of hypophosphatemia (<2.5 mg/dL) peaked at week 2 and then progressively decreased through month 12. Levels of intact fibroblast growth factor 23 decreased rapidly during the first 3 months after transplantation in both PTH strata and remained less than 40 pg/mL thereafter.
Conclusions
Persistent hyperparathyroidism is common after kidney transplantation. Further studies should determine if persistent hyperparathyroidism or its treatment influences long-term posttransplantation clinical outcomes.
The prevalence of posttransplant hyperparathyroidism was 86% at 12 months (PTH >65 pg/ml) but only 40% (PTH >130 mg/dL) in the absence of cinacalcet, vitamin D sterols, or parathyroidectomy. Intact fibroblast growth factor 23 decreased rapidly to G40 pg/ml by 3 months posttransplant. Supplemental digital content is available in the text.
Kidney transplantation is the preferred treatment for end-stage renal disease (ESRD) because it reverses many complications of chronic kidney disease (CKD), improves recipients' quality of life, and prolongs survival.1 Advances in immune suppression, which vastly enhanced short-term allograft outcomes, have enabled the transplant community to focus more attention on managing the nonimmune aspects of the posttransplant period to optimize recipients' long-term health.
Disordered mineral metabolism is a common complication of CKD that begins early in the course of disease and progressively worsens as patients approach ESRD.2 In its most severe form, disordered mineral metabolism in ESRD is characterized by hyperphosphatemia, hypocalcemia, deficiencies of 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D, and markedly elevated levels of parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF23).3-8 Individually and in aggregate, these alterations are associated with increased risks of cardiovascular disease, fracture, and death.8-12 Although restoring normal kidney function by transplanting a healthy kidney might be expected to fully reverse disordered mineral metabolism due to ESRD, most existing data suggest that transplantation only partially corrects certain alterations.13-18 Furthermore, presence of a healthy allograft that can respond to the hormonal effects of lingering elevations in PTH and FGF23 levels can precipitate de novo alterations in mineral metabolism, including hypercalcemia and hypophosphatemia.19,20 Posttransplant hypercalcemia and hypophosphatemia present clinicians with management challenges because they may jeopardize graft function and bone health and exacerbate fracture and cardiovascular risk.20-22
The postkidney transplant period should be considered a unique phase in the natural history of disordered mineral metabolism associated with CKD that requires dedicated investigation.14 Few studies have systematically studied mineral metabolism in the posttransplant period. Among those that did, most previous studies were small, single-center, brief, and failed to measure a comprehensive panel of mineral metabolites.23-28 As a result, the frequency of hypercalcemia, hypophosphatemia, and persistent hyperparathyroidism during the first year after kidney transplantation remain incompletely characterized. We conducted the current prospective, observational study to examine the evolution of persistent hyperparathyroidism and associated alterations in mineral metabolism during the first year after kidney transplantation.
MATERIALS AND METHODS
Study Design and Population
We performed a prospective, observational cohort study in 12 kidney transplant centers across the United States (Lists of Study Team Members, SDC, http://links.lww.com/TP/B170). Individuals aged 18 years or older who were admitted for kidney transplantation were eligible to participate if they were currently undergoing hemodialysis treatment for ESRD, had at least 1 documented measurement of plasma PTH greater than 65 pg/mL within the previous 6 months, and a plasma PTH greater than 65 pg/mL at the time of admission for transplantation. Exclusion criteria included multiple organ transplantation or history of medical conditions known to cause hypercalcemia, such as primary hyperparathyroidism, active malignancy, or granulomatous diseases. The study was approved by the institutional review boards of the participating clinical centers, and all participants provided written informed consent.
Two hundred forty-six individuals met the study's eligibility criteria and underwent kidney transplantation. They were enrolled in 2 strata, according to their pretransplant baseline plasma PTH level that was measured on admission for transplantation: low PTH (>65 to ≤ 300 pg/mL) or high PTH (>300 pg/mL). To reflect the general US kidney transplant population, the protocol prespecified enrollment of up to 90 living donor recipients among the study's 246 total participants. Baseline laboratory values for the study were collected from participants after they were admitted to the hospital for transplantation.
The duration of clinical observation was 12 months, during which serum and plasma samples were collected at weeks 1, 2, 4, and 8 and at months 3, 6, 9, and 12 after transplantation to measure mineral metabolites and allograft function. Concomitant use of cinacalcet, vitamin D sterols, and other medications were ascertained at each study visit. To minimize confounding that would be introduced by poor allograft function, the protocol specified withdrawal of participants with sustained reductions in estimated glomerular filtration rate (eGFR, via the Modification of Diet in Renal Disease equation29) of less than 30 mL/min per 1.73 m2 for greater than 75 consecutive days after transplantation. Other reasons for removal from the study included withdrawal of consent or loss to follow-up.
Assays
Serum calcium, phosphate, and creatinine and plasma PTH and FGF23 were measured at each study visit. Serum 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D, bone-specific alkaline phosphatase (BALP), procollagen type 1 N-terminal propeptide (P1NP), C-terminal telopeptide type 1, osteocalcin, and urine N-terminal telopeptide corrected to the simultaneous urine creatinine were measured at baseline and at months 3, 6, and 12. All laboratory tests were performed in real time by a central laboratory (Covance Central Laboratory Services, Indianapolis, IN). Intact PTH was measured using a 2-site sandwich immunoassay on an ADVIA Centaur Immunoassay System (Siemens Medical Solutions USA, Malvern, PA). The FGF23 was measured using an intact enzyme-linked immunosorbent assay (ELISA; Immutopics International, San Clemente, CA). Both 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D were measured with a radioimmunoassay (DiaSorin, Stillwater, MN). The BALP was measured with a one-step immunoenzymatic assay (Beckman Coulter Inc, Brea, CA), P1NP with a radioimmunoassay (Orion Diagnostica, Espoo, Finland), C-terminal telopeptide type 1 with an ELISA (Alere Scarborough, Inc, Scarborough, ME), osteocalcin with an electrochemiluminescence immunoassay (Roche Diagnostics, Indianapolis, IN), and N-terminal telopeptide with an ELISA (IDS, Herlev, Denmark).
