

Abstract
Enteropathogenic Escherichia coli (EPEC) is a food-borne pathogen that causes infantile diarrhea worldwide. EPEC decreases the activity and surface expression of the key intestinal Cl−/HCO3− exchanger SLC26A3 [downregulated in adenoma (DRA)], contributing to the pathophysiology of early diarrhea. Little is known about the mechanisms governing membrane recycling of DRA. In the current study, Caco-2 cells were used to investigate DRA trafficking under basal conditions and in response to EPEC. Apical Cl−/HCO3− exchange activity was measured as DIDS-sensitive 125I− uptake. Cell surface biotinylation was performed to assess DRA endocytosis and exocytosis. Inhibition of clathrin-mediated endocytosis by chlorpromazine (60 μM) increased apical Cl−/HCO3− exchange activity. Dynasore, a dynamin inhibitor, also increased function and surface levels of DRA via decreased endocytosis. Perturbation of microtubules by nocodazole revealed that intact microtubules are essential for basal exocytic (but not endocytic) DRA recycling. Mice treated with colchicine showed a decrease in DRA surface levels as visualized by confocal microscopy. In response to EPEC infection, DRA surface expression was reduced partly via an increase in DRA endocytosis and a decrease in exocytosis. These effects were dependent on the EPEC virulence genes espG1 and espG2. Intriguingly, the EPEC-induced decrease in DRA function was unaltered in the presence of dynasore, suggesting a clathrin-independent internalization of surface DRA. In conclusion, these studies establish the role of clathrin-mediated endocytosis and microtubules in the basal surface expression of DRA and demonstrate that the EPEC-mediated decrease in DRA function and apical expression in Caco-2 cells involves decreased exocytosis.
Keywords: DRA recycling, EPEC infection, endocytosis, exocytosis, clathrin, microtubules
downregulated in adenoma (DRA), or SLC26A3, is an integral membrane transporter expressed on the apical membrane of epithelial cells, including the small intestine and colon. Mutations in the SLC26A3 gene are associated with congenital chloride diarrhea (CLD), characterized by voluminous diarrhea with high fecal chloride content (28). DRA knockout mice exhibit severe diarrhea and a phenotype similar to that of CLD patients (32). Emerging studies indicate that posttranslational modifications of DRA protein may underlie the pathophysiology of diarrheal diseases. For example, CLD-causing mutations induce diarrhea by causing loss of functional DRA protein at the plasma membrane, possibly via its misfolding and mistrafficking (25, 36). Studies from our laboratory have shown that enteropathogenic Escherichia coli (EPEC) infection decreases DRA function and surface levels (7), which may underlie early diarrhea; however, the cellular recycling mechanisms of DRA under normal or diseased conditions are not well understood. Thus, investigation of the cellular pathways responsible for maintaining appropriate DRA levels on the apical membrane is important for understanding disease pathogenesis and development of novel antidiarrheal therapies.
Unlike other enteric pathogens, EPEC is modestly invasive and does not produce enterotoxins, such as heat-labile toxin, from enterotoxigenic E. coli. The attachment of EPEC to host cells occurs in three stages: 1) initial localized adherence or nonintimate attachment, 2) translocation of virulence factors into the host cell via a type 3 secretion system (T3SS), leading to cytoskeletal disruption, and 3) intimate attachment to host cells and formation of actin pedestals and attaching/effacing lesions (16). Initial nonintimate attachment via adhesins expressed by EPEC is followed by T3SS-dependent intimate adherence to host intestinal epithelial cells (IECs), resulting in the typical attaching/effacing lesions, formation of actin pedestals, and loss of microvilli. The profound loss of the absorptive surface microvilli contributes greatly to the diarrheal phenotype, particularly later in EPEC infection. Other secretion apparatuses, such as the type 2 secretion system, which is well characterized in toxin-producing bacteria, are also present in EPEC. However, a role for the type 2 secretion system in the rapid onset of diarrhea has not been demonstrated. Rapid onset of diarrhea, within 3 h after EPEC ingestion, suggests that other mechanisms of dysregulation in water and solute absorption are at play (12). Recent studies from our group and others have shown that early diarrhea during EPEC infection may result from a direct inhibition of ion transport processes involved in solute and electrolyte movement (7, 13). These studies have also shown that the EPEC translocation machinery T3SS and the translocated virulence factors EspF, EspG1, and EspG2 play a major role in the downregulation of both Na+/H+ exchanger type 3 (NHE3) and DRA by reducing apical membrane levels of these transporters (7, 13, 15). Earlier studies from our group showed that EspG1 and EspG2, known to disrupt the host microtubule network, are essential for EPEC-induced reduction of apical DRA levels and Cl−/HCO3− exchange activity (7). However, the molecular basis and underlying cellular events responsible for altered DRA recycling are not defined. Also, how EPEC modulates these events has not been investigated. Our current studies demonstrate, for the first time, the involvement of clathrin-dependent pathways in DRA endocytosis in IECs and further implicate EPEC-induced host microtubule disruption in alteration of exocytosis of DRA.
MATERIALS AND METHODS
Materials.
Eagle's minimum essential medium (EMEM), Dulbecco's modification of EMEM (DMEM-F12), Caco-2 and T-84 cells, and Lactobacillus acidophilus strain LA4357 were obtained from American Type Culture Collection (Manassas, VA). Radionuclide 125I (NaI) was procured from Perkin Elmer (Boston, MA); 4,4′-diisothiocyanostilbene-2,2′ disulfonic acid (DIDS), the pharmacological inhibitors dynasore and chloropromazine, anti-actin antibody, and colchicine from Sigma-Aldrich (St. Louis, MO); sulfo-NHS-SS-biotin, sulfo-NHS-acetate, and reduced glutathione (GSH) from Thermo Scientific (Rockford, IL); and nocodazole from Biomol (Plymouth Meeting, PA). Affinity-purified DRA antibody was custom-synthesized against the COOH-terminal amino acid (745–764) sequence INTNGGLRNRVYEPVETKF of SLC26A3 (accession no. BC025671) by the Research Resources Center at the University of Illinois at Chicago. All other chemicals were at least reagent grade and were purchased from Sigma or Fisher Scientific (Pittsburgh, PA).
Cell culture.
The human colonic adenocarcinoma cell line Caco-2 was grown in EMEM supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 μg/ml gentamicin, and 20% fetal bovine serum in 5% CO2-95% air at 37°C on plastic support. T-84 cells were cultured in 2.5 mM l-glutamine, 5% FBS, and antibiotic-supplemented DMEM-F12 medium. Cells were plated on 24-well plates at a density of 2 × 104 cells/well. Fully differentiated confluent monolayers (10–12 days postplating) were used for the uptake studies, biotinylation experiments, and lipid raft studies. Cell monolayers grown on Transwell inserts (5,000 cells/well) for 12 days were used for immunofluorescence staining.
Assessment of Cl−/HCO3− exchange activity.
