

Abstract
Carotenoid cleavage oxygenases (CCOs) are non-heme, Fe(II)-dependent enzymes that participate in biologically important metabolic pathways involving carotenoids and apocarotenoids, including retinoids, stilbenes, and related compounds. CCOs typically catalyze the cleavage of non-aromatic double bonds by dioxygen (O2) to form aldehyde or ketone products. Expressed only in vertebrates, the RPE65 sub-group of CCOs catalyzes a non-canonical reaction consisting of concerted ester cleavage and trans-cis isomerization of all-trans-retinyl esters. It remains unclear whether the former group of CCOs functions as mono- or di-oxygenases. Additionally, a potential role for O2 in catalysis by the RPE65 group of CCOs has not been evaluated to date. Here, we investigated the pattern of oxygen incorporation into apocarotenoid products of Synechocystis apocarotenoid oxygenase. Reactions performed in the presence of 18O-labeled water and 18O2 revealed an unambiguous dioxygenase pattern of O2 incorporation into the reaction products. Substitution of Ala for Thr at position 136 of apocarotenoid oxygenase, a site predicted to govern the mono- versus dioxygenase tendency of CCOs, greatly reduced enzymatic activity without altering the dioxygenase labeling pattern. Reevaluation of the oxygen-labeling pattern of the resveratrol-cleaving CCO, NOV2, previously reported to be a monooxygenase, using a purified enzyme sample revealed that it too is a dioxygenase. We also demonstrated that bovine RPE65 is not dependent on O2 for its cleavage/isomerase activity. In conjunction with prior research, the results of this study resolve key issues regarding the utilization of O2 by CCOs and indicate that dioxygenase activity is a feature common among double bond-cleaving CCOs.
Keywords: carotenoid, enzyme, enzyme catalysis, enzyme mechanism, enzyme structure, retinal metabolism, retinoid, retinol
Introduction
Carotenoid cleavage oxygenases (CCOs)3 are non-heme iron-dependent enzymes found in all kingdoms of life that participate in the metabolism of carotenoids and related compounds. Since their initial discovery in bacteria and plants, tremendous progress has been made in elucidating the biological substrates of these enzymes (1–3). Most characterized CCOs catalyze the oxidative cleavage of carotenoid carbon-carbon double bonds to yield aldehyde and/or ketone-containing apocarotenoid products (Fig. 1). Such activity was possibly the earliest to evolve in this family because apocarotenoids, particularly retinal, mediate photoreception by the ancient type 1 opsin protein family, members of which carry out many important physiological processes in phototactic and phototrophic organisms (4, 5). Another group of bacterial and fungal members has evolved to cleave carbon double bonds of stilbenes such as resveratrol and lignin-related phenylpropanoids (6, 7). The RPE65 class of CCOs, found only in vertebrates, underwent catalytic neofunctionalization during its split from a β-carotene oxygenase (BCO)-2-like ancestor to acquire the non-oxidative retinyl ester cleavage/isomerase activity central to the visual cycle that generates 11-cis-retinal required for visual pigment (type II opsin) formation (Fig. 1, A and B) (8, 9).
FIGURE 1.

Phylogenic and enzymatic relationships among CCOs. A, unrooted maximum likelihood phylogenic tree of select CCO family members. The tree termini are colored according the substrate specificity of the associated taxon/clade. The BCO1, BCO2, and RPE65 clades are composed of sequences from Mus musculus, Rattus norvegicus, Homo sapiens, and Bos taurus. The altitudes of the triangles associated with these clades are proportional to the evolutionary diversity among the examined taxa. Bootstrap values from 1000 pseudo-replicates are displayed as percentages beside the associated branches. The scale bar indicates the average number of site substitutions over the indicated distance. Members of the CCO family whose reaction mechanisms have previously been examined by isotope labeling experiments are shown in bold. Abbreviations are as follows: dNinaB, Drosophila melanogaster NinaB; gNinaB. Galleria mellonella NinaB; ACO. Synechocystis sp. PCC 6803 ACO; NSC. Nostoc sp. PCC 7120 carotenoid oxygenase; NOV2, Novosphingobium aromaticivorans CCO 2; IEM, Pseudomonas nitroreducens isoeugenol monooxygenase; umRCO1, Ustilago maydis resveratrol cleavage oxygenase 1; CAO1, Neurospora crassa carotenoid oxygenase 1; LSD1, Pseudomonas paucimobilis lignostilbene dioxygenase 1; umBCO1, U. maydis β-carotene cleavage oxygenase 1; fCARX, Fusarium fujikuroi carotenoid oxygenase; VP14, Zea mays viviparous 14; atCCD1, Arabidopsis thaliana carotenoid cleavage dioxygenase 1. B, reactions catalyzed by CCOs considered in this study. Synechocystis ACO, a prototypical CCO member, specifically cleaves all-trans-8′-apocarotenol at the 15,15′-double bond position to generate all-trans-retinal (RAL) and 8′-hydroxy-15′-apocarotenal (C10-apocarotenal). RPE65, an atypical CCO member, catalyzes a coupled ester cleavage/isomerization of all-trans-retinyl esters (atRE) rather than oxidative carotenoid cleavage. This reaction produces 11-cis-retinol (11cROL), a key intermediate in the regeneration of visual chromophore required for vertebrate vision. NOV2 from Novosphingobium cleaves resveratrol at the interphenyl double bond to produce 3,5-DHBA and 4-HBA. Wavy red lines indicate the location of the scissile bond in each reaction.
The spin-forbidden reaction of O2 with alkene substrates of CCOs is made kinetically favorable through its reductive activation by the non-heme iron(II) centers of these enzymes (10). Of central importance to understanding the mechanism of O2 activation by CCOs and its reaction with substrates is knowledge of whether one or both of the oxygen atoms from O2 is/are incorporated into the reaction products. In the former case, the non-O2-derived oxygen atom found in the reaction is expected to originate from an active site-bound water molecule. A number of studies using isotopically labeled O2 and H2O were carried out to address this question for CCOs, but the issue still remains debatable (11–15). A factor complicating the interpretation of such experiments is the exchange of oxygen incorporated into the nascent products with that of bulk water during the sample workup and analysis. This makes dioxygenases appear to be monooxygenases (16, 17). An initial study on the catalytic mechanism of chicken BCO1 that suggested it was a monooxygenase (11) was criticized for its use of conditions that allowed substantial solvent back-exchange (17). More recent studies of human BCO1 (12) and Arabidopsis CCD1 (14), in which solvent exchange was carefully monitored, demonstrated dioxygenase labeling patterns for these CCOs. By contrast, labeling experiments performed on resveratrol and isoeugenol-cleaving CCOs, NOV2 (13) and isoeugenol monooxygenase (15), from the non-carotenogenic bacteria Novosphingobium aromaticivorans and Pseudomonas nitroreducens, respectively, suggested a monooxygenase pattern of oxygen incorporation for both enzymes. Thus, the labeling studies performed to date seem to indicate that perhaps both monooxygenase and dioxygenase catalytic mechanisms may exist within the CCO enzyme family (18), a possibility previously suggested by a theoretical study of the Synechocystis ACO catalytic mechanism (19).
In contrast to oxidative carotenoid cleavage by CCOs, a basic consideration of the ester cleavage/isomerization chemistry catalyzed by RPE65 does not suggest an obvious role for O2 in the retinoid isomerization reaction (Fig. 1B). Additionally, substrate and water isotope labeling studies do not indicate direct incorporation of O2-derived oxygen into the 11-cis-retinol product of the isomerization reaction (20–23). However, the close structural similarity of the RPE65 and carotenoid-cleaving CCO Fe(II) centers, together with the evolutionary relatedness of these proteins, raise the possibility that certain features of their catalytic modes could be shared, including a requirement for O2 (24). Recently, a hypothetical role for O2 in RPE65-catalyzed retinoid isomerization that does not involve its permanent incorporation into the reaction products has been proposed (25).