Statistical Analysis
For descriptive analyses of demographics and laboratory characteristics, continuous variables are reported as means ± SD or SE, and categorical variables as frequencies and proportions.
The primary, prespecified measure of interest was the rate of persistent hyperparathyroidism, defined as PTH greater than 65 pg/mL at months 3, 6, 9, and 12 among “untreated” participants. For the primary measure of interest, participants were considered “treated” if they had undergone parathyroidectomy at any time before a scheduled study visit or received off-label, posttransplant treatment with cinacalcet or active vitamin D sterols within 3 months before a scheduled study visit. The secondary, prespecified measure of interest was the rate of persistent hyperparathyroidism among all participants, including those who were treated. For this analysis, treated participants were considered as having persistent hyperparathyroidism in addition to those with PTH greater than 65 pg/mL. In a secondary analysis, we defined persistent hyperparathyroidism as PTH greater than 130 pg/mL, which was 2 times the laboratory's upper limit of normal. For each of these analyses, we calculated the rates and 95% CIs of persistent hyperparathyroidism at months 3, 6, 9, and 12.
We used a similar approach to calculate rates of hypercalcemia (albumin-corrected serum calcium > 10.2 mg/dL) and hypophosphatemia (serum phosphate < 2.5 mg/dL) at each time point among the untreated population. We also analyzed values of PTH, FGF23, albumin-corrected serum calcium, eGFR, and serum phosphate among untreated participants. We used repeated measures modeling for all statistical comparisons. For each analyte, we compared the values between the high versus low PTH strata, and between baseline versus peak or nadir values within each stratum. We also estimated the difference between the high and low PTH strata with 95% CIs and calculated a P value for the comparison.
We performed logistic regression analyses to assess predictors of persistent hyperparathyroidism (PTH > 65 pg/mL) and hypercalcemia (>10.2 mg/dL) at the 3, 6, 9, and 12 month visits. Candidate factors that we considered included age (< or ≥ 65 years), sex, race, baseline PTH stratum, diabetes, dialysis vintage, pretransplant cinacalcet use, pretransplant vitamin D sterol use, baseline-corrected serum calcium (≤10.2 or > 10.2 mg/dL), baseline serum phosphate (<2.5, 2.5 to < 5.1, or ≥ 5.1 mg/dL), and kidney transplant donor type (living or deceased). Baseline PTH stratum (forced into model) and factors that met the stay criterion (P < 0.10) were maintained in the final model. We calculated Spearman correlation coefficients to describe the relationships between FGF23 and PTH, PTH and 1,25-dihydroxyvitamin D, FGF23 and 1,25-dihydroxyvitamin D, and FGF23 and phosphate.
Nominal P values were generated with no adjustment for multiplicity. No imputation methods were used for missing data. Analyses were performed using SAS version 9.2 and later (SAS Institute, Cary, NC).
RESULTS
Two hundred forty-six individuals from 12 kidney transplant centers across the United States were enrolled in 1 of 2 strata, according to their pretransplant baseline plasma PTH level: low PTH (>65 to ≤ 300 pg/mL) or high PTH (>300 pg/mL). Demographics and baseline characteristics of the study population overall and by PTH strata are presented in Table 1. Key laboratory values at baseline are presented in Table 2. In addition to higher PTH, the high PTH stratum had higher baseline levels of FGF23, serum phosphate, and BALP, and lower albumin-corrected serum calcium compared to the low PTH stratum.
TABLE 1.
Demographics and baseline characteristics overall and by strata of parathyroid hormone

TABLE 2.
Baseline biochemical characteristics overall and by strata of parathyroid hormone

The disposition of the 246 participants who enrolled in the study is presented in Figure 1. Twenty-two participants withdrew before month 3. Among the 224 who continued, 41 received treatment with off-label cinacalcet (Sensipar/Mimpara; Amgen Inc, Thousand Oaks, CA) or active vitamin D sterols or underwent parathyroidectomy (which was performed in 1 participant in each stratum) by month 3. Among these 41 participants, 23 remained on treatment through month 12; 12 subsequently discontinued treatment, and 5 withdrew from the study before month 12. Among the 183 participants who went untreated during the first 3 months, 7 initiated treatment between months 3 and 6, and 32 withdrew from the study before month 12, leaving 144 who completed the full year of follow-up without undergoing parathyroidectomy or ever receiving treatment with off-label cinacalcet or active vitamin D sterols.
FIGURE 1.

Disposition of all enrolled study participants over time. “Untreated” patients were those who did not undergo parathyroidectomy and did not receive off-label treatment with cinacalcet or vitamin D sterols after transplantation and before the scheduled visit. aOf the 8 who stopped treatment, 1 returned to treatment by Month 12. bOf the 7 who initiated treatment, 1 terminated study early between months 9 and 12.