Cl−/HCO3− exchange activity was determined by measurement of DIDS-sensitive 125I− uptake in base-loaded cells as previously described by us with minor modification of the 36Cl− uptake method. Use of 125I− to measure Cl−/HCO3− exchange activity was validated in our previous study (31). For uptake studies with EPEC, cells were pretreated with specific inhibitors for 30 min and then infected with EPEC for 30 min in the presence or absence of the inhibitor. After infection, cell monolayers were incubated with loading buffer containing 20 mM HEPES (pH 8.5) for 30 min at room temperature. Cells were rapidly washed with 1 ml of tracer-free mannitol uptake buffer containing 260 mM mannitol and 20 mM Tris-MES (pH 7.0). Cells were then incubated with the uptake buffer with or without 600 μM DIDS containing 1.0 μCi of 3 mM 125I (NaI, specific activity 17.4 Ci/mg) for 5 min. Uptake was stopped by rapid washing of the cells with ice-cold PBS (pH 7.2). Finally, cells were solubilized by incubation with 0.5 N NaOH for 4 h, and protein concentration was measured by the Bradford method (2). Radioactivity was measured with a liquid scintillation analyzer (Tri-Carb 1600 TR, Packard Instruments, Perkin Elmer). Cl−/HCO3− exchange activity was assessed as DIDS-sensitive 125I− uptake, and specific activity was expressed as nanomoles per milligram of protein per 5 min.
EPEC culture and cell infection.
The following EPEC strains were used: 1) wild-type and 2) ΔespG/ΔespG2. Bacterial strains were grown at 37°C aerobically in 5 ml of Luria broth in the presence of appropriate antibiotics. On the day of infection the overnight bacterial culture was subcultured in 10 ml DMEM-F12 medium supplemented with 0.5% mannose, to promote formation of T3SS as previously described (8, 24). Caco-2 monolayers were infected at multiplicity of infection of 1:100.
Cell lysates.
After treatment with dynasore (80 μM) or following EPEC infection, cells were washed with ice-cold 1× PBS, and total protein was extracted by suspension of the cell pellet in lysis buffer [20 mM Tris·HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and 1 mM EGTA] supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN) and phosphatase inhibitor (Sigma). For biotinylation studies, cells were lysed by sonication and centrifuged at 13,000 rpm for 10 min at 4°C. Protein concentration of the supernatant was determined by the Bradford method (2).
Western blotting.
To examine the expression levels of DRA, equal amounts (75 μg/sample) of whole cell lysates were solubilized in SDS-gel loading buffer [250 mM Tris·HCl (pH 6.8), 10% SDS, 250 μM DTT, 50% glycerol, and 0.2% bromophenol blue] and boiled for 5 min. Proteins were loaded onto a 7.5% SDS-polyacrylamide gel and transblotted to a nitrocellulose membrane. After incubation in blocking buffer (1× PBS and 5% nonfat dry milk), the membrane was probed with affinity-purified anti-DRA antibody (1:100 dilution) or GAPDH antibody (1:3,000 dilution; Sigma) in 1× PBS and 2.5% nonfat dry milk overnight at 4°C. The membrane was washed with wash buffer (1× PBS and 0.1% Tween 20) and probed with horseradish peroxidase-conjugated goat anti-rabbit antibody (1:2,000 dilution) and detected by enhanced chemiluminescence (Bio-Rad, Hercules, CA).
Cell surface biotinylation.
Cell surface biotinylation studies were performed in Caco-2 monolayers utilizing sulfo-NHS-SS-biotin (1.5 mg/ml; Pierce, Rockford, IL) in borate buffer [in mM: 154 NaCl, 7.2 KCl, 1.8 CaCl2, and 10 H3BO3, (pH 9.0)] (7). Surface proteins were labeled with biotin for 60 min. The process was performed at 4°C to prevent cellular recycling. NeutrAvidin-agarose beads were used to pull down biotinylated antigens. Biotinylated fractions were boiled in gel loading buffer containing 100 μM DTT. Released proteins were subjected to SDS-PAGE and transferred to nitrocellulose membranes. Western blotting was used to compare surface DRA with total cellular DRA.
Endocytic internalization assay.
To measure the extent of endocytosis, Caco-2 cells were plated at a density of 1 × 105 cells on six-well plates. The cell surface was labeled with sulfo-NHS-SS-biotin (1.5 mg/ml; Pierce) in borate buffer [in mM: 154 NaCl, 7.2 KCl, 1.8 CaCl2, and 10 H3BO3 (pH 9.0)] at 4°C for 60 min (19). After surface biotinylation, cells were treated with dynasore or vehicle (DMSO) at 37°C for 60 min. For studies with EPEC, cells were infected with EPEC for 60 min at 37°C. Immediately after treatment, the cells were rinsed twice with ice-cold 1× PBS at 4°C. Excess surface biotin was cleaved by 150 mM reduced glutathione (GSH) in 1× PBS, and the biotinylated freshly endocytosed proteins were protected from cleavage by GSH. All steps except treatment were carried out on ice. Cells were solubilized in lysis buffer, and biotinylated proteins were retrieved and assayed for endocytosed DRA as described above.
Exocytic insertion into the plasma membrane.
To measure exocytic insertion of DRA into the plasma membrane, NHS-reactive sites on the Caco-2 cell surface were masked by pretreatment with sulfo-NHS-acetate at 4°C for 60 min, followed by quenching in 1 M glycine, as previously described (19). To disrupt microtubules, cells were kept at 4°C for 1 h. Microtubules were then allowed to recover in control monolayers by incubation at 37°C for 3 h, while test monolayers were returned to 37°C in the presence of the microtubule-disrupting agent nocodazole (66 μM) to maintain microtubule disruption (10). For EPEC studies, cells were infected at 37°C for 60 min with wild-type EPEC, which is able to disrupt microtubules, and the ΔespG1/ΔespG2 mutant, which is unable to disrupt microtubules (7, 9). After microtubule disruption and recovery, cells were subjected to cell surface biotinylation as described above. The biotinylated fractions, which represent the newly inserted membrane proteins, were precipitated with NeutrAvidin-agarose beads, and the precipitate was subjected to SDS-PAGE and Western blotting with anti-DRA antibody.
Isolation of detergent-insoluble and -soluble fractions.
Caco-2 cell lysates prepared in detergent-free buffer were subjected to ultracentrifugation at 100,000 g for 30 min at 4°C. The resulting total membrane pellet was resuspended in a detergent-based MES buffer [50 mM MES (pH 6.5), 60 mM NaCl, 3 mM EGTA, 5 mM MgCl2, 1% Triton X-100, and 1× Complete protease inhibitor cocktail] and incubated at 4°C on a rotary shaker for 30 min. The mixture was then ultracentrifuged at 100,000 g for 30 min at 4°C to separate detergent-soluble and -insoluble membranes. The supernatant was referred to as the detergent-soluble fraction. The pellet was resuspended in a RIPA lysis buffer [5 mM HEPES (pH 7.4), 150 mM NaCl, 10 mM EDTA, 1 mM DTT, 1% Triton X-100, 0.1% SDS, and 1× Complete protease inhibitor cocktail] and designated the detergent-insoluble fraction (1, 31). The detergent-soluble and -insoluble fractions obtained after treatment of Caco-2 cells with dynasore (or infection with EPEC) were analyzed by Western blotting.
Immunofluorescence staining in Caco-2 cells.
Caco-2 cells grown on 12-well Transwell inserts were treated from the apical side with dynasore for 60 min. Cells were then washed twice in 1× PBS containing 1 mM CaCl2 (pH 7.4) and fixed with paraformaldehyde. Cells were then permeabilized with 0.08% saponin, washed with 1× PBS-CaCl2, and blocked in 5% normal goat serum (NGS) for 2 h at room temperature. Monolayers were then incubated with rabbit anti-human DRA antibody (1:100 dilution) for 2 h and washed with 1× PBS containing CaCl2 and saponin. Cells were incubated in Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (1:100 dilution; Invitrogen) and rhodamine-phalloidin (1:60 dilution; Invitrogen) for 60 min at room temperature. The immunostained inserts were mounted on glass slides with SlowFade Gold antifade reagent. Images were obtained with a ×63 water-immersion objective on a confocal microscope (LSM 510 META, Zeiss). Beams at 488 and 534 nm from an Ag/Kr laser and 361 nm from a UV laser were used for excitation of fluorophores. Green and red fluorescence emissions were detected using LP 505 and LP 585 filters, respectively. A series of images were taken in the z-axis plane, and the z stack was used to make orthogonal sections in the xz plane. Images were processed using ZenLite imaging software (Carl Zeiss) (6).