In this study, we carried out isotope labeling studies on highly purified and active preparations of Synechocystis ACO and Novosphingobium NOV2 with assay protocols that minimized solvent oxygen back-exchange. These experiments unambiguously showed that both enzymes, like human BCO1 and Arabidopsis CCD1, are dioxygenases. A T136A substitution in the ACO sequence that was predicted to open an occluded coordination site for solvent binding, thus potentially favoring monooxygenase chemistry, did not alter the dioxygenase-labeling pattern (19). Using ACO as an internal control, we demonstrated that O2 is not required for RPE65-mediated conversion of all-trans-retinyl esters into 11-cis-retinol, in agreement with a recently proposed mechanism of retinoid isomerization (26). These data provide compelling evidence for a common dioxygenase mechanism among all double bond-cleaving CCOs.
Materials and Methods
Phylogeny Inference
The CCO amino acid sequences of interest were retrieved from the NCBI protein database. Atomic coordinates for CCOs of known structure, Synechocystis ACO, bovine RPE65, and Zea mays VP14 (PDB accession codes, 4OU9, 4RSC, and 3NPE, respectively) were obtained from the Protein Data Bank. A structure-based sequence alignment was first generated using the program MUSTANG (27). This structure-based alignment was then used as a profile for sequence-based alignment of the remaining proteins using MUSCLE (28). A standard sequence-based alignment of the proteins was also generated with MUSCLE to assess the influence of the alignment methodology on the inferred tree topology. Columns containing gaps were removed from the alignment. The CCO phylogeny was inferred based on the maximum likelihood optimality criterion as implemented in PhyML (29). The LG substitution matrix with an estimation of the γ-parameter (four categories) and invariant sites resulted in the highest log-likelihood tree as assessed by ProtTest (30). Improvements to the starting neighbor-joining tree were carried out by both nearest neighbor interchange and surface pruning and regrafting rearrangements. Both tree topology and branch lengths were optimized. Tree robustness was assessed by analysis of 1000 bootstrap pseudo-replicates. The presented maximum likelihood tree had a log likelihood value of −14321.6 (Fig. 1A). An identical tree topology was obtained when the procedure was repeated with the pure sequence-based alignment.
Protein Expression and Purification
Synechocystis ACO was expressed and purified as described previously (31). The ACO T136A point mutant was generated with a QuikChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA) and confirmed by DNA sequencing. T136A-ACO was expressed and purified identically to the wild-type protein. The coding sequence of NOV2 from N. aromaticivorans DSM 12444 (GI: 499765715) was synthesized and cloned into the pET3a expression vector (Novagen) without any fusion tags. The integrity of this expression plasmid was confirmed by sequencing. The plasmid was transformed into the T7 express BL21 Escherichia coli strain (New England Biolabs, Ipswich, MA) for protein expression studies. One-liter cultures containing 100 μg of ampicillin/ml of LB media were grown at 37 °C to an A600 nm of 0.5–0.8 when the temperature was lowered to 28 °C and additional ampicillin (100 μg/ml) was added. Protein expression relied on leaky T7 promoter activity. After overnight growth (12–16 h), cells were harvested by centrifugation and stored at −80 °C. NOV2 was purified in the same manner as ACO (31) by ammonium sulfate fractionation and gel filtration chromatography. Purified protein samples (≥95% pure as judged by SDS-PAGE analysis) were flash-frozen and stored at −80 °C or placed on ice for immediate use.
Enzymatic Assays and Rate Determination for Background Oxygen Exchange
ACO activity studies were conducted according to previously established methods (31). NOV2 enzymatic activity was assayed with resveratrol as a substrate in a 20 mm HEPES-NaOH, pH 7.0. NOV2 steady-state kinetics were assessed by monitoring the reduction of resveratrol absorbance at 304 nm over time using a Flexstation 3 plate reader device (Molecular Devices, Sunnyvale, CA). Reactions were run at 28 °C in 200 μl of 10 mm BisTris-HCl, pH 7.0, containing 0.8 μg of purified NOV2 and resveratrol (Sigma, 99% purity) at eight concentrations ranging from 62.5 to 0.49 μm separated by 2-fold dilutions. Absorbance measurements were taken every 15 s for a total of 10 min. Initial reaction velocities were determined from the linear portions of the reaction profiles. Absorbance was related to absolute concentrations by simultaneous absorbance recordings of resveratrol standards. For NOV2 oxygen labeling studies, resveratrol was delivered in DMSO to a HEPES-NaOH, pH 7.0, buffer system containing purified NOV2, and the reaction was carried out at 28 °C with 500 rpm shaking in a Thermomixer (Eppendorf, Hauppauge, NY), as described in detail below. Following the incubation, products were directly extracted by ethyl acetate and analyzed by HPLC (see below). Product amounts were quantified by plotting peak areas of known quantities of standards. To determine the background oxygen exchange for ACO- and NOV2-catalyzed reaction products, the exchange reactions were performed in H218O. For ACO reaction products, the reaction mixture consisted of 192 μl of H218O (99% 18O atom, Sigma), 4 μl of 1 m HEPES-NaOH, pH 7.0, 1 μl of 10% (w/v) Triton X-100, and 3 μl of 1 μg/μl freshly purified ACO. Then either authentic all-trans-retinal (TRC, Toronto, Canada, > 95% purity) or HPLC-purified 8′-hydroxy-15′-apocarotenal generated in-house by a large scale ACO-catalyzed reaction were added to the reaction mixture. The reaction mixture then was incubated at 28 °C with 500 rpm shaking in a Thermomixer. Samples (20 μl) were removed from the mixture at 0, 1, 2, 5, 15, 20, and 30 min and placed into tubes containing 200 μl of hexane followed by vigorous shaking. Then the extracted mixtures were centrifuged in a bench top centrifuge at 15,000 rpm for 2 min. Organic phases were collected and analyzed by LC-MS (see below). Two additional experiments were conducted in the same manner but with 1% (w/v) BSA and/or 5% (v/v) glacial acetic acid to evaluate protein or pH-facilitated oxygen exchange. Background oxygen exchange for the NOV2 reaction products was determined in a similar manner. Briefly, the reaction mixture contained 150 μl of H218O (97% 18O atom, Sigma), 3 μl of 1 m HEPES-NaOH, pH 7.0, and 2.3 μl of freshly purified NOV2 at 4.4 μg/μl. Authentic 4-hydroxybenzaldehyde (4-HBA) or 3,5-dihydroxybenzaldehyde (3,5-DHBA) (Sigma, >95% purity for both compounds) dissolved in DMSO was added to the reaction mixture. Samples (20 μl) were removed from the mixture at 0, 2, 5, 10, and 15 min, placed into tubes containing 200 μl of ethyl acetate, dried under argon, and then redissolved in acetonitrile for MS analysis. As with the apocarotenoid product experiment, reactions containing 1% (w/v) BSA or 5% (v/v) glacial acetic acid were used as controls.
Isotope Labeling Study in H218O
For the labeling study in H218O, cleavage reactions were performed in the same manner as described above. Specifically, 3 μl of purified ACO at 1 μg/μl were added to a reaction mixture containing 192 μl of H218O (99% 18O atom, Sigma), 4 μl of 1 m HEPES-NaOH, pH 7.0, and 1 μl of 10% (w/v) Triton X-100. The cleavage reaction was initiated by adding 5 μl of 4 mm all-trans-8′-apocarotenol dissolved in ethanol. The reaction mixture was incubated in a Thermomixer at 28 °C with 500 rpm shaking for 5 min. Then the reaction was quenched with 200 μl of methanol, and 400 μl of diethyl ether/hexanes (4:1, v/v) was added to extract the cleavage products. For the NOV2-catalyzed reaction, 2 μl of 25 mm resveratrol in DMSO was added to a 200-μl reaction mixture consisting of 194 μl of H218O (97% 18O atom, Sigma), 4 μl of 1 m HEPES-NaOH, pH 7.0, and 2.3 μl of 4.4 μg/μl purified NOV2, and this reaction was allowed to proceed for 5 min at 28 °C with 500 rpm shaking. The 4-HBA and 3,5-DHBA products of the reaction were then extracted with 500 μl of ethyl acetate and immediately purified by normal phase HPLC as described below. Peaks corresponding to each cleavage product were collected in glass tubes, dried under argon, and dissolved in acetonitrile. Both NOV2 products were separated by HPLC (see below). ACO and NOV2 reaction products were further analyzed by mass spectrometry (MS) as outlined below.