The primary reasons for withdrawal from the study included protocol-specified discontinuation for impaired allograft function (eGFR < 30 mL/min per 1.73 m2 for > 75 consecutive days) in 18 participants, loss to follow-up in 19, withdrawal of consent in 15, and death in 9. Three participants in the high PTH stratum (1.2%) experienced permanent loss of allograft function, defined as the need to reinitiate dialysis for greater than 30 consecutive days. The mean (± SD) eGFR in the overall population throughout months 3 to 12 was 52.5 ± 16.4 mL/min per 1.73 m2. Throughout the study, the difference in eGFR levels between the high and low PTH strata was minimal (1.5 mL/min per 1.73 m2; 95% confidence interval [95% CI], −1.6 to 4.5 mL/min per 1.73 m2; P = NS).
Parathyroid Hormone
Mean (± SE) posttransplant plasma levels of PTH among untreated participants are presented in Figure 2A. In the high PTH stratum, mean PTH levels decreased sharply during the first 3 months after transplantation (615.2 ± 31.0 pg/mL at baseline to 202.9 ± 18.2 pg/mL by month 3; P < 0.001), and then declined more gradually through month 12. In the low PTH stratum, PTH increased slightly from baseline to month 6 (193.8 ± 5.8 to 222.5 ± 94.3 pg/mL by month 6; P = NS) before gradually decreasing. In both strata, however, mean PTH remained substantially elevated at month 12 (high PTH stratum: 146.3 ± 13.4; low PTH stratum: 118.1 ± 7.0 pg/mL).
FIGURE 2.

Posttransplant levels of PTH among untreated participants, defined as those who did not undergo parathyroidectomy at any time after transplantation and did not receive off-label treatment with cinacalcet or vitamin D sterols within 3 months before the scheduled study visit. A, Mean (± standard error [SE]) PTH (pg/mL) levels over time by PTH stratum among untreated participants. B, Rate of persistent hyperparathyroidism (HPT) (PTH > 65 pg/mL) among untreated participants at months 3, 6, 9, and 12. C, Distribution of normal PTH (PTH ≤ 65 pg/mL) and persistent HPT using 2 PTH cutpoints (>65 to ≤ 130; > 130 pg/mL) among untreated participants at months 3, 6, 9, and 12.
The primary, prespecified measure of interest was the rate of persistent hyperparathyroidism, defined as PTH greater than 65 pg/mL, at months 3, 6, 9, and 12 among “untreated” participants, which included those who had not undergone parathyroidectomy at any time before a scheduled visit, or had not received off-label, posttransplant treatment with cinacalcet or active vitamin D sterols within 3 months before a scheduled study visit (Figure 2B). The rates (95% CI) among all untreated participants were 89.5% (85.0-94.1%) at month 3, 86.8% (81.5-92.1%) at month 6, 83.1% (77.2-89.0%) at month 9, and 86.2% (80.2-92.1%) at month 12.
When considering all participants who received treatment as having persistent hyperparathyroidism, the rates of persistent hyperparathyroidism were even higher: 91.5% (87.8-95.3%) at month 3, 89.2% (84.9-93.6%) at month 6, 86.2% (81.2-91.1%) at month 9, and 88.8% (83.9-93.6%) at month 12. The rates of persistent hyperparathyroidism among untreated participants remained high even when higher PTH cut points were used to define persistent hyperparathyroidism (Figure 2C).
In a multivariable logistic regression analysis using backward selection, the identified independent predictors of hyperparathyroidism at month 3 after transplant (PTH > 65 pg/mL) were greater dialysis vintage (odds ratio 1.44 per each year; 95% CI, 1.05-1.98; P = 0.025), and pretransplant treatment with vitamin D sterols, which was associated with significantly lower risk (odds ratio, 0.29; 95% CI, 0.10-0.87; P = 0.028). Pretransplant cinacalcet use may be associated with higher likelihood of hyperparathyroidism at month 3 after transplantation (odds ratio, 8.20; 95% CI, 0.99-67.94; P = 0.051). Similar analyses at months 6, 9, and 12 yielded inconsistent results with no single factor consistently predicting likelihood of hyperparathyroidism at all time points; however, the absolute rates of persistent hyperparathyroidism were numerically higher among participants who were treated with cinacalcet before transplantation (94-100% at the different time points during follow-up) than in those who did not receive pretransplant cinacalcet (78-85%).
Calcium
Mean (±SE) posttransplant levels of albumin-corrected serum calcium among untreated participants are presented in Figure 3A. Levels increased significantly from baseline to week 4 in both the high PTH stratum (9.6 ± 0.1 to 10.2 ± 0.1 mg/dL; P < 0.001) and the low PTH stratum (9.8 ± 0.1 to 10.0 ± 0.1 mg/dL; P < 0.001), and remained elevated above baseline through month 12. Results were qualitatively similar using uncorrected serum calcium levels (data not shown).
FIGURE 3.

Posttransplant levels of corrected serum calcium among untreated participants, defined as those who did not undergo parathyroidectomy and did not receive off-label treatment with cinacalcet or vitamin D sterols after transplantation and before the scheduled visit. A, Mean (±SE) total corrected serum calcium (mg/dL) levels over time by PTH stratum. B, Rate (95% CI) of hypercalcemia (corrected serum calcium > 10.2 mg/dL) over time by PTH stratum.