Animal protocol.
Eight-week-old C57BL/6 mice (20–25 g body wt) were acclimatized for 1 wk prior to the experiment on a standard mouse pellet diet with free access to water. The animal experiments were conducted according to the guidelines of the Animal Care Committee of the University of Illinois at Chicago and the Jesse Brown Veterans Affairs Medical Center, which approved this study. Mice were treated with the microtubule-disrupting agent colchicine (3 mg/kg body wt ip) for 24 h. Then the mice were euthanized, and a segment of distal colon was dissected, flushed with PBS, and used for ex vivo biotinylation studies and immunofluorescence.
Immunofluorescence staining in tissue sections.
Distal colon sections from control and colchicine-treated mice were snap-frozen in optimal cutting temperature embedding medium (Tissue-Tek OCT compound, Sakura), and 5-μm sections were cut utilizing a cryostat.
Tissues were fixed with 1% paraformaldehyde, permeabilized using 0.5% NP-40, and blocked in 2.5% NGS for 1.5 h at room temperature and then incubated with primary antibodies [rabbit anti-DRA (1:100 dilution) and mouse anti-villin (1:100 dilution)] in 1% NGS for 2 h at room temperature. Finally, sections were incubated with secondary antibodies (Alexa Fluor 568-conjugated goat anti-rabbit IgG and Alexa Fluor 488-conjugated goat anti-mouse IgG) and mounted on glass slides with SlowFade with DAPI. Microscopy was performed using a laser-scanning confocal microscope (LSM 710, Zeiss) equipped with a ×63 water-immersion objective.
Statistical analysis.
Values are means ± SE and represent data from three to five independent experiments. One-way ANOVA with Tukey's multiple-comparison test was used for statistical analysis. For experiments where only two independent groups were compared with each other, unpaired Student's t-test was used as a statistical parameter. Differences between control and treated groups were considered significant at P < 0.05.
RESULTS
Role of dynamin-dependent endocytic pathways in regulating apical Cl−/HCO3− exchange activity.
Clathrin- and caveolin-dependent pathways are well-defined cellular endocytic routes previously shown to be involved in the basal and regulated internalization of ion transporters (21, 26, 27, 33, 37). The endocytosis inhibitor dynasore inhibits dynamin, a 100-kDa GTPase required to pinch-off coated vesicles from the plasma membrane, and, thereby, prevents both clathrin- and caveolin-dependent endocytosis (23). To investigate the cellular endocytic pathway responsible for apical Cl−/HCO3− activity, postconfluent differentiated Caco-2 cells were serum-starved overnight and treated with 40–120 μM dynasore in serum-free medium for 1 h. Inhibition of endocytosis with 40 μM dynasore showed no effect on apical Cl−/OH−(HCO3−) exchange activity. However, at 80 and 120 μM dynasore, an approximately twofold stimulation of apical Cl−/HCO3− activity was observed (Fig. 1A). These results support the notion that apical Cl−/OH−(HCO3−) exchange activity is regulated by a dynamin-dependent endocytic pathway in IECs. Next, the effect of 80 μM dynasore on Cl−/OH−(HCO3−) exchange activity was investigated in the human colonic epithelial cell line T-84. As shown in Fig. 1B, similar to the effects observed in Caco-2 cells, 80 μM dynasore increased apical Cl−/OH−(HCO3−) exchange activity (∼2-fold) in T-84 cells. These results suggest that the effect of dynamin inhibition on Cl−/OH−(HCO3−) exchange is not cell line-specific.
Fig. 1.

Stimulation of apical Cl−/OH−(HCO3−) exchange activity by dynasore. A: overnight serum-starved Caco-2 cells were treated with 40–120 μM dynasore for 60 min, and Cl−/HCO3− exchange activity was measured as DIDS (600 μM)-sensitive 125I− uptake in base-loaded Caco-2 cells. Values are means ± SE of 4 different experiments performed in triplicate. *P < 0.01 and **P < 0.006 vs. control. B: overnight serum-starved T-84 cells were treated with 80 μM dynasore for 60 min, and Cl−/HCO3− exchange activity was measured as DIDS (600 μM)-sensitive 125I− uptake in base-loaded cells. Values are means ± SE of 3 experiments performed in triplicate. **P < 0.006 vs. control.
Apical Cl−/HCO3− exchange activity is regulated by the clathrin-dependent pathway.
To specifically examine the role of the clathrin-mediated pathway, Caco-2 cells were treated with chlorpromazine, which is known to inhibit clathrin-mediated endocytosis (CME) without affecting other endocytic pathways (35). Similar to dynasore, chlorpromazine treatment (60 μM for 1 h) resulted in an increase (∼2-fold) in apical Cl−/OH−(HCO3−) exchange activity (Fig. 2). These data indicate that Cl−/OH−(HCO3−) exchange activity in Caco-2 cells is regulated by clathrin-dependent endocytosis. Previous studies and our data (not shown) demonstrated that Caco-2 cells do not express the two major caveolin isoforms caveolin-1 and caveolin-2 (34). Therefore, the role of caveolin-dependent DRA recycling in Caco-2 cells was not examined.
Fig. 2.
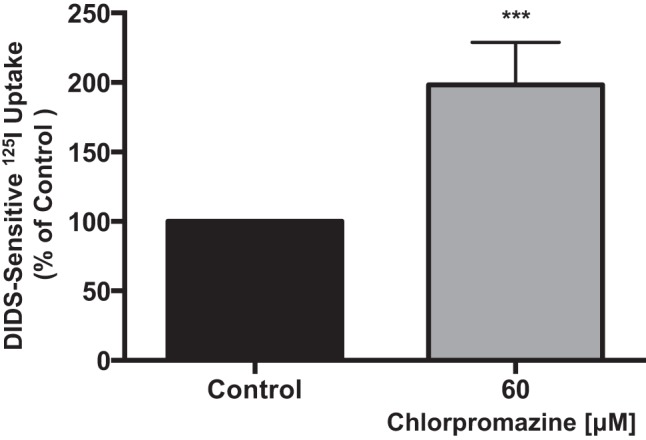
Apical Cl−/HCO3− exchange activity is increased by inhibition of clathrin-mediated endocytosis. Overnight serum-starved Caco-2 cells were treated with 60 μM chlorpromazine for 60 min, and Cl−/HCO3− exchange activity was measured as DIDS (600 μM)-sensitive 125I− uptake in base-loaded Caco-2 cells. Values are means ± SE of 4 different experiments performed in triplicate. ***P < 0.0007 vs. control.
Dynasore increases apical surface expression of DRA via decreased endocytosis in Caco-2 cells.