Sample Deoxygenation and Isotope Labeling Study in 18O2
Sample deoxygenation was performed in 3-ml screw-capped glass vials with a gas-tight Teflon septum. A vial containing 1 ml of 20 mm HEPES-NaOH, pH 7.0, was flushed with ultrapure argon (Airgas, Cleveland, OH) with constant stirring. This deoxygenation treatment was carried out for different times to evaluate its efficiency. Oxygen supplementation was accomplished by flushing the argon-purged buffer solution with O2 (Airgas) for 3 min. To test ACO activity in argon-purged or oxygen-supplemented buffer, 5 μl of Triton X-100 (10% w/v) and 7.5 μl of freshly purified ACO at 2 μg/μl was injected into the treated reaction solutions with an airtight syringe (Hamilton, Reno, NV). The reaction was initiated by injecting 10 μl of 4 mm all-trans-8′-apocarotenol in ethanol and allowed to proceed for 3 min at room temperature with 800 rpm shaking. Then 1.5 ml of methanol was injected to quench the reaction followed by addition of 1 ml of diethyl ether/hexanes (4:1, v/v) to extract the products. The organic phase was collected for LC/MS analysis. For the cleavage reaction performed in an 18O2 atmosphere, 30 μg of purified ACO and 25 μl of substrate at 4 mm in ethanol were used, and 18O2 was introduced into the argon-purged reaction vial. The reaction was carried out for 10 min, followed by product extraction and LC-MS analysis. Both the deoxygenation and NOV2-catalyzed reaction in the presence of 18O2 were conducted in the same way as for ACO. Following a 15-min argon purge of the reaction solution, 5 μl of 20 mg/ml NOV2 and 10 μl of 25 mm resveratrol in DMSO were injected to initiate the reaction, which was carried out for 3 min under an 18O2 atmosphere. Reaction products were extracted with 500 μl of ethyl acetate and immediately dried by argon gas to remove the organic solvent and co-extracted water. The dried sample then was redissolved in ethyl acetate, purified by HPLC, and further analyzed by MS.
HPLC and Mass Spectrometric Analyses of Cleavage Products
HPLC-MS analyses of the ACO-catalyzed cleavage products of all-trans-8′-apocarotenol were performed according to previously established methods (31). Products of NOV2-catalyzed resveratrol cleavage were extracted with ethyl acetate, dried under argon, redissolved in ethyl acetate, and injected onto a normal phase Zorbax Sil column (5 μm, 4.6 × 250 mm) (Agilent, Santa Clara, CA). Separation of the reaction products was achieved with hexanes/ethyl acetate (3:2, v/v) as the mobile phase at a flow rate of 1.4 ml/min. Peaks eluted at ∼4.2 min (4-HBA) and ∼5.3 min (3,5-DHBA) were individually collected, dried under argon, and dissolved in acetonitrile. Purified products from the isotope labeling experiments were directly injected into the APCI source of an LXQ linear ion trap mass spectrometer (Thermo Scientific, Waltham, MA) with acetonitrile and 10 mm ammonium formate. MS data were analyzed with the Xcalibur 2.0.7 software package. Incorporation of 18O was quantified by comparing relative ion intensities within the isotopic distribution window.
RPE Microsome Deoxygenation and Isomerization Activity Assays
Bovine RPE microsomal membranes enriched in RPE65 were prepared as described previously (32). The concentration of RPE65 in microsomal preparations was determined by quantitative densitometry on SDS-polyacrylamide gels. Deoxygenation of RPE microsomes was carried out in the same manner as for ACO and NOV2. Briefly, 0.5 ml of RPE microsomes in a 3-ml screw-capped glass vial with a gas-tight Teflon septum was purged with ultrapure argon. Afterward, 2.5 μl of 20 μm all-trans-retinol in N,N-dimethylformamide was injected into the vial, and the reaction and product analyses were performed as described previously (32). O2 supplementation was accomplished by flushing the sample with O2 gas for 3 min before adding the substrate. The efficiency of O2 depletion/supplementation of RPE microsomes was assessed by monitoring the progress of ACO-catalyzed all-trans-8′-apocarotenol cleavage in the microsomal reaction mixture. Specifically, 7.5 μl of purified ACO at a concentration of 1 mg/ml were injected into the RPE65 reaction mixture after O2 depletion/supplementation followed by injection of the apocarotenoid substrate. Triton X-100 was omitted from these control experiments as it interferes with RPE65 activity. The reaction and analysis were carried out as indicated above.
Protein Crystallization, Structural Determination, and Analysis
Crystallization of T136A-ACO was conducted as described previously for the wild-type protein (31). Briefly, 1.5 μl of purified enzyme at 10 mg/ml in 20 mm HEPES-NaOH, pH 7.0, containing 0.02% (w/v) Triton X-100 was mixed with a reservoir mixture containing 0.1 m BisTris propane-HCl, pH 6.0, 21–23% (w/v) sodium polyacrylate 2100, and 0.2 m NaCl in a 1:1 ratio. Crystallization was carried out by the hanging-drop vapor-diffusion method at 8 °C. Rod-shaped crystals typically appeared within 2 weeks. Mature crystals were directly harvested and flash-cooled in liquid nitrogen before x-ray exposure. Diffraction data were collected at the NE-CAT 24-ID-E beamline of the Advanced Proton Source. The diffraction data were indexed, integrated, and scaled with the XDS package (33). Mutant T136A-ACO crystals were isomorphous to previously reported orthorhombic wild-type ACO crystals, and their structures were determined by rigid body refinement in REFMAC5 (34) using PDB entry 4OU9 as the starting model. Manual adjustments to the structure were made with COOT (35), and restrained refinement was carried out in REFMAC5. Structures were validated using MOLPROBITY (36) and the wwPDB structure validation server (37). A summary of the x-ray data and refinement statistics is shown in Table 1. All structural figures were prepared with PyMOL (Schrödinger, New York, NY).
TABLE 1.
X-ray crystallographic data collection and refinement statistics for T136A-ACO
| Data collection | |
| Beamline | NECAT 24-ID-E |
| Wavelength (Å) | 0.9793 |
| Space group | P212121 |
| Unit cell parameters (Å) | a = 118.76 |
| b = 125.96 | |
| c = 203.91 | |
| Resolution (Å) | 48.6–2.8 (2.9–2.8)a |
| Unique reflections | 75,385 (11,999) |
| Completeness (%) | 99.8 (99.1) |
| Multiplicity | 3.7 (3.8) |
| 〈I/σI〉 | 7.1 (0.73) |
| RmeasI (%)b | 25.1 (219.6) |
| CC½ (%)b | 98 (18.8) |
| Refinement | |
| Resolution (Å) | 48.6–2.8 |
| No. of observations | 71,479 |
| Rwork/Rfree (%)c | 22.5/26.4 |
| No. of atoms | |
| Protein | 15,090 |
| Water | 44 |
| Metal/ion | 4 Fe2+, 2 Cl− |
| B-factors (Å2) | |
| Protein | 63 |
| Water | 38 |
| Metal/ion | 49(Fe), 56(Cl) |
| Root mean square deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.40 |
| Ramachandran plotd | |
| Favored/outliers (%) | 96/0 |
| PDB accession code | 5E47 |
a Values in parentheses are those for the highest resolution shell of data.
b Data are as calculated in XDS.
c Data are as in REFMAC.
d Data are as assessed with MolProbity.