Rates of hypercalcemia (albumin-corrected serum calcium > 10.2 mg/dL) among untreated participants are presented in Figure 3B. By week 1 after transplantation, the rate (95% CI) of hypercalcemia was 30.6% (22.1-39.2%) in the high PTH stratum and 21.7% (13.9-29.5%) in the low PTH stratum. The rate of hypercalcemia increased to a peak of 48.2% (37.6-58.9%) at week 8 in the high PTH stratum, and then steadily decreased through month 12. In the low PTH stratum, the rate of hypercalcemia peaked at 29.0% (19.8-38.3%) at week 2, and then steadily decreased through month 12.
In multivariable logistic regression analyses, the identified independent predictors of posttransplant hypercalcemia (corrected calcium >10.2 mg/dL), at months 3, 6, 9, and 12 were high versus low baseline PTH stratum (all P values ≤ 0.05; Table S1, SDC, http://links.lww.com/TP/B170), and baseline corrected serum calcium greater than 10.2 mg/dL (all P values < 0.01; Table S1, SDC, http://links.lww.com/TP/B170). Although the absolute rates of posttransplant hypercalcemia were numerically higher among participants who were treated with cinacalcet before transplantation (24-49% at the different time points during follow-up) than in those who did not receive pretransplant cinacalcet (13-32%), pretransplant cinacalcet use was not an independent predictor of posttransplant hypercalcemia at any time point.
Fibroblast Growth Factor 23
Mean (±SE) posttransplant plasma levels of intact FGF23 among untreated participants are presented in Figure 4A. In both strata, FGF23 levels decreased rapidly during the first 3 months after transplantation: from 492.1 ± 25.7 at baseline to 34.1 ± 10.7 pg/mL by month 3 in the high PTH stratum (P < 0.001), and from 355.6 ± 29.3 at baseline to 16.9 ± 1.3 pg/mL by month 3 in the low PTH stratum (P < 0.001). Values remained stable thereafter and were consistently less than 40 pg/mL in both strata.
FIGURE 4.
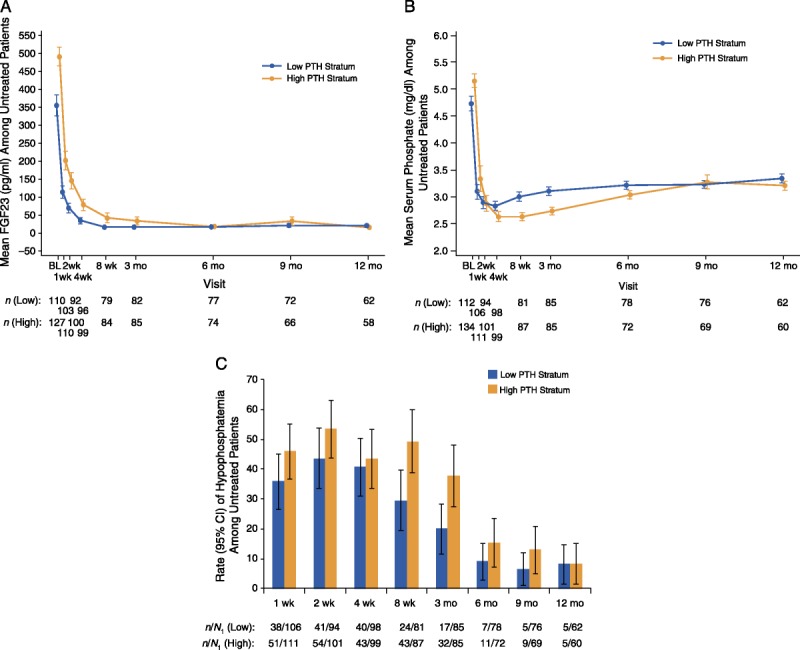
Posttransplant levels of FGF23 and serum phosphate among untreated participants, defined as those who did not undergo parathyroidectomy and did not receive off-label treatment with cinacalcet or vitamin D sterols after transplantation and before the scheduled visit. A, Mean (±SE) plasma FGF23 (pg/mL) levels over time by PTH stratum. B, Mean (± SE) serum phosphate (mg/dL) levels over time by PTH stratum. C, Rate (95% CI) of hypophosphatemia (serum phosphate < 2.5 mg/dL) over time by PTH stratum.
Phosphate
Mean (±SE) posttransplant levels of serum phosphate among untreated participants are presented in Figure 4B. Serum phosphate decreased from baseline within the first 4 weeks after transplantation in both the high PTH stratum (5.2 ± 0.1 to 2.6 ± 0.1 mg/dL; P < 0.001) and the low PTH stratum (4.7 ± 0.1 to 2.8 ± 0.1 mg/dL; P < 0.001). Levels increased slightly thereafter and remained stable through month 12 at 3.3 ± 0.1 mg/dL in the overall study population.
Rates of hypophosphatemia (serum phosphate < 2.5 mg/dL) among untreated participants are presented in Figure 4C. By week 2 after transplantation, the rate (95% CI) of hypophosphatemia was 53.5% (43.7-63.2%) in the high PTH stratum and 43.6% (33.6-53.6%) in the low PTH stratum. Thereafter, rates of hypophosphatemia progressively decreased through month 12.