To examine if the increase in Cl−/OH−(HCO3−) exchange activity in the presence of CME inhibitors correlates with an increase in surface levels of DRA, Caco-2 monoloayers were treated with 80 μM dynasore for 1 h, and surface levels of DRA were assessed by a cell surface biotinylation assay. Treatment with dynasore significantly increased DRA surface levels compared with control. As shown in Fig. 3A, densitometric analysis of protein bands showed a 40–50% increase in DRA surface levels in dynasore-treated cells compared with controls. There was no change in total cellular DRA levels. Immunofluorescence studies (Fig. 3B) showed that DRA (shown in green) was increased on the apical membrane (shown in the xz plane) in Caco-2 cells treated for 1 h with 80 μM dynasore compared with untreated controls. These data are consistent with an increase in apical Cl−/OH−(HCO3−) exchange activity in the presence of the endocytosis inhibitor dynasore, further establishing the role of CME in basal DRA recycling. To determine whether the increase in apical Cl−/OH−(HCO3−) exchange activity and surface expression of DRA in Caco-2 cells treated with dynasore was indeed a result of inhibition of DRA endocytosis, we performed an endocytic internalization assay (reverse cell surface biotinylation) in treated and untreated Caco-2 cells. As shown in Fig. 3C, densitometric analysis of protein bands showed that dynasore significantly decreased basal DRA endocytosis by 50–60% compared with control, thus confirming the role of the clathrin-dependent pathway in basal DRA recycling.
Fig. 3.

Dynasore increases apical surface expression of downregulated in adenoma (DRA) via decreased endocytosis. A: overnight serum-starved Caco-2 cells were treated with 80 μM dynasore for 60 min and subjected to cell surface biotinylation. Biotinylated proteins were pulled down with NeutrAvidin-agarose beads, and surface and total fractions were resolved on a 7.5% SDS-polyacrylamide gel. Representative blot was probed with rabbit anti-DRA antibody. Biotinylated fraction represents surface DRA expression. Results are shown as ratio of surface to total DRA. Values are means ± SE of 3 different experiments. *P < 0.05 vs. control. B: apical surface of Caco-2 cell monolayers grown on Transwell filter inserts was treated with 80 μM dynasore for 60 min. Cells were fixed with paraformaldehyde and incubated with anti-DRA antibody (1:100 dilution) and then with Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (1:100 dilution) and rhodamine-phalloidin (1:60 dilution) for 60 min. DRA is shown in green and actin in red. Orthogonal xz images were obtained with a Zeiss LSM 510 confocal microscope. C: overnight serum-starved Caco-2 monolayers were used for reverse cell surface biotinylation at 4°C and then incubated with 80 μM dynasore or vehicle (DMSO) at 37°C for 30 min. Excess biotin was cleaved by glutathione. Biotinylated proteins were pulled down with NeutrAvidin-agarose beads, and biotinylated and total fractions were resolved on a 7.5% SDS-polyacrylamide gel. Western blot was probed with rabbit anti-DRA antibody. Biotinylated fraction represents the endocytosed pool of DRA. Results are shown as ratio of endocytosed to total DRA. Values are means ± SE of 3 different experiments. *P < 0.05 vs. control.
Dynasore increases association of DRA with lipid rafts.
Previous studies showed that partitioning of DRA in the membrane lipid raft fraction increases DRA functional activity (22, 31). We next examined if DRA is preferentially associated with lipid raft domains in response to treatment with dynasore. Caco-2 cells grown on plastic support were treated with dynasore or vehicle (DMSO), lysed, and ultracentrifuged to obtain the total membrane pellet. Detergent-soluble (nonraft) and detergent-insoluble (raft) fractions were separated by solubilization in a Triton X-100-based buffer at 4°C. As shown in Fig. 4, dynasore treatment increased DRA association with lipid rafts by ∼1.5-fold compared with controls, suggesting that inhibition of DRA endocytosis increases DRA levels on the plasma membrane and its association with lipid raft domains, enhancing its function.
Fig. 4.
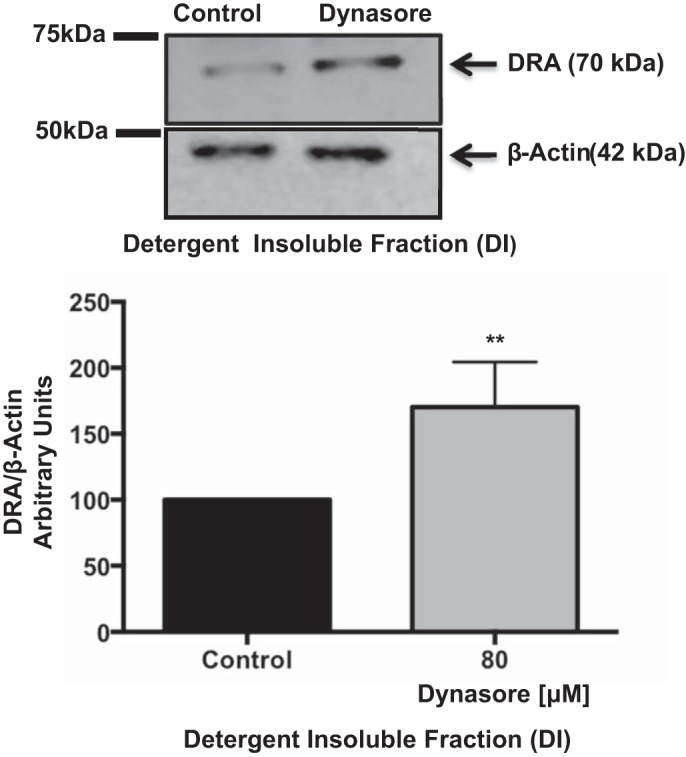
Dynasore increased DRA association with lipid rafts. Overnight serum-starved Caco-2 monolayers were treated with 80 μM dynasore for 60 min. Membrane pellets obtained from total cell lysate by ultracentrifugation were solubilized at 4°C in a Triton X-100 detergent buffer to separate lipid raft [detergent-insoluble (DI)] and nonraft (detergent-soluble) fractions. DI fractions were resolved on a 7.5% SDS-polyacrylamide gel and analyzed by Western blotting for DRA and β-actin expression. Representative blot is shown. Data were quantified by densitometric analysis of results from 4 separate immunoblots. Results are shown as ratio of DRA to β-actin. Values are means ± SE. **P < 0.006 vs. control.
Role of microtubules in regulating DRA endocytosis and exocytosis.
Cellular recycling of membrane proteins involves movement of endocytic and exocytic vesicles along cytoskeletal components (3, 5). Previous studies utilizing colchicine, a microtubule-disrupting drug, showed a decrease in DRA function in colchicine-treated Caco-2 cells (7). Thus it was logical to examine the role of microtubules in endocytic and exocytic routes of DRA recycling. To elucidate the importance of intact microtubules in DRA endocytosis, Caco-2 cells were subjected to surface biotinylation followed by disruption and recovery of microtubules in the presence or absence of the microtubule-disrupting agent nocodazole (66 μM). The biotinylated fractions pulled down with NeutrAvidin beads represent the endocytosed proteins. As shown in Fig. 5A, there was no significant change in DRA endocytosis in Caco-2 cells with microtubule disruption compared with cells with recovered (intact) microtubules. Thus our studies suggest that microtubules are not involved in endocytosis of DRA in Caco-2 cells.
Fig. 5.