Results
Assessment of Apocarotenoid Solvent Back-exchange
ACO-catalyzed cleavage of 8′-apocarotenol generates two aldehyde products, all-trans-retinal (RAL) and a second C10-apocarotenal molecule (Fig. 1B). To quantify the solvent exchange rates of both aldehyde groups, RAL and the C10-apocarotenoid were incubated in a 200-μl ACO reaction mixture containing 3 μg of ACO with 18O-water (99% 18O atom) used as the reaction solvent. The incubation was quenched, and the apocarotenoids were extracted with hexane at different time intervals (0–30 min). The exchange rate was determined by quantifying the ratio of target compound and its 18O-labeled counterpart that was generated by the oxygen exchange. Virtually no exchange was observed within the time course of this experiment as found previously (Fig. 2, A and B) (38). However, the exchange rates for both products were significantly increased by supplementing the reaction mixture with 1% (w/v) BSA or by lowering the pH with 5% (v/v) acetic acid, both of which can catalyze the oxygen exchange reaction (Fig. 2, A and B). For both apocarotenoids, about 80% of the carbonyl 16O atoms were substituted by 18O after 30 min of incubation in an acidic environment with 1% (w/v) BSA. Therefore, our data indicated that solvent back-exchange in our experimental conditions would not be a significant complicating factor in the interpretation of the ACO labeling experiments.
FIGURE 2.
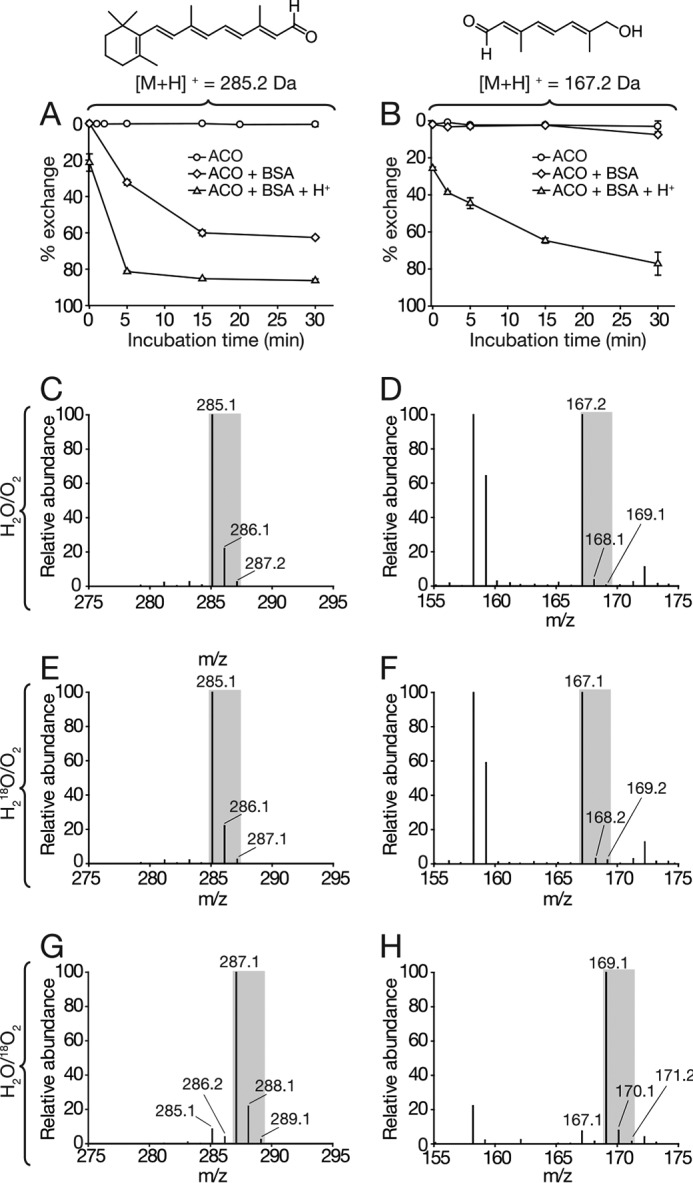
In vitro isotope-labeling analysis of the ACO-catalyzed reaction. Assessment of solvent back-exchange rate for RAL (A) and C10-apocarotenal (B). Authentic RAL or C10-apocarotenal was added to a buffered reaction system containing H218O (99% 18O) and purified ACO, and the mixture was incubated at 28 °C with 500 rpm shaking. Samples were collected at the indicated time points. Solvent back-exchange rates for RAL and C10-apocarotenal were quantified as the ion peak area ratios of the 16O- and 18O-labeled species. Note the increased exchange rates induced by addition of 1% (w/v) BSA and/or 5% (v/v) acetic acid. Error bars represent S.D. from duplicate measurements. Mass spectra for RAL (C) and C10-apocarotenal (D) generated under standard H216O/16O2 conditions. E and F, mass spectra for the apocarotenoid products generated in an H218O/16O2 environment showed isotope distributions similar to those of the control spectra shown in C and D, which demonstrated a lack of 18O incorporation from the labeled water into the products. G and H, by contrast, spectra for both apocarotenoids generated in an H216O/18O2 milieu showed a 2-Da shift in their isotope distribution patterns, attributable to 18O incorporation into both products. The data are quantitated in Table 2.
Apocarotenoid Labeling Studies in the Presence of H218O
To assess the origin of the carbonyl oxygen atoms in the nascent apocarotenoid products of the ACO reaction, activity assays were performed in the presence of H218O/16O2, H216O/18O2, or H216O/16O2, the latter serving as a control. In all experiments, two peaks corresponding to RAL and the C10-apocarotenal product were detected and identified based on their optical absorbance spectra (Fig. 3A). The pseudomolecular masses [M + H]+ of m/z equal to 285 and 167 for products generated in H216O/16O2 were assigned to protonated RAL and C10 products, respectively (Fig. 2, C and D). For reactions carried out in an H218O/16O2 environment, identical masses were observed for both products indicating that oxygen atoms in the RAL and C10-apocarotenal aldehyde groups originated from O2 rather than water (Fig. 2, E and F). As with the background exchange experiments performed in the presence of BSA, high concentrations of the enzyme might facilitate carbonyl oxygen solvent exchange. To test this possibility, reactions were carried out in H218O using 1 mg of ACO (0.5% w/v protein). Here, both products showed a remarkable increase in their m/z + 2 counterparts, suggesting that 16O atoms in the aldehyde products were replaced by 18O from water (Table 2). These data strongly suggest that oxygen atoms in the RAL and C10-apocarotenal aldehyde groups originated from O2 rather than water.
FIGURE 3.

HPLC analysis of ACO-catalyzed reaction products and the influence of O2 depletion and supplementation on ACO enzymatic activity. A, HPLC trace of ACO-catalyzed reaction products generated from cleavage of all-trans-8′-apocarotenol under an 18O2-gas environment. The peak assignments were made based on their elution times and their characteristic absorbance spectra as shown to the right of the HPLC. Absorbance maxima for 8′-apocarotenol, RAL, and C10-carotenal were 425, 360, and 327 nm, respectively. mAU, milliabsorbance units. B, ACO activity assays were performed either without argon pretreatment to displace O2 (control) or after a 10- or 15-min argon purge (white bars) and compared with ACO samples undergoing the same treatments but subjected to 3 min of O2 gas supplementation as a final step (gray bars). Error bars represent S.D.s calculated from measurements performed in triplicate.
TABLE 2.
Summary of isotope labeling results for ACO
| Compound |
16O2-H218O system |
18O2-H216O system |
||
|---|---|---|---|---|
| 16O-Labeled | 18O-Labeled | 16O-Labeled | 18O-Labeled | |
| All-trans-retinal | 97.7% (71.8%)a | 2.2% (28.2%) | 7.8% | 92.2% |
| C10-apocarotenal | 97.9% (89.4%) | 2.1% (10.6%) | 6.8% | 93.2% |
a Numbers in parentheses are the results obtained in a reaction system containing 1 mg of ACO.
Apocarotenoid Labeling Studies in the Presence of 18O2
To further confirm the origin of the aldehyde oxygen atoms, additional labeling studies were carried out in an 18O2-enriched environment. An efficient method for removal of atmospheric O2 in the reaction solutions was first developed. This procedure efficiently displaced dissolved O2 in the reaction mixture within 10–15 min, as shown by a drastic reduction in ACO-catalyzed RAL formation (Fig. 3B). Importantly, this RAL reduction could be restored by introducing O2 back into the solution (Fig. 3B). In fact, O2 supplementation caused an ∼2.5-fold enhancement in ACO activity attributable to the higher concentration of dissolved O2 in the reaction solutions.