Other Analytes
Mean (±SE) levels of serum 1,25-dihydroxyvitamin D levels increased from baseline to month 12 in both strata (high PTH stratum: 27.5 ± 1.3 to 54.7 ± 3.1 pg/mL; low PTH stratum, 25.9 ± 1.1 to 55.9 ± 2.1 pg/mL; Figure S1, SDC, http://links.lww.com/TP/B170). Mean (±SE) levels of serum 25-hydroxyvitamin D levels decreased slightly from baseline to month 12 in both strata (high PTH stratum: 32.5 ± 1.5 to 28.5 ± 2.0 ng/mL; low PTH stratum: 30.8 ± 1.5 to 30.5 ± 1.4 ng/mL). The BALP was higher at baseline in the high PTH stratum; the levels increased from baseline through months 3 to 6 before declining by month 12 in both groups. P1NP was initially higher in the high PTH group and decreased substantially over time through month 12. Procollagen type 1 N-terminal propeptide was relatively stable through month 12 in the low PTH stratum. Other markers of bone turnover were initially higher in the high PTH stratum, decreased substantially over time in both strata, and were similar across the strata by month 12 (Figure S2, SDC, http://links.lww.com/TP/B170).
Relationships Between Analytes
Higher FGF23 correlated with higher PTH during the first 3 months after transplantation (month 3, r = 0.33, P < 0.01), but the association weakened over time (month 12, r = 0.09; P = 0.34). Higher FGF23 correlated with lower 1,25-dihydroxyvitamin D at month 3 (r = −0.37; P <0.01) and month 6 after transplantation (r = −0.28; P <0.01), but there was no significant association by month 12. Between week 4 and month 3 after transplantation, higher FGF23 correlated with lower serum phosphate (range of r = −0.18 to −0.28; P <0.01), but thereafter, they were not correlated. PTH did not correlate with 1,25-dihydroxyvitamin D at any time point.
DISCUSSION
In this prospective, multicenter, observational study based in the United States, we demonstrate rates of persistent hyperparathyroidism in excess of 80% throughout the first year after kidney transplantation, despite the recipients' healthy allograft function. Even when considering a higher PTH threshold greater than 130 pg/mL, more than 40% of participants demonstrated persistent hyperparathyroidism throughout the first year. High posttransplant PTH levels were accompanied by increased serum calcium and decreased serum phosphate levels, and high rates of hypercalcemia and hypophosphatemia. These data demonstrate that secondary hyperparathyroidism due to CKD is only partially corrected by kidney transplantation and that rates of persistent hyperparathyroidism remain extremely high during the first year after transplantation.
The results of our prospective, multicenter study extend the findings of the few previous, single-center studies that investigated posttransplant mineral metabolism.30,31 Although one might have expected more severe pretransplant hyperparathyroidism to be associated with more severe hyperparathyroidism after transplantation, by stratifying transplant recipients according to their baseline pretransplant PTH levels, we were able to show minor differences in PTH and other mineral metabolites after the initial 3 months after transplantation, and high rates of persistent hyperparathyroidism in both strata. These results suggest that clinical surveillance for persistent hyperparathyroidism and hypercalcemia should not be reserved exclusively for individuals with high pretransplant PTH levels. We also observed reductions in markers of bone turnover in the early posttransplant period, especially among recipients with high pretransplant PTH levels. Although this may suggest improved bone health in response to transplantation, the clinical implications of these changes in bone biomarkers in kidney transplant recipients require further study.
Study participants who were undergoing treatment with cinacalcet before transplantation had high rates of persistent hyperparathyroidism and hypercalcemia after transplantation. We speculate that the rates of posttransplant hyperparathyroidism and hypercalcemia were higher in this subgroup because pretransplant treatment with cinacalcet likely served as a marker of more severe hyperparathyroidism at baseline. It is important to emphasize that rates of posttransplant hyperparathyroidism and hypercalcemia were also high even among those participants who were untreated with cinacalcet before transplantation. Thus, rates of persistent hyperparathyroidism and hypercalcemia were high, regardless of cinacalcet exposure before transplant.
In contrast to persistently elevated PTH levels, FGF23 levels decreased precipitously during the first 3 months after transplantation and remained low thereafter, regardless of the individuals' baseline PTH level. Although FGF23 levels fell rapidly, we found that higher FGF23 levels were associated with lower serum phosphate through the first 3 months after transplantation and with lower 1,25-dihydroxyvitamini D levels through month 6 after transplantation, consistent with the known physiological effects of FGF23. These results align with previous studies that implicated persistently elevated FGF23 levels as a contributing cause of hypophosphatemia due to renal phosphate wasting and 1,25-dihydroxyvitamin D deficiency during the early posttransplant period.19,32-37 Most of these studies also demonstrated similarly rapid and sustained reductions in FGF23 levels after the initial posttransplant period.30,34,37,38 The finding that FGF23 normalizes far more rapidly than PTH suggests fundamental differences in the cellular biology of FGF23- and PTH-secreting cells that enable more rapid involution of the FGF23 response to CKD. It is also noteworthy that FGF23 decreased despite persistently elevated PTH, which stimulates FGF23 production.37,39-42 Perhaps subtle but protracted PTH-mediated renal phosphate wasting in transplant recipients induces negative phosphate balance that is appropriately sensed by osteocytes, which reduce FGF23 production in an effort to conserve phosphate, despite the stimulatory effects of high PTH. Indeed, serum phosphate levels tend to be approximately 0.5 mg/dL lower in kidney transplant recipients than CKD patients with comparably reduced eGFR.37 Phosphate balance studies have not been performed in this population, but such studies would help determine whether PTH-induced phosphate wasting is a novel mechanism that contributes to high fracture rates in kidney transplant recipients.43,44
Although the prospective design, large sample size, PTH stratification, and frequent measurements of a comprehensive panel of mineral metabolites are strengths of the current study, we acknowledged certain limitations. The criteria we used to define persistent hyperparathyroidism, hypercalcemia, and hypophosphatemia may not comport with contemporary practice across all transplant centers. Our results also may not generalize to transplant centers outside the United States. We did not collect data on use of phosphate supplementation after transplantation. Early withdrawal from the study led to missing data points and decreased the number of evaluable participants at specific time points. However, many of the withdrawals from our relatively large study were mandated by the protocol to minimize confounding by impaired allograft function. Since the estimates of persistent hyperparathyroidism rates excluded individuals with poor allograft function, which would increase rather than decrease PTH levels, our results likely underestimate the true prevalence of persistent hyperparathyroidism.