Role of microtubules in basal DRA endocytosis and exocytosis. A: Caco-2 monolayers were incubated with sulfo-NHS-SS-biotin, and microtubules were disrupted and incubated at 4°C. Microtubule recovery upon return to 37°C was prevented by addition of nocodazole to the medium or was allowed to proceed in the absence of nocodazole. Total protein lysates were incubated with NeutrAvidin-agarose beads to pull down biotinylated proteins. Total and biotinylated fractions were run on a 7.5% SDS-polyacrylamide gel and then immunoblotted with anti-DRA antibody. DRA bands in the representative blot were quantified by densitometric analysis. Results are shown as ratio of endocytosed to total DRA. Values are means ± SE of results from 3 different blots. B: cell surface Caco-2 cells were masked with sulfo-NHS-acetate. Cells were then incubated at 4°C to disrupt microtubules and allowed to recover, or not, in the absence or presence of nocodazole. Amount of newly exocytosed protein was assessed by cell surface biotinylation. Immunoblot was probed with anti-DRA antibody and quantitated by densitometric analysis. Results are shown as ratio of endocytosed to total DRA. Values are means ± SE of results from 3 different blots. ***P < 0.001 vs. recovered. C: Caco-2 cells were subjected to cold exposure + nocodazole treatment to induce microtubule disruption. I: cells were stained for α-tubulin (red) to confirm microtubule disruption and recovery. II: Caco-2 cells were immunostained for localization of DRA (green) and Alexa Fluor 568-conjugated phalloidin (actin; red), and DAPI was used to label nuclei (blue) in cells with recovered and disrupted microtubules.
Having established that intact microtubules are not essential for DRA endocytosis, we examined whether the decrease in DRA surface expression by microtubule-disrupting agents such as colchicine resulted from decreased DRA exocytosis. A previously described exocytosis assay was performed in postconfluent Caco-2 cells (19). Cells were incubated with sulfo-NHS-acetate to mask cell surface proteins. Microtubules were disrupted in the cold and then allowed to recover at 37°C in the presence or absence of nocodazole (66 μM). The newly exocytosed proteins were assessed by cell surface biotinylation. As shown in Fig. 5B, disruption of microtubules reduced DRA exocytosis compared with cells with intact microtubules. The role of microtubules in surface expression of DRA was further supported by our immunofluorescence studies carried out in Caco-2 monolayers. Images in Fig. 5C clearly suggest that disruption of microtubules significantly decreased DRA expression on the apical membrane and that recovery of microtubules restored surface DRA in Caco-2 cells. Taken together, these results indicate that intact microtubules are important for basal exocytic recycling and apical expression of DRA in Caco-2 cells.
Disruption of microtubules in vivo reduces DRA surface levels.
To examine whether disruption of microtubules in vivo resulted in a similar decrease in DRA surface levels, 8-wk-old C57BL/6 mice were treated with the microtubule-disrupting agent colchicine (3 mg/kg body wt ip) for 24 h. Ex vivo surface biotinylation was performed in intact treated and untreated mouse distal colonic segments. As shown in Fig. 6A, disruption of microtubules with colchicine resulted in a significant decrease in apical surface levels of DRA (∼40%). This observation was supported by immunofluorescence studies in Fig. 6B, which showed decreased surface levels of DRA with increased accumulation in subapical pools in colchicine-treated mice compared with controls. Combined with our in vitro results, these data further support the hypothesis that disruption of microtubules decreases DRA surface expression, perhaps via decreased exocytic insertion of DRA.
Fig. 6.

Disruption of microtubules in vivo reduces DRA surface expression. A: ex vivo biotinylation was performed in colons from control and colchicine-treated (3 mg/kg) mice 24 h after administration of the drug. Total lysates from control and treated mice were incubated with NeutrAvidin-agarose beads overnight at 4°C to pull down biotinylated proteins. Biotinylated and total fractions were run on a 7.5% gel and then transferred to a nitrocellulose membrane. Membrane was probed with anti-DRA antibody, and protein bands were quantified by densitometric analysis. Results are shown as ratio of surface (apical) to total DRA. Values are means ± SE of results from 3 different experiments. ***P < 0.006 vs. control. B: immunofluorescent staining [DRA (green) and villin (red)] in colonic tissue sections from control and colchicine-treated mice. Nuclei were stained with SlowFade with DAPI (blue). A representative image from 3 different animals for each treatment is shown. Images were obtained using a confocal microscope (LSM 710, Zeiss) with a ×63 objective. Control and colchicine images were obtained in the same plane.
EPEC infection decreases DRA surface levels via increased endocytosis.
Having established the mechanisms of DRA recycling under physiological conditions, we next examined the DRA recycling pathways implicated in decreasing its surface expression in response to EPEC infection. First, cell surface and endocytic internalization assay studies (Fig. 7, A and B) were utilized to investigate whether EPEC-induced reduction of DRA surface levels is a result of increased DRA endocytosis in Caco-2 cells infected with EPEC. Consistent with previous studies (7), EPEC infection of Caco-2 monolayers decreased surface expression of DRA as early as 60 min (∼50%) compared with controls (Fig. 7A). Decreased DRA surface levels were partly associated with increased endocytosis, as reverse biotinylation studies showed that EPEC significantly increased DRA endocytosis (35–45%) compared with uninfected control cells (Fig. 7B). Also, an increase in endocytosis upon EPEC infection was not observed (Fig. 7C) when the experiment was performed on ice to block cellular recycling, providing further proof that the EPEC-mediated decrease in cell surface levels of DRA is a result of increased endocytic recycling.
Fig. 7.

Enteropathogenic Escherichia coli (EPEC) decreases DRA surface expression partly via increased DRA endocytosis in Caco-2 cells. A–C: cells infected with EPEC for 60 min were subjected to cell surface biotinylation (A), reverse cell surface biotinylation (B), or reverse biotinylation on ice (negative control, n = 5; C). NeutrAvidin-agarose beads were used to pull down biotinylated proteins. Biotinylated and total protein fractions were run on a 7.5% SDS-polyacrylamide gel. Blot was immunostained with rabbit anti-DRA antibody. Biotinylated fractions (A and B) represent apical and endocytosed pool of DRA, respectively. Densitometric analysis presented below each blot is representative of 3 separate experiments. Results are shown as ratio of apical to total DRA (A) and endocytosed to total DRA (B and C). Values are means ± SE of 3 different experiments. *P < 0.05 and **P < 0.004 vs. control.
Inhibition of dynamin does not alter the EPEC-mediated decrease in apical Cl−/OH−(HCO3−) exchange activity.
Since our above-described results showed that basal DRA endocytosis is clathrin-dependent in Caco-2 cells and that EPEC modulates DRA surface levels via increased endocytosis, we next examined the role of CME in EPEC-induced inhibition of Cl−/OH−(HCO3−) exchange activity. Serum-starved Caco-2 monolayers were pretreated for 30 min with dynasore (80 μM) and then infected with EPEC for 30 min in the presence or absence of dynasore. Dynasore failed to block EPEC-mediated inhibition of Cl−/OH−(HCO3−) exchange activity (Fig. 8). Our results suggest that inhibition of apical Cl−/OH−(HCO3−) exchange in Caco-2 cells by EPEC infection is unaltered by blocking dynamin activity.
Fig. 8.
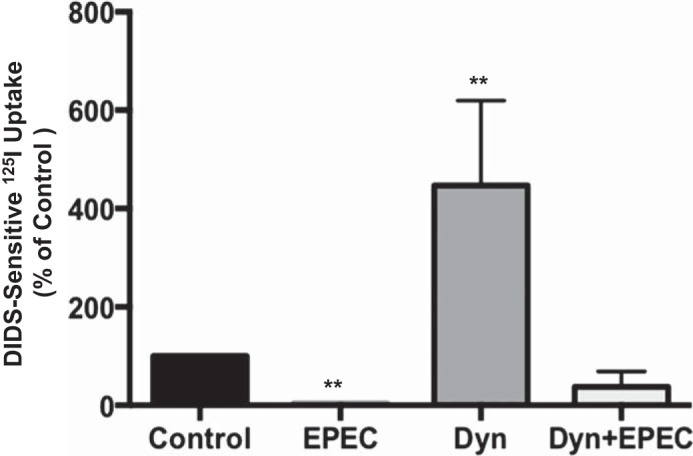
Dynamin inhibition does not alter EPEC-induced reduction of apical Cl−/HCO3− exchange activity in Caco-2 cells. Overnight serum-starved Caco-2 cells were preincubated with 80 μM dynasore (Dyn) for 30 min and then infected with EPEC for 30 min in the presence or absence of the inhibitor. Apical Cl−/HCO3− exchange activity was measured as DIDS (600 μM)-sensitive 125I− uptake in base-loaded cells. Values are means ± SE of 4 experiments performed in triplicate. **P < 0.001 vs. control.