Labeling studies in an 18O2-enriched environment were performed by forcing 18O2 back into the reaction mixture following the deoxygenation treatment. In this case, the isotopic distributions of both reaction products were shifted by 2 Da (Fig. 2, G and H), indicating the formation of 18O-labeled products. Only small amounts of 16O-labeled RAL and C10-apocarotenal (<10%) were detected under these conditions, likely generated by residual 16O-oxygen in the reaction mixture and low levels of background solvent exchange (Table 2). Taken together, these labeling studies in 18O-water and 18O2 environments unequivocally demonstrate that ACO is a dioxygenase.
Preparation of Highly Purified and Active NOV2
NOV2 is a stilbene oxygenase member of the CCO family that, among other substrates, cleaves resveratrol to form 4-HBA and 3,5-DHBA (Fig. 1B). Previously, this CCO was characterized as a monooxygenase, indicating the possibility that different groups of CCOs could adopt different oxygenation mechanisms (13). Thus, examination of the catalytic properties of NOV2 activity could reveal the determinants of mono- versus dioxygenase activity in CCOs. The previous oxygen labeling study employed crude cell lysates from NOV2-expressing E. coli due to a substantial loss of activity after NOV2 was subjected to purification procedures. Excess E. coli protein in these labeling experiments could have significantly promoted solvent back-exchange in the aldehyde products, which would obfuscate the mono- versus dioxygenase assignment. To circumvent this limitation, we developed an expression and purification method for NOV2, based upon the protocol used for ACO that enabled the production of a highly pure and active protein sample (Fig. 4, A and B). NOV2 expressed under reduced temperatures via leaky T7 promoter activity accumulated to very high levels in the cytosolic fraction of E. coli. A monodisperse and >95% pure preparation of NOV2 was obtained by a simple purification scheme consisting of ammonium sulfate fractionation and gel filtration chromatography (Fig. 4, A and B). With this homogeneous enzyme sample, we carried out a steady-state kinetic study of NOV2 that revealed a kcat value similar to that of ACO (Fig. 4C) (31, 39). Notably, NOV2 purified in this manner retained its activity for several weeks when stored on ice or at −80 °C.
FIGURE 4.

NOV2 purification and enzymatic characterization. A, gel filtration chromatogram showing a symmetrical peak (indicated by the arrow) for NOV2 obtained from ammonium sulfate fractionation. AU, absorbance unit. Comparison of the elution volume of this NOV2 peak with those of standards indicates NOV2 is a monomeric protein with a molecular mass of ∼54 kDa. AU, absorbance unit. B, SDS-PAGE analysis of pooled gel filtration fractions constituting the main NOV2 peak. Proteins were visualized by Coomassie R-250 staining. C, steady-state kinetics of purified NOV2. Error bars represent standard deviations computed from triplicate measurements. Uncertainty estimates for the kinetic parameters are standard errors computed from the curve-fitting algorithm in SigmaPlot.
Assessment of the Solvent Back-exchange Rate for 4-HBA and 3,5-DHBA
To measure solvent back-exchange in the benzaldehyde products of NOV2-catalyzed resveratrol cleavage, authentic 4-HBA and 3,5-DHBA were incubated in a 150-μl H218O reaction mixture in the presence of 10 μg of purified NOV2 for different periods of time. The aldehyde oxygen of 4-HBA remained unchanged after 15 min of incubation (Fig. 5A). By contrast, the carbonyl oxygen in 3,5-DHBA was rather susceptible to solvent exchange with ∼20% of oxygen replaced by water-derived 18O during a 2-min incubation (Fig. 5B). Addition of 1% (w/v) BSA into the reaction mixture significantly promoted the exchange for 3,5-DHBA but not for 4-HBA. Lowering the pH also drastically accelerated the oxygen substitution for both products with nearly 80% of the original oxygen atoms replaced by those from water (Fig. 5, A and B). These results suggested that the oxygen back-exchange would not significantly cloud the interpretation of product oxygen labeling in the NOV2-catalyzed reaction.
FIGURE 5.
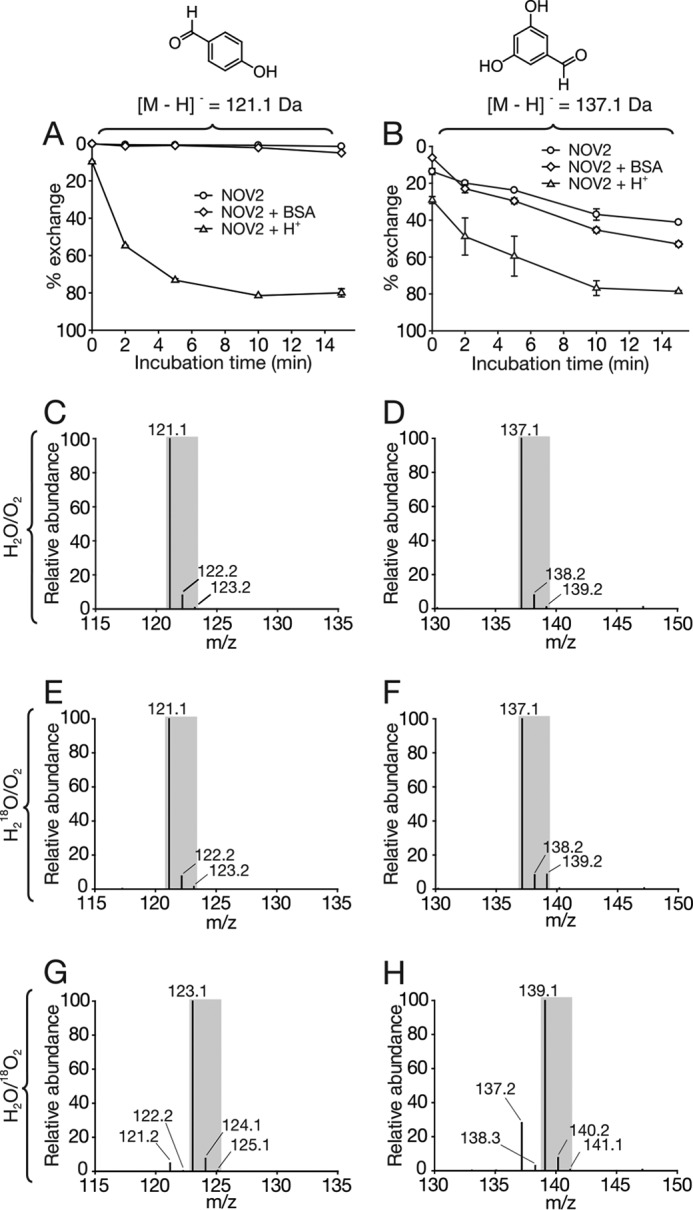
In vitro isotope-labeling analysis of the NOV2-catalyzed reaction. Assessment of solvent back-exchange rates for 4-HBA (A) and 3,5-DHBA (B). Authentic 4-HBA and 3,5-DHBA were added to a buffered reaction system containing H218O (97% 18O) and purified NOV2, and the mixture was incubated at 28 °C with 500 rpm shaking. Samples were collected at the indicated time points. Solvent back-exchange rates for 4-HBA and 3,5-DHBA were quantified as the ion peak area ratios of the 16O- and 18O-labeled species. Note the greater susceptibility of 3,5-DHBA to solvent back-exchange compared with that of 4-HBA, as well as the increased exchange rate for both compounds in the presence of 1% (w/v) BSA or 5% (v/v) acetic acid. Error bars represent S.D.s from experiments performed in duplicate. Mass spectra for 4-HBA (C) and 3,5-DHBA (D) generated under standard H216O/16O2 conditions. E and F, mass spectra for the cleavage products generated in an H218O/16O2 environment showed isotope distributions similar to those of the control spectra in C and D, which demonstrated a lack of 18O incorporation from the labeled water into the products. G and H, by contrast, spectra for the two products generated in an H216O/18O2 environment showed a 2-Da shift in their isotope distribution patterns, attributable to 18O incorporation into both compounds. A quantitative analysis of the data is presented in Table 3.