We also did not measure levels of 24,25-dihydroxyvitamin D, which is a marker of vitamin D catabolism. Levels of 24,25-dihydroxyvitamin D are lower in patients with CKD than in those with normal renal function.45 It would have been interesting to determine how 24,25-dihydroxyvitamin D levels evolve after transplantation in parallel with changes in FGF23 and 1,25-dihydroxyvitamin D, which stimulate expression of Cyp24A1, which encodes the 24-hyrdoxylase that degrades vitamin D.46 Although we measured bone turnover markers, we did not perform bone imaging and did not investigate whether persistent hyperparathyroidism was associated with more severe bone loss or increased risk of fractures. Furthermore, we did not examine if the presence or severity of persistent hyperparathyroidism influenced long-term allograft and patient outcomes. Previous studies yielded conflicting results.25,47,48
Treatment of kidney transplant recipients with cinacalcet can improve biochemical alterations in mineral metabolism, including hypercalcemia, hypophosphatemia, and hyperparathyroidism,49-54 but there is controversy regarding the broader utility and cost-effectiveness of cinacalcet treatment after kidney transplantation. In a recent, randomized, placebo-controlled study of 114 kidney transplant recipients, 12 months of cinacalcet treatment significantly increased the proportion of patients who maintained a mean corrected serum calcium concentration less than 10.2 mg/dL, increased serum phosphate, and decreased PTH levels compared to placebo.55 Although the study reported no adverse safety signals, it was a relatively small study of brief duration. Importantly, this study and all previous studies of cinacalcet during the posttransplant period focused exclusively on biochemical endpoints, and cinacalcet is not approved for use in kidney transplant recipients. Given the high rates of persistent posttransplant hyperparathyroidism that we observed, additional trials are needed to determine whether the biochemical effects of cinacalcet on mineral metabolites can translate into improved long-term clinical outcomes after kidney transplantation.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Holly Tomlin and Tim Peoples for medical writing and journal formatting assistance and Yumi Kubo for statistical analysis assistance (all employees and stockholders, Amgen, Inc).
Footnotes
This study was funded and supported by Amgen Inc.
Disclosures: M.W.: Scientific advisor and clinical trial executive committee member for Amgen. M.R.W.: Scientific advisor and clinical trial executive committee member for Amgen Inc. N.K.: Clinical trial investigator for Amgen-sponsored studies. R.B.M.: Past (>1 year) clinical trial investigator for Amgen-sponsored studies and consultant for Amgen. J.V.V.: Clinical trial advisory board member for Amgen Inc. H.D.: Employee and stockholder of Amgen. S.Y.: Employee and stockholder of Amgen Inc. F.V.: Research grants from Amgen Inc.
M.W., M.R.W., N.K., R.B.M., J.V.V., and F.V. were clinical investigators in the study described in this article. H.D. performed statistical analyses. All authors participated in interpreting the data. M.W. wrote the first draft, and all authors revised the article for important intellectual content and approved the final draft for submission.
The primary data from this study were presented as a poster at the 51st European Renal Association/European Dialysis and Transplant Association Congress from May 31 to June 3, 2014.
Supplemental digital content (SDC) is available for this article. Direct URL citations appear in the printed text, and links to the digital files are provided in the HTML text of this article on the journal's Web site (www.transplantjournal.com).
REFERENCES
- 1.US Renal Data System (USRDS). USRDS Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States Vol 7.2 Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Disease; 2013. [Google Scholar]
- 2. Goodman WG, Quarles LD. Development and progression of secondary hyperparathyroidism in chronic kidney disease: lessons from molecular genetics. Kidney Int. 2008; 74: 276– 288. [DOI] [PubMed] [Google Scholar]
- 3. Cunningham J, Locatelli F, Rodriguez M. Secondary hyperparathyroidism: pathogenesis, disease progression, and therapeutic options. Clin J Am Soc Nephrol. 2011; 6: 913– 921. [DOI] [PubMed] [Google Scholar]
- 4. Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011; 121: 4393– 4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ganesh SK, Stack AG, Levin NW, et al. Association of elevated serum PO(4), Ca x PO(4) product, and parathyroid hormone with cardiac mortality risk in chronic hemodialysis patients. J Am Soc Nephrol. 2001; 12: 2131– 2138. [DOI] [PubMed] [Google Scholar]
- 6. Larsson T, Nisbeth U, Ljunggren O, et al. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003; 64: 2272– 2279. [DOI] [PubMed] [Google Scholar]
- 7. Moe SM, Chertow GM, Coburn JW, et al. Achieving NKF-K/DOQI bone metabolism and disease treatment goals with cinacalcet HCl. Kidney Int. 2005; 67: 760– 771. [DOI] [PubMed] [Google Scholar]