EPEC infection reduces DRA association with lipid rafts.
We next investigated the effect of EPEC infection on the association of DRA with membrane lipid raft domains. Figure 9A clearly shows that infection with EPEC resulted in a ∼40% decrease in DRA association with lipid rafts compared with control. These results indicate that the EPEC-mediated increase in DRA endocytosis parallels a decrease in function, surface expression, and lipid raft association. Furthermore, when cells were infected with EPEC in the presence or absence of dynasore (Fig. 9B), changes in DRA association with lipid rafts paralleled changes in apical Cl−/OH−(HCO3−) exchange activity, as mentioned above (Fig. 4).
Fig. 9.

EPEC infection reduces DRA association with lipid rafts. A: Caco-2 monolayers were infected with EPEC for 60 min. Total membrane pellet was obtained by ultracentrifugation. Lipid rafts were separated by solubilization in detergent buffer at 4°C. Lipid raft [detergent-insoluble (DI)] fractions were resolved on a 7.5% SDS-polyacrylamide gel and immunoblotted with anti-DRA and anti-actin antibodies. Results are shown as ratio of DRA to β-actin. Values are means ± SE of results from 4 different experiments. **P < 0.001 vs. control. B: Caco-2 monolayers were infected with EPEC in the presence or absence of dynasore for 60 min. Total membrane pellet was obtained from control and EPEC-infected cell lysates by ultracentrifugation. Lipid rafts were separated by solubilization in a Triton X-100 detergent buffer at 4°C. Lipid raft (DI) fractions were resolved on a 7.5% SDS-polyacrylamide gel and immunoblotted with anti-DRA and anti-actin antibodies. Results are shown as ratio of DRA to β-actin. Values are means ± SE of results from 4 different experiments. **P < 0.001 vs. control.
EPEC effectors EspG1 and EspG2 are essential for the EPEC-mediated decrease in DRA exocytosis.
Previous studies from our group showed that the EPEC-mediated decrease in DRA activity was dependent on the effector molecules EspG1 and EspG2, which are known to disrupt the host microtubule network (7). Since our studies in uninfected Caco-2 cells indicated that microtubules are involved in DRA exocytosis, we investigated their role in the EPEC-mediated decrease in DRA surface expression. To elucidate whether EPEC-mediated disruption of microtubules is implicated in the decreased surface levels of DRA in Caco-2 cells, we performed a biotin-based exocytosis assay in cells infected with wild-type EPEC and ΔespG1 and ΔespG2 mutants for 60 min. The amount of exocytosed DRA was significantly reduced (∼60%) following infection with wild-type EPEC compared with ΔespG1/ΔespG2 (Fig. 10), suggesting that the EPEC-mediated decrease in DRA exocytosis is due to disruption of microtubules, which are essential for DRA surface expression.
Fig. 10.
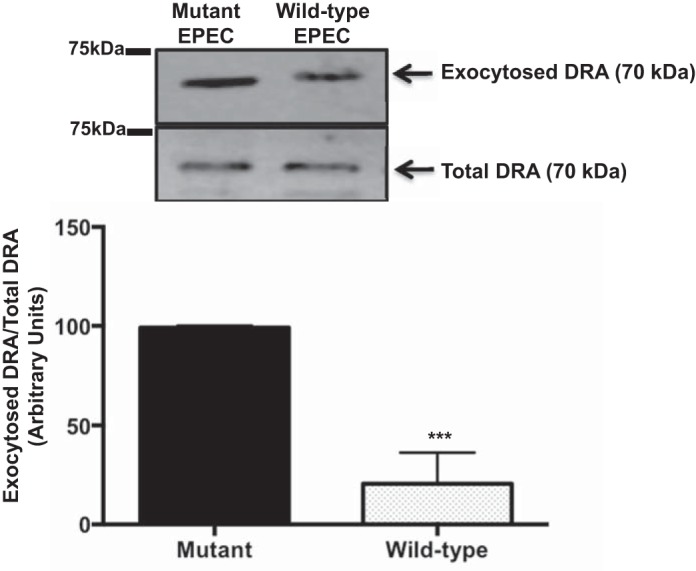
EPEC effectors EspG1 and EspG2 are essential for EPEC-mediated decrease in DRA exocytosis. Surface NHS-reactive sites on Caco-2 cells grown on plastic support were masked with sulfo-NHS-acetate at 4°C for 60 min. Cells were then infected with wild-type EPEC or the ΔespG1/G2 mutant for 60 min. Amount of protein exocytosed to the surface was measured by cell surface biotinylation. Biotinylated proteins were pulled down with NeutrAvidin-agarose beads and resolved on a 7.5% SDS-polyacrylamide gel. Immunoblot was probed with anti-DRA antibody and analyzed by densitometry. Results are shown as ratio of exocytosed to total DRA. Values are means ± SE of results from 3 different blots. ***P < 0.005 vs. mutant.
DISCUSSION
Membrane proteins, channels, and ion transporters undergo constitutive and regulated endocytic retrieval followed by delivery to the plasma membrane to maintain steady surface levels. Previous studies in HEK 293 cells stably transfected with DRA have shown that DRA undergoes basal endocytic recycling (17). These studies show that, once internalized, DRA enters the subapical pool of early endosomes, where it is sorted to recycling endosomes to be returned to the apical membrane (18, 21, 22). However, the role of clathrin- and caveolin-mediated pathways in the recycling of DRA in IECs under basal conditions has not been fully investigated. Our studies showed that the endocytosis inhibitor dynasore increased DRA surface expression in Caco-2 cells. Dynasore is a specific inhibitor of dynamin, a 100-kDa GTPase that is required to pinch-off coated vesicles from the plasma membrane, and blocks both clathrin- and caveolin-mediated endocytosis (23). Since dynamin regulates clathrin- and caveolin-dependent endocytic routes, we performed functional studies utilizing an inhibitor specific to CME (14). Chlorpromazine is a cationic drug that, when used at 50–100 μM, inhibits CME (35). Chlorpromazine prevents translocation and assembly of clathrin and adaptor proteins (required for recruitment of cargo and clathrin) to the site of endocytosis at the plasma membrane. Our results show a significant increase in apical Cl−/HCO3− exchange activity in Caco-2 cells treated with chlorpromazine, suggesting that Cl−/HCO3− exchange activity is dependent on dynamin- and clathrin-mediated pathways. The role of caveolin-dependent pathways in regulating Cl−/HCO3− exchange activity in Caco-2 cells was ruled out, as our data (not shown) and previous studies (34) demonstrated that Caco-2 cells do not express caveolin-1 and caveolin-2.