NOV2 Labeling Studies in the Presence of H218O
The NOV2-catalyzed resveratrol cleavage reaction was performed in H216O/16O2 and H218O/16O2 and H216O/18O2. HPLC analysis of the products obtained in H216O/16O2 showed two peaks corresponding to 4-HBA and 3,5-DHBA. The identity of each product was confirmed by their absorbance spectra (Fig. 6A). Subsequent MS analysis of the products from H216O/16O2 showed that the pseudomolecular masses of m/z of 121 and 137 could be assigned to deprotonated 4-HBA and 3,5-DHBA ([M − H]−), respectively (Fig. 5, C and D). The product isotopic distribution patterns were similar when the reaction was performed in H218O/16O2 (97% 18O) (Fig. 5, E and F, compared with C and D) with a slightly higher content of the m/z = 139 species (∼10%) for 3,5-DHBA that was attributable to solvent back-exchange. These labeling results suggested that the carbonyl oxygen atoms in the final products originated from molecular oxygen rather than water.
FIGURE 6.

HPLC analysis of NOV2-catalyzed reaction products and the influence of O2 depletion and supplementation on NOV2 enzymatic activity. A, HPLC trace of NOV2-catalyzed reaction products generated from cleavage of resveratrol. The peak assignments were made based on their elution times, in comparison with authentic standards, and their characteristic absorbance spectra are shown to the right of the HPLC. Absorbance maxima for resveratrol, 4-HBA, and 3,5-DHBA were 304, 271, and 267 nm, respectively. mAU, milliabsorbance unit. B, NOV2 activity assays were performed either without argon pretreatment to displace O2 (control) or after a 10- or 15-min argon purge (white bars) and compared with NOV2 samples undergoing the same treatments but subjected to 3 min of O2 gas supplementation as a final step (gray bars). Error bars represent S.D.s calculated from measurements performed in triplicate.
NOV2 Labeling Studies in the Presence of 18O2
18O2 labeling experiments for NOV2 were carried out in deoxygenated buffer in the same manner as described above for ACO. Removal of atmospheric O2 resulted in a dramatic reduction in 3,5-DHBA production by NOV2 that could be restored by reintroduction of O2 gas (Fig. 6B). MS analysis of products generated in the 18O2 atmosphere during the 1-min reaction revealed a 2-dalton shift for the 4-HBA molecule compared with the mass observed from reactions carried out in standard O2 (Fig. 5G). Similarly, the isotopic distribution patterns revealed 78.1% of 3,5-DHBA molecules become 18O-labeled (Fig. 5H). Because 3,5-DHBA has a readily exchangeable aldehyde oxygen, it is likely that this unlabeled 3,5-DHBA species (21.9%) was generated by solvent back-exchange. Extension of the assay incubation time to 2 min only slightly reduced the percentage of 3,5-DHBA carrying an 18O label (Table 3). However, when the sample was dried in a SpeedVac under heat (∼15 min) rather than under a stream of argon (∼2 min), the unlabeled 3,5-DHBA species substantially increased to 35% (Table 3), likely due to the elevated temperature and increased time of exposure to water. Therefore, this indicated that seemingly minor differences in sample preparation can have a significant impact on the experimental outcome. Taken together, the high incorporation rate of 18O from H216O/18O2 and 16O from H218O/16O2 into both products demonstrated that NOV2 is a dioxygenase.
TABLE 3.
Summary of isotope labeling results for NOV2
| Compound |
16O2-H218O system |
18O2-H216O system |
||
|---|---|---|---|---|
| 16O-Labeled | 18O-Labeled | 16O-Labeled | 18O-Labeled | |
| 4-HBA | 98.7% | 1.3% | 4.5% | 95.5% |
| 3,5-DHBA | 92.2% | 7.8% | 21.9% | 78.1% |
| (24.1%)a | (75.9%)a | |||
| (35.1%)b | (65.9%)b | |||
a Reaction products were dried by argon stream after a 2-min reaction.
b Reaction products were dried by SpeedVac method after a 2-min reaction.
Activity and Structure of T136A-ACO
A density functional theory (DFT) study of the ACO reaction mechanism found that Thr-136 could promote a dioxygenase-type mechanism by limiting access of water to the iron center (19). The non-heme iron center of wild-type ACO adopts an octahedron-like structure with four coordination sites occupied by His ligands and a fifth, trans to His-183, occupied by solvent. The sixth site is occluded by the methyl group of Thr-136, positioned ∼4.6 Å away from the Fe(II) atom. This arrangement creates a hydrophobic microenvironment around the sixth coordination site that likely impedes its accessibility to water (40). DFT calculations indicated that the coordinated solvent is displaced by O2 during catalysis, which could potentially bind in a side-on or end-on fashion. A structure-based sequence alignment revealed that most CCOs contain a hydrophobic or semi-hydrophobic residue (Thr, Val, or Ile) at this position (Fig. 7, A and B). A reduction in the bulkiness at this site could create enough space for water to coordinate iron and allow it to participate in the reaction. In fact, VP14, one of the few CCOs possessing a non-bulky Ala residue at this position (Fig. 7A), was demonstrated to bind water at this site (41). If the ACO reaction occurs through an epoxide intermediate, an iron-coordinated solvent molecule could act as a nucleophile to open the epoxide, which would give rise to a monooxygenase labeling pattern.
FIGURE 7.
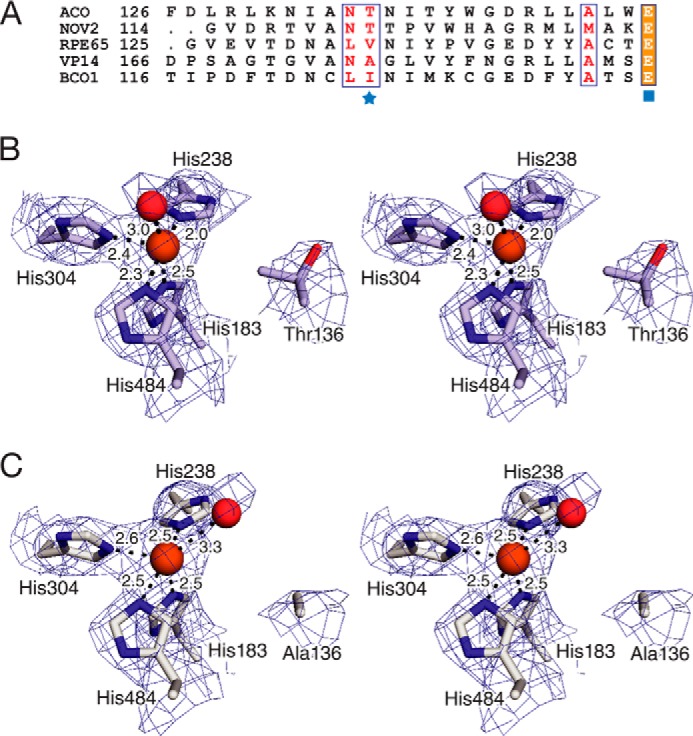
Active site structures of wild-type and T136A ACO. A, structure-based sequence alignment of selected CCOs (45, 46). The position in the sequences homologous to Thr-136 of Synechocystis ACO (Novosphingobium NOV2, bovine RPE65, maize VP14, and human BCO1) is marked with a blue star. A conserved Glu residue that participates in second shell iron coordination is marked by a blue square. B, structure of the wild-type ACO Fe(II)-center showing a 5-coordinate partially filled octahedral geometry. C, structure of the T136A-ACO Fe(II)-center that has a structure more consistent with trigonal bipyramidal geometry because of a change in the binding position of the coordinated solvent. Notably, no extra electron density that would indicate the presence of a coordinated solvent molecule between Ala-136 and iron was observed. Blue mesh represents 2mFo − DFc electron density contoured at one root mean square deviation and computed without inclusion of the iron-bound solvent molecules in the structural models. Bond lengths (in Å) between Fe(II) and nitrogen atoms that form the 4-His coordination shell are shown. Iron and the iron-bound water molecules are depicted as orange and red spheres, respectively. B and C are walleye stereoviews.