- 8. Isakova T, Xie H, Yang W, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011; 305: 2432– 2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alam MU, Kirton JP, Wilkinson FL, et al. Calcification is associated with loss of functional calcium-sensing receptor in vascular smooth muscle cells. Cardiovasc Res. 2009; 81: 260– 268. [DOI] [PubMed] [Google Scholar]
- 10. Floege J, Ketteler M. Vascular calcification in patients with end-stage renal disease. Nephrol Dial Transplant. 2004; 19(Suppl 5): V59– V66. [DOI] [PubMed] [Google Scholar]
- 11. Foley RN, Parfrey PS. Cardiovascular disease and mortality in ESRD. J Nephrol. 1998; 11: 239– 245. [PubMed] [Google Scholar]
- 12. Wolf M, Molnar MZ, Amaral AP, et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol. 2011; 22: 956– 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Apostolou T, Damianou L, Kotsiev V, et al. Treatment of severe hypercalcemia due to refractory hyperparathyroidism in renal transplant patients with the calcimimetic agent cinacalcet. Clin Nephrol. 2006; 65: 374– 377. [DOI] [PubMed] [Google Scholar]
- 14. Evenepoel P, Claes K, Kuypers D, et al. Natural history of parathyroid function and calcium metabolism after kidney transplantation: a single-centre study. Nephrol Dial Transplant. 2004; 19: 1281– 1287. [DOI] [PubMed] [Google Scholar]
- 15. Evenepoel P, Kuypers D, Maes B, et al. Persistent hyperparathyroidism after kidney transplantation requiring parathyroidectomy. Acta Otorhinolaryngol Belg. 2001; 55: 177– 186. [PubMed] [Google Scholar]
- 16. Evenepoel P, Lerut E, Naesens M, et al. Localization, etiology and impact of calcium phosphate deposits in renal allografts. Am J Transplant. 2009; 9: 2470– 2478. [DOI] [PubMed] [Google Scholar]
- 17. Hamdy NA. Calcium and bone metabolism pre- and post-kidney transplantation. Endocrinol Metab Clin North Am. 2007; 36: 923– 935 viii. [DOI] [PubMed] [Google Scholar]
- 18. Heaf J, Tvedegaard E, Kanstrup IL, et al. Bone loss after renal transplantation: role of hyperparathyroidism, acidosis, cyclosporine and systemic disease. Clin Transplant. 2000; 14: 457– 463. [DOI] [PubMed] [Google Scholar]
- 19. Bhan I, Shah A, Holmes J, et al. Post-transplant hypophosphatemia: tertiary ‘hyper-phosphatoninism’? Kidney Int. 2006; 70: 1486– 1494. [DOI] [PubMed] [Google Scholar]
- 20. Evenepoel P. Recovery versus persistence of disordered mineral metabolism in kidney transplant recipients. Semin Nephrol. 2013; 33: 191– 203. [DOI] [PubMed] [Google Scholar]
- 21. Benavente D, Chue CD, Moore J, et al. Serum phosphate measured at 6 and 12 months after successful kidney transplant is independently associated with subsequent graft loss. Exp Clin Transplant. 2012; 10: 119– 124. [DOI] [PubMed] [Google Scholar]
- 22. Connolly GM, Cunningham R, McNamee PT, et al. Elevated serum phosphate predicts mortality in renal transplant recipients. Transplantation. 2009; 87: 1040– 1044. [DOI] [PubMed] [Google Scholar]
- 23. Mazzaferro S, Pasquali M, Taggi F, et al. Progression of coronary artery calcification in renal transplantation and the role of secondary hyperparathyroidism and inflammation. Clin J Am Soc Nephrol. 2009; 4: 685– 690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meneghini M, Regalia A, Alfieri C, et al. Calcium and osteoprotegerin levels predict the progression of the abdominal aortic calcifications after kidney transplantation. Transplantation. 2013; 96: 42– 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perrin P, Caillard S, Javier RM, et al. Persistent hyperparathyroidism is a major risk factor for fractures in the five years after kidney transplantation. Am J Transplant. 2013; 13: 2653– 2663. [DOI] [PubMed] [Google Scholar]
- 26. Al-Gabri S, Zadrazil J, Krejcí K, et al. Changes in bone mineral density and selected metabolic parameters over 24 months following renal transplantation. Transplant Proc. 2005; 37: 1014– 1019. [DOI] [PubMed] [Google Scholar]
- 27. Sprague SM, Belozeroff V, Danese MD, et al. Abnormal bone and mineral metabolism in kidney transplant patients—a review. Am J Nephrol. 2008; 28: 246– 253. [DOI] [PubMed] [Google Scholar]
- 28. Tuchman S, Kalkwarf HJ, Zemel BS, et al. Vitamin D deficiency and parathyroid hormone levels following renal transplantation in children. Pediatr Nephrol. 2010; 25: 2509– 2516. [DOI] [PubMed] [Google Scholar]
- 29. Levey AS, Coresh J, Greene T, et al. Using standardized serum creatinine values in the Modification of Diet in Renal Disease Study equation for estimating glomerular filtration rate. Ann Intern Med. 2006; 145: 247– 254. [DOI] [PubMed] [Google Scholar]
- 30. Barros X, Torregrosa JV, Martínez de Osaba MJ, et al. Earlier decrease of FGF-23 and less hypophosphatemia in preemptive kidney transplant recipients. Transplantation. 2012; 94: 830– 836. [DOI] [PubMed] [Google Scholar]
- 31. Reinhardt W, Bartelworth H, Jockenhövel F, et al. Sequential changes of biochemical bone parameters after kidney transplantation. Nephrol Dial Transplant. 1998; 13: 436– 442. [DOI] [PubMed] [Google Scholar]
- 32. Sánchez Fructuoso AI, Maestro ML, Calvo N, et al. Role of fibroblast growth factor 23 (FGF23) in the metabolism of phosphorus and calcium immediately after kidney transplantation. Transplant Proc. 2012; 44: 2551– 2554. [DOI] [PubMed] [Google Scholar]