Further elucidation of the mechanisms of basal DRA endocytosis in Caco-2 cells revealed that the association of DRA with lipid rafts correlates with dynasore-stimulated apical Cl−/HCO3− exchange activity. Lipid rafts are cholesterol- and sphingolipid-rich membrane microdomains that serve as key docking complexes for signaling molecules and adaptor and membrane proteins influencing membrane protein trafficking (4). Previous studies from our group showed that DRA is associated with lipid rafts in IECs and that neuropeptide Y stimulated DRA function in Caco-2 cells by increasing its association with lipid raft fractions (31). Studies from Lissner et al. (22) also indicate that DRA is associated with lipid rafts in an intracellular fraction, such as recycling vesicles, and that its exocytic insertion into the apical plasma membrane depends on raft integrity and phosphoinositide 3-kinase. Therefore, it appears that a dynasore-mediated decrease in endocytosis causes retention of DRA on the plasma membrane, leading to increased association with lipid rafts, which results in enhanced activity.
Earlier studies from our laboratory showed decreased apical Cl−/HCO3− exchange activity in Caco-2 cells treated with the microtubule-disrupting agent colchicine (7). These studies were complemented by our current confocal microscopy data showing reduced apical surface DRA in Caco-2 cells with disrupted microtubules. Disruption of microtubules by cold exposure and nocodazole treatment did not significantly alter the endocytosed pool of DRA compared with control cells with recovered microtubules. However, there was a decrease in DRA exocytosis in the presence of nocodazole compared with cells with intact microtubules. These data suggest that microtubule disruption decreases DRA surface expression via a decrease in DRA exocytosis, rather than an increase in DRA endocytosis. Our results are in agreement with previous studies showing that disruption of microtubules by nocodazole retards the apical delivery of plasma membrane proteins such as aminopeptidase N in Caco-2 cells (6). Our data regarding the role of intact microtubules in DRA exocytosis and surface expression were further supported by in vivo studies showing a significant decrease in apical DRA levels in colchicine-treated mice. These results are consistent with previous findings in an in vivo study that NHE3 and NHE regulatory factor are mistargeted to the basolateral membrane in proximal tubules of colchicine-treated rats (30).
The decrease in surface levels of DRA by EPEC infection could result from an increase in DRA endocytosis and/or a decrease in DRA exocytosis. Our findings from biochemical and confocal microscopy studies in Caco-2 cells show a significant increase in DRA endocytosis in EPEC-infected cells compared with uninfected cells. However, infection of Caco-2 cells with EPEC resulted in a decrease in apical Cl−/HCO3− exchange activity in the presence and absence of dynasore. These results were puzzling but consistent with recent studies carried out in T-84 cells that showed failure of dynasore to block EPEC-mediated internalization of CFTR (29). Therefore, it appears that the EPEC-mediated decrease in apical Cl−/HCO3− exchange activity is independent of dynamin-mediated cellular recycling events. Perhaps EPEC acts at a point in DRA endocytosis prior to the involvement of dynamin, or it prevents assembly of endocytic machinery independent of dynamin. Therefore, it will be interesting to perform future studies to address this complex cellular trafficking. In addition to an increase in DRA endocytosis, EPEC infection of Caco-2 cells also significantly decreased DRA levels in the exocytosed pool. Furthermore, this decrease was found to be dependent on the virulence factors EspG1 and EspG2, as significantly less DRA exocytosis was observed in Caco-2 cells infected with wild-type EPEC, which is able to cause microtubule disruption, than in cells infected with the ΔespG1/ΔespG2 mutant, which does not disrupt IEC microtubules. Therefore, the integrity of the microtubular network in EPEC-mediated modulation of DRA exocytosis appears to be critical. Our findings are consistent with previous in vivo studies utilizing Citrobacter rodentium-infected mice (the equivalent murine model for human EPEC infection), showing the mislocalization of the water channels aquaporin-2 and aquaporin-3 from the plasma membrane to the cytoplasm partially via an EspG-dependent mechanism (11).
Moreover, we demonstrated that EPEC infection significantly redistributes DRA from lipid rafts to detergent-soluble nonraft membrane fractions, which may influence its function in the apical membrane. Similar to these observations, an in vivo study showed that EPEC causes redistribution of the tight junction proteins occludin and claudin-1 from detergent-insoluble raft fractions to detergent-soluble fractions, causing a loss of tight junction barrier function (20). However, it remains unknown whether lipid raft dissociation of DRA in response to EPEC infection is a result of increased endocytosis or a consequence of decreased reinsertion of DRA into the plasma membrane. This question warrants further investigation. Collectively, these data indicate that EPEC infection modulates DRA recycling partly via increased endocytic retrieval of DRA and partly via decreased DRA exocytosis dependent on disruption of the host microtubular network. These data highlight the importance of membrane transporter recycling events in diarrhea induced by EPEC infection. In summary, our results demonstrate, for the first time, novel mechanisms of DRA cellular recycling under physiological conditions and in response to EPEC infection. Further elucidation of these pathways will enhance our understanding of the mechanisms underlying the pathophysiology of EPEC-induced diarrhea and aid in the identification of novel targets for the treatment of diarrheal disorders.
GRANTS
These studies were supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-54016 (P. K. Dudeja), DK-81858 (P. K. Dudeja), DK-92441 (P. K. Dudeja), DK-71596 (W. A. Alrefai), DK-097043 (G. A. Hecht), DK-98170 (R. K. Gill), and DK-96254 (S. Saksena) and Department of Veterans Affairs, Veteran Heath Administration, Office of Research and Development, Biomedical Laboratory Research and Development Grant BX002011, a Senior Research Career Scientist Award (P. K. Dudeja), and Grants BX000152 (W. A. Alrefai) and BX000785 (G. A. Hecht).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.G., S.S., R.K.G., W.A.A., and P.K.D. developed the concept and designed the research; T.G., A.K., and S.P. performed the experiments; T.G., A.K., and S.P. analyzed the data; T.G., A.K., S.P., S.S., and R.K.G. interpreted the results of the experiments; T.G. prepared the figures; T.G. drafted the manuscript; T.G., S.S., R.K.G., K.H., W.A.A., G.A.H., and P.K.D. edited and revised the manuscript; W.A.A. and P.K.D. approved the final version of the manuscript.
REFERENCES
- 1.Annaba F, Sarwar Z, Kumar P, Saksena S, Turner JR, Dudeja PK, Gill RK, Alrefai WA. Modulation of ileal bile acid transporter (ASBT) activity by depletion of plasma membrane cholesterol: association with lipid rafts. Am J Physiol Gastrointest Liver Physiol 294: G489–G497, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254, 1976. [DOI] [PubMed] [Google Scholar]
- 3.Cao X, Surma MA, Simons K. Polarized sorting and trafficking in epithelial cells. Cell Res 22: 793–805, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dart C. Lipid microdomains and the regulation of ion channel function. J Physiol 588: 3169–3178, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duffield A, Caplan MJ, Muth TR. Protein trafficking in polarized cells. Int Rev Cell Mol Biol 270: 145–179, 2008. [DOI] [PubMed] [Google Scholar]
- 6.Eilers U, Klumperman J, Hauri HP. Nocodazole, a microtubule-active drug, interferes with apical protein delivery in cultured intestinal epithelial cells (Caco-2). J Cell Biol 108: 13–22, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gill RK, Borthakur A, Hodges K, Turner JR, Clayburgh DR, Saksena S, Zaheer A, Ramaswamy K, Hecht G, Dudeja PK. Mechanism underlying inhibition of intestinal apical Cl/OH exchange following infection with enteropathogenic E. coli. J Clin Invest 117: 428–437, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glotfelty LG, Hecht GA. Enteropathogenic E. coli effectors EspG1/G2 disrupt tight junctions: new roles and mechanisms. Ann NY Acad Sci 1258: 149–158, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glotfelty LG, Zahs A, Hodges K, Shan K, Alto NM, Hecht GA. Enteropathogenic E. coli effectors EspG1/G2 disrupt microtubules, contribute to tight junction perturbation and inhibit restoration. Cell Microbiol 16: 1767–1783, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glotfelty LG, Zahs A, Iancu C, Shen L, Hecht GA. Microtubules are required for efficient epithelial tight junction homeostasis and restoration. Am J Physiol Cell Physiol 307: C245–C254, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guttman JA, Samji FN, Li Y, Deng W, Lin A, Finlay BB. Aquaporins contribute to diarrhoea caused by attaching and effacing bacterial pathogens. Cell Microbiol 9: 131–141, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Hecht G. Microbes and microbial toxins: paradigms for microbial-mucosal interactions. VII. Enteropathogenic Escherichia coli: physiological alterations from an extracellular position. Am J Physiol Gastrointest Liver Physiol 281: G1–G7, 2001. [DOI] [PubMed] [Google Scholar]
- 13.Hecht G, Hodges K, Gill RK, Kear F, Tyagi S, Malakooti J, Ramaswamy K, Dudeja PK. Differential regulation of Na+/H+ exchange isoform activities by enteropathogenic E. coli in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 287: G370–G378, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Hinshaw JE. Dynamin and its role in membrane fission. Annu Rev Cell Dev Biol 16: 483–519, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodges K, Alto NM, Ramaswamy K, Dudeja PK, Hecht G. The enteropathogenic Escherichia coli effector protein EspF decreases sodium hydrogen exchanger 3 activity. Cell Microbiol 10: 1735–1745, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai Y, Rosenshine I, Leong JM, Frankel G. Intimate host attachment: enteropathogenic and enterohaemorrhagic Escherichia coli. Cell Microbiol 15: 1796–1808, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamprecht G, Baisch S, Schoenleber E, Gregor M. Transport properties of the human intestinal anion exchanger DRA (down-regulated in adenoma) in transfected HEK293 cells. Pflügers Arch 449: 479–490, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Lamprecht G, Heil A, Baisch S, Lin-Wu E, Yun CC, Kalbacher H, Gregor M, Seidler U. The down regulated in adenoma (dra) gene product binds to the second PDZ domain of the NHE3 kinase A regulatory protein (E3KARP), potentially linking intestinal Cl−/HCO3− exchange to Na+/H+ exchange. Biochemistry 41: 12336–12342, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Lee-Kwon W, Kawano K, Choi JW, Kim JH, Donowitz M. Lysophosphatidic acid stimulates brush border Na+/H+ exchanger 3 (NHE3) activity by increasing its exocytosis by an NHE3 kinase A regulatory protein-dependent mechanism. J Biol Chem 278: 16494–16501, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Zhang Q, Wang C, Li N, Li J. Invasion of enteropathogenic Escherichia coli into host cells through epithelial tight junctions. FEBS J 275: 6022–6032, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Lissner S, Hsieh CJ, Nold L, Bannert K, Bodammer P, Sultan A, Seidler U, Graeve L, Lamprecht G. The PDZ-interaction of the intestinal anion exchanger downregulated in adenoma (DRA; SLC26A3) facilitates its movement into Rab11a-positive recycling endosomes. Am J Physiol Gastrointest Liver Physiol 304: G980–G990, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Lissner S, Nold L, Hsieh CJ, Turner JR, Gregor M, Graeve L, Lamprecht G. Activity and PI3-kinase dependent trafficking of the intestinal anion exchanger down-regulated in adenoma (DRA) depend on its PDZ interaction and on lipid rafts. Am J Physiol Gastrointest Liver Physiol 299: G907–G920, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10: 839–850, 2006. [DOI] [PubMed] [Google Scholar]
- 24.McNamara BP, Koutsouris A, O'Connell CB, Nougayrede JP, Donnenberg MS, Hecht G. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J Clin Invest 107: 621–629, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moseley RH, Hoglund P, Wu GD, Silberg DG, Haila S, de la Chapelle A, Holmberg C, Kere J. Downregulated in adenoma gene encodes a chloride transporter defective in congenital chloride diarrhea. Am J Physiol Gastrointest Liver Physiol 276: G185–G192, 1999. [DOI] [PubMed] [Google Scholar]
- 26.Musch MW, Arvans DL, Wang Y, Nakagawa Y, Solomaha E, Chang EB. Cyclic AMP-mediated endocytosis of intestinal epithelial NHE3 requires binding to synaptotagmin 1. Am J Physiol Gastrointest Liver Physiol 298: G203–G211, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Musch MW, Arvans DL, Wu GD, Chang EB. Functional coupling of the downregulated in adenoma Cl−/base exchanger DRA and the apical Na+/H+ exchangers NHE2 and NHE3. Am J Physiol Gastrointest Liver Physiol 296: G202–G210, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norio R, Perheentupa J, Launiala K, Hallman N. Congenital chloride diarrhea, an autosomal recessive disease: genetic study of 14 Finnish and 12 other families. Clin Genet 2: 182–192, 1971. [PubMed] [Google Scholar]
- 29.Ohland CL, DeVinney R, MacNaughton WK. Escherichia coli-induced epithelial hyporesponsiveness to secretagogues is associated with altered CFTR localization. Cell Microbiol 14: 447–459, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Sabolic I, Herak-Kramberger CM, Ljubojevic M, Biemesderfer D, Brown D. NHE3 and NHERF are targeted to the basolateral membrane in proximal tubules of colchicine-treated rats. Kidney Int 61: 1351–1364, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Saksena S, Tyagi S, Goyal S, Gill RK, Alrefai WA, Ramaswamy K, Dudeja PK. Stimulation of apical Cl−/HCO3−(OH−) exchanger, SLC26A3 by neuropeptide Y (NPY) is lipid raft dependent. Am J Physiol Gastrointest Liver Physiol 299: G1334–G1343, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schweinfest CW, Spyropoulos DD, Henderson KW, Kim JH, Chapman JM, Barone S, Worrell RT, Wang Z, Soleimani M. slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. J Biol Chem 281: 37962–37971, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Sun TX, Van Hoek A, Huang Y, Bouley R, McLaughlin M, Brown D. Aquaporin-2 localization in clathrin-coated pits: inhibition of endocytosis by dominant-negative dynamin. Am J Physiol Renal Physiol 282: F998–F1011, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Ullrich N, Caplanusi A, Brone B, Hermans D, Lariviere E, Nilius B, Van Driessche W, Eggermont J. Stimulation by caveolin-1 of the hypotonicity-induced release of taurine and ATP at basolateral, but not apical, membrane of Caco-2 cells. Am J Physiol Cell Physiol 290: C1287–C1296, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Vercauteren D, Vandenbroucke RE, Jones AT, Rejman J, Demeester J, De Smedt SC, Sanders NN, Braeckmans K. The use of inhibitors to study endocytic pathways of gene carriers: optimization and pitfalls. Mol Ther 18: 561–569, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wedenoja S, Pekansaari E, Hoglund P, Makela S, Holmberg C, Kere J. Update on SLC26A3 mutations in congenital chloride diarrhea. Hum Mutat 32: 715–722, 2011. [DOI] [PubMed] [Google Scholar]
- 37.Young A, Gentzsch M, Abban CY, Jia Y, Meneses PI, Bridges RJ, Bradbury NA. Dynasore inhibits removal of wild-type and ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR) from the plasma membrane. Biochem J 421: 377–385, 2009. [DOI] [PubMed] [Google Scholar]