To test these hypotheses, we generated a T136A version of ACO, in which the sixth coordination site was expected to be water-accessible, and we examined its catalytic activity and labeling pattern. Cleavage of 8′-apocarotenol by this mutant enzyme was severely impaired with minimal product formation observed using the standard reaction protocol. The poor activity could nevertheless be overcome by increasing the amount of enzyme used in the assay. In reactions containing 100 μg of T136A-ACO, RAL formation after a 10-min incubation was ∼50% that formed by wild-type protein over the same period of time (Fig. 8A). Using these modified conditions, we carried out a labeling study using T136A-ACO in the presence of H218O. We observed that the RAL and C10-apocarotenal products generated by T136A-ACO were labeled primarily with 16O, similar to those generated by the wild-type enzyme (Fig. 8, B and C compared with D and E). In both cases, the reaction products showed increased 18O labeling compared with those produced under standard assay conditions by wild-type protein. This elevation is attributable to an increase in nonspecific protein-catalyzed oxygen exchange caused by the large quantities of enzyme used in these experiments.
FIGURE 8.
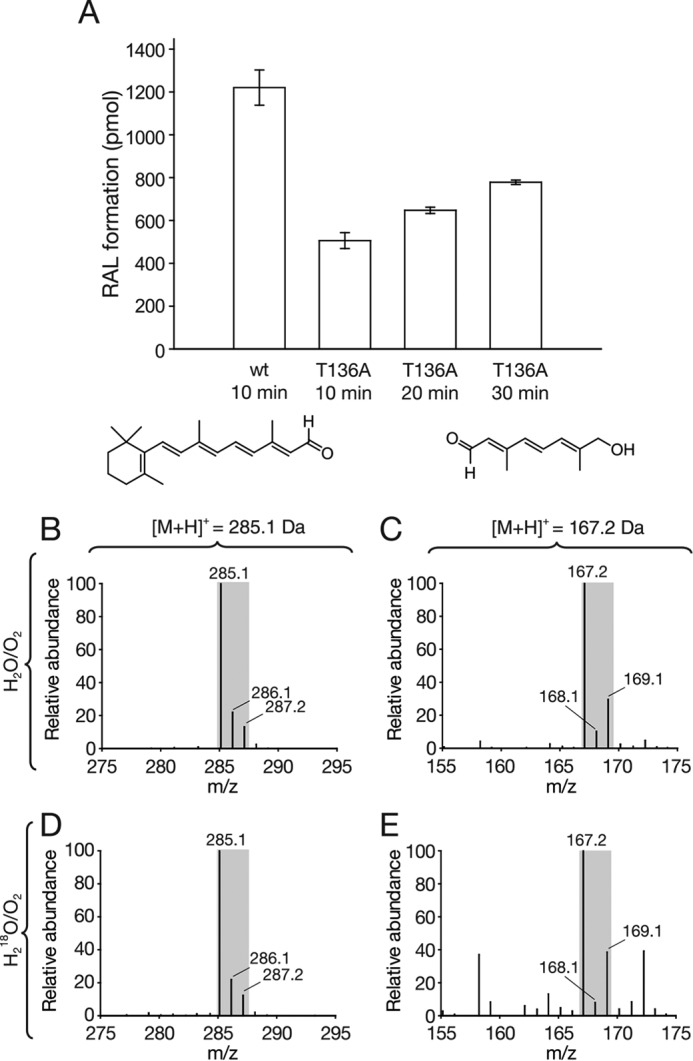
Activity and in vitro isotope-labeling analysis of T136A-ACO. A, activity results of T136A and wild-type ACO. 100 μg of purified protein were used in each reaction. Enzyme activity was assessed by following the formation of RAL over time. Note that the substrate is completely consumed by the wild-type protein within the 1st min of the reaction; therefore, product formation in this graph cannot be used to compare the activity levels of wild-type and T136A ACO. Error bars represent S.D.s calculated from triplicate measurements. B and C, mass spectra of apocarotenoid products generated by wild-type ACO in an H218O/16O2 environment. Note the increased levels of 18O-labeled RAL (m/z = 287.2) and C10-apocarotenal (m/z = 169.1) generated as a result of the high protein concentration used in the assay. D and E, mass spectra of apocarotenoid products generated by T136A-ACO in an H218O/16O2 environment were highly similar to those in B and C, which indicates preservation of dioxygenase activity in the ACO point mutant.
A crystal structure of T136A ACO confirmed that this substitution could theoretically allow for the binding of water to the coordination site trans to His-304. The ∼6-Å gap separating the Ala-136 methyl side chain and the iron center could easily accommodate an iron-bound solvent molecule with minimal steric hindrance (Fig. 7C). However, rather than a 6-coordinate octahedral geometry with water molecules occupying the vacant sites, we instead observed a 5-coordinate trigonal bipyramidal structure with a single water molecule bound to the iron. Additionally, there was a slight but consistent increase (average 0.26 Å) in the Fe-His bond lengths compared with the wild-type protein (Fig. 7, B and C). Together, these data demonstrate that the Thr-136 side chain is not a factor dictating the dioxygenase-labeling pattern of ACO; however, it does appear to be critical for overall catalytic activity and structural integrity of the iron center.
Influence of O2 on RPE65 Retinoid Isomerase Activity
RPE65 is an atypical CCO that catalyzes ester cleavage and isomerization of all-trans-retinyl esters instead of oxidative cleavage of carotenoid substrates (Fig. 1B) (42). Although not necessarily expected based on the chemistry being performed, the question of whether O2 is required for this important reaction has not been experimentally evaluated. Exploration of this possibility is certainly warranted given the close phyletic relationship of RPE65 to the oxygen-utilizing BCO1 and BCO2 enzymes (Fig. 1A), as well as the presumed structural similarity of their iron centers. To address this question, we compared RPE65 isomerase activity in native RPE microsomes before and after sample deoxygenation. To evaluate the extent of O2 removal from the microsome-containing sample we measured, in parallel, the apocarotenoid oxygenase activity of ACO that was exogenously added to the RPE65 reaction mixture, serving as an internal control. Importantly, ACO activity in the RPE65 reaction system was comparable with that found under standard reaction conditions (Fig. 9A). Similar to what was observed under standard conditions (Fig. 3B), ACO activity was markedly impaired following argon treatment to remove O2 with a 3.4-fold reduction in turnover number. The activity was fully rescued and in fact augmented by reintroduction of O2 into the reaction system (Fig. 9B). ACO thus served as a reliable indicator of the oxygen concentration in the RPE65 reaction system. When RPE65 activity was measured following the same deoxygenation treatment, the amount of 11-cis-retinol formed was only marginally decreased (∼14%), and the reduced activity could not be restored by reintroduction of O2 into the reaction mixture (Fig. 9B). Moreover, O2 supplementation of the reaction mixture without prior deoxygenation actually depressed the activity by ∼30% in contrast to the ∼34% activity boost seen for ACO (Fig. 9B). This loss of RPE65 activity in the presence of excess O2 could be caused by oxidation of the iron cofactor, which is required to be its ferrous form to be catalytically competent (43). Importantly, the total number of substrate turnover events was 3.6 times higher for RPE65 compared with ACO, which rules out the possibility that a reduced O2 requirement by RPE65, due to less overall catalytic activity, could explain its insensitivity to O2 concentration (Table 4). Collectively, these results support an O2-independent mechanism for RPE65-catalyzed retinoid isomerization.
FIGURE 9.
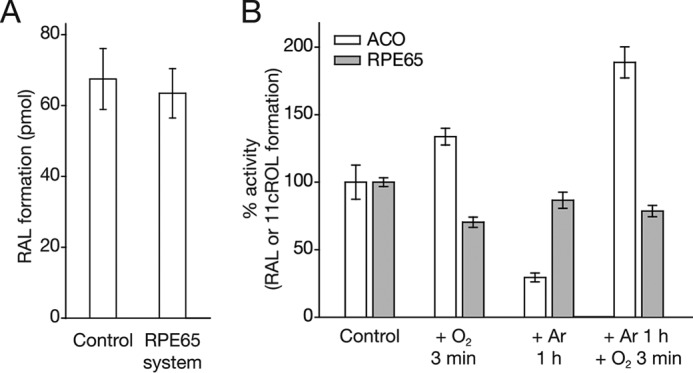
Influence of O2 levels on the retinoid isomerase activity of RPE65. A, ACO enzymatic activity, as monitored by the formation of RAL from 8′-apocarotenol, is maintained when the reaction is carried out in the RPE65 retinoid isomerization assay mixture. B, influence of O2 depletion or supplementation on ACO and RPE65 enzymatic activity. Whereas ACO activity is markedly affected by manipulations in O2 concentration within the reaction buffer, RPE65 activity is indifferent to either O2 supplementation or depletion. Each group was either supplemented for 3 min with O2, purged with argon for 1 h, or supplemented with O2 for 3 min following a 1-h argon purge. Error bars represent S.D.s calculated from triplicate measurements.
TABLE 4.
Enzymatic turnover number of ACO and RPE65 before and after deoxygenation treatment
| Enzyme | kcat (untreated) | kcat (argon- treated) | Total no. of turnovers per molecule (untreated reactions) |
|---|---|---|---|
| ACO | 1.09 ± 0.13 | 0.32 ± 0.03 | 5.5 |
| RPE65 | 0.33 ± 0.01 | 0.28 ± 0.02 | 20 |
Discussion
The question of whether CCOs are monooxygenases or dioxygenases, although seemingly straightforward to determine experimentally, has remained contentious despite several published studies addressing the subject. Major difficulties and problems associated with these studies include their use of crude cell extracts as a source of enzymatic activity, the low activity of CCO enzymes in general, inclusion of only one of the two cleavage reaction products for analyses of isotopic labeling, and most importantly, solvent back-exchange of the aldehyde cleavage products facilitated by both high protein concentrations and long incubation times. Taken at face value, discrepancies between these studies might also indicate that both monooxygenases and dioxygenases could exist within the CCO family. With these issues in mind, we set out to examine the oxygen-labeling pattern of the well characterized prototypical CCO Synechocystis ACO as well as a distantly related stilbene-cleaving bacterial CCO called NOV2 that was previously classified as a monooxygenase.
Capitalizing on our recent success in generating a highly purified native ACO with robust and durable enzymatic activity, we found that this enzyme exhibits a dioxygenase pattern of O2 incorporation into its reaction products. This result is consistent with a previous computational study on the ACO reaction mechanism, which favored a dioxetane-based mechanism that would cause both oxygen atoms of O2 to appear in the reaction products (19). A key prediction from this theoretical study was that the ability of a vacant site in the iron coordination sphere to bind water could govern whether the reaction occurs through a dioxetane or an epoxide intermediate, which could give rise to either dioxygenase- or monooxygenase-labeling patterns in the reaction products, respectively. This site, located trans to His-304, is blocked in ACO by the hydrophobic methyl group of Thr-136. We replaced Ala for Thr at this position to potentially remove the steric and electrostatic barriers to solvent binding at the sixth coordination site. Notably, VP14 has a naturally occurring Ala residue at the corresponding position in its sequence, and the reported crystal structure contains bound solvent at the coordination site in question (41). Despite a major reduction in catalytic activity caused by the T136A substitution, the dioxygenase-labeling pattern for the enzyme was maintained. The crystal structure of T136A-ACO showed that the iron center remained five-coordinate just like the native enzyme even though the coordination geometry adopted a trigonal bipyramidal structure due to a shift in the coordinated solvent molecule. Thus, the dioxygenase activity of ACO is resistant to changes in solvent coordination potential of its iron center. Given the reported ability of the VP14 iron center to accept water in its sixth coordination site, it would be of interest to examine the oxygen-labeling pattern of this enzyme which, despite some claims to the contrary, has not yet been properly investigated.
Our labeling experiments with the stilbene-cleaving CCO, NOV2, previously described as a monooxygenase (13), clearly demonstrated a dioxygenase pattern of oxygen incorporation into its benzaldehyde reaction products. What are the causes for this discrepancy in experimental outcomes? Quantification of oxygen labeling in the prior study indicated a substantial amount of both 4-HBA (69%) and 3,5-DHBA (35%) were labeled with 18O when the reaction was performed in an 18O2/H216O environment. Such a labeling pattern could be obtained through a monooxygenase-generated epoxide intermediate that is opened in a non-regioselective manner to yield a mixture of 16O and 18O in each product. Importantly, because the samples were not deoxygenated prior to initiation of the reaction, a significant amount of product could have been labeled with atmospheric 16O2 thus lowering the incorporation of 18O2 into the products. However, this explanation for the labeling results is in conflict with the labeling pattern observed in the same study from reactions performed in H218O/16O2 in which very little (∼10%) 4-HBA was labeled with 18O. These prior experiments employed crude NOV2-containing cell lysate as an enzyme source, and contaminating proteins could have exacerbated solvent back-exchange. The reactions in this study were also carried out for 15 min, a period of time that, under our reaction conditions, allowed substantial solvent back-exchange to occur for 3,5-DHBA. Thus it appears likely that the prior analyses were confounded by unappreciated solvent back-exchange. This difficulty was circumvented in the present experiments by the use of highly purified and active NOV2, which enabled the use of low protein concentrations and short reaction times. Back-exchange was further minimized by employing water-free normal phase HPLC conditions for product purification and MS analyses together with rapid solvent removal from the product-containing extracts.
Our understanding of carotenoid cleavage by CCOs would greatly benefit from a genuine high resolution CCO-substrate structure, which has yet to be reported. A major difficulty in obtaining such a complex relates to the extreme hydrophobicity of carotenoid substrates, which limits their aqueous solubility and prevents stoichiometric formation of enzyme-substrate complexes for structural studies. However, hydroxylated stilbene substrates of the lignostilbene-cleaving CCOs, such as resveratrol, exhibit much greater water solubility and could be more amenable to structural studies. Our demonstration that NOV2 is a dioxygenase indicates that this enzyme may serve as a reliable model system for studying the general mechanism of alkene cleavage by CCOs. We have described a straightforward expression and purification procedure that results in ample quantities of purified and active NOV2 for structural and spectroscopic studies of the alkene cleavage reaction.
Taken together with prior studies, our demonstration of dioxygenase activity in a primitive cyanobacterial carotenoid-cleaving CCO, as well as the lignostilbene-cleaving CCO, NOV2, strongly suggest that all alkene-cleaving CCOs are dioxygenases that effect the double bond cleavage reaction by a common catalytic mechanism. It should be noted that a dioxygenase or monooxygenase-labeling pattern does not necessarily imply a specific mechanism of O2 incorporation into the substrate. For example, DFT calculations on carotenoid cleavage by ACO have shown that a dioxygenase-labeling pattern could be achieved through either dioxetane or epoxide reaction intermediates (19). Future biophysical studies are needed to delineate the precise mode of catalysis by alkene-cleaving CCOs.
In contrast to other CCOs, we have shown here that RPE65 does not rely on O2 to catalyze ester cleavage/isomerization of all-trans-retinyl esters. This result is not unexpected given that transformation of all-trans-retinyl esters into 11-cis-retinol does not entail any redox chemistry. Moreover, the hydroxyl oxygen atom in the 11-cis-retinol product has been directly shown to originate from water (20). However, it has recently been proposed that O2 may play a catalytic role in retinoid/carotenoid isomerization by bonding with C12 of the retinoid backbone to generate a temporary C11–C12 single bond that would allow free bond rotation to a cis-like state followed by oxygen dissociation to restore the polyene conjugation in an 11-cis configuration (25). Even though O2 does not stoichiometrically participate in this proposed reaction, a major reduction in O2 concentration, as accomplished by our deoxygenation procedure, should still greatly slow the rate of retinoid isomerization, which is not what we observed. The lack of activity reduction after deoxygenation also cannot be explained by potential tight binding of O2 to the iron center as our prior structural and spectroscopic studies of RPE65 did not reveal such a stable Fe-O2 complex (20, 44). Most importantly, a recent structural determination of RPE65