- 33. Sirilak S, Chatsrisak K, Ingsathit A, et al. Renal phosphate loss in long-term kidney transplantation. Clin J Am Soc Nephrol. 2012; 7: 323– 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Evenepoel P, Meijers BK, de Jonge H, et al. Recovery of hyperphosphatoninism and renal phosphorus wasting one year after successful renal transplantation. Clin J Am Soc Nephrol. 2008; 3: 1829– 1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Economidou D, Dovas S, Papagianni A, et al. FGF-23 levels before and after renal transplantation. J Transplant. 2009; 2009: 379082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pande S, Ritter CS, Rothstein M, et al. FGF-23 and sFRP-4 in chronic kidney disease and post-renal transplantation. Nephron Physiol. 2006; 104: 23– 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012; 82: 737– 747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yilmaz MI, Sonmez A, Saglam M, et al. Longitudinal analysis of vascular function and biomarkers of metabolic bone disorders before and after renal transplantation. Am J Nephrol. 2013; 37: 126– 134. [DOI] [PubMed] [Google Scholar]
- 39. Lavi-Moshayoff V, Wasserman G, Meir T, et al. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol. 2010; 299: F882– F889. [DOI] [PubMed] [Google Scholar]
- 40. López I, Rodríguez-Ortiz ME, Almadén Y, et al. Direct and indirect effects of parathyroid hormone on circulating levels of fibroblast growth factor 23 in vivo. Kidney Int. 2011; 80: 475– 482. [DOI] [PubMed] [Google Scholar]
- 41. Rhee Y, Bivi N, Farrow E, et al. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone. 2011; 49: 636– 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wesseling-Perry K, Harkins GC, Wang HJ, et al. The calcemic response to continuous parathyroid hormone (PTH)(1–34) infusion in end-stage kidney disease varies according to bone turnover: a potential role for PTH(7–84). J Clin Endocrinol Metab. 2010; 95: 2772– 2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ball AM, Gillen DL, Sherrard D, et al. Risk of hip fracture among dialysis and renal transplant recipients. JAMA. 2002; 288: 3014– 3018. [DOI] [PubMed] [Google Scholar]
- 44. Sukumaran Nair S, Lenihan CR, Montez-Rath ME, et al. Temporal trends in the incidence, treatment and outcomes of hip fracture after first kidney transplantation in the United States. Am J Transplant. 2014; 14: 943– 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bosworth CR, Levin G, Robinson-Cohen C, et al. The serum 24,25-dihydroxyvitamin D concentration, a marker of vitamin D catabolism, is reduced in chronic kidney disease. Kidney Int. 2012; 82: 693– 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Alshayeb H, Showkat A, Wall BM, et al. Activation of FGF-23 mediated vitamin D degradative pathways by cholecalciferol. J Clin Endocrinol Metab. 2014; 99: E1830– E1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bleskestad IH, Bergrem H, Leivestad T, et al. Parathyroid hormone and clinical outcome in kidney transplant patients with optimal transplant function. Clin Transplant. 2014; 28: 479– 486. [DOI] [PubMed] [Google Scholar]
- 48. Molnar MZ, Kovesdy CP, Mucsi I, et al. Association of pre-kidney transplant markers of mineral and bone disorder with post-transplant outcomes. Clin J Am Soc Nephrol. 2012; 7: 1859– 1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cohen JB, Gordon CE, Balk EM, et al. Cinacalcet for the treatment of hyperparathyroidism in kidney transplant recipients: a systematic review and meta-analysis. Transplantation. 2012; 94: 1041– 1048. [DOI] [PubMed] [Google Scholar]
- 50. Courbebaisse M, Diet C, Timsit MO, et al. Effects of cinacalcet in renal transplant patients with hyperparathyroidism. Am J Nephrol. 2012; 35: 341– 348. [DOI] [PubMed] [Google Scholar]
- 51. Aalten J, Wetzels JF, Hoitsma AJ. Continuation of cinacalcet immediately after renal transplantation: a prospective cohort study. Clin Nephrol. 2010; 74: 433– 439. [DOI] [PubMed] [Google Scholar]
- 52. Paschoalin RP, Torregrosa JV, Barros X, et al. Cinacalcet de novo in persistent hypercalcemia after kidney transplantation secondary to hyperparathyroidism: long-term follow-up and effect of withdrawal. Transplant Proc. 2012; 44: 2376– 2378. [DOI] [PubMed] [Google Scholar]
- 53. Torregrosa JV, Bergua C, Martinez de Osaba MJ, et al. Evolution of secondary hyperparathyroidism after kidney transplantation in patients receiving cinacalcet on dialysis. Transplant Proc. 2009; 41: 2396– 2398. [DOI] [PubMed] [Google Scholar]
- 54. Torregrosa JV, Morales E, Díaz JM, et al. Cinacalcet for hypercalcaemic secondary hyperparathyroidism after renal transplantation: A multicentre, retrospective, 3-year study. Nephrology (Carlton). 2014; 19: 84– 93. [DOI] [PubMed] [Google Scholar]
- 55. Evenepoel P, Cooper K, Holdaas H, et al. A randomized study evaluating cinacalcet to treat hypercalcemia in renal transplant recipients with persistent hyperparathyroidism. Am J Transplant. 2014; 14: 2545– 2555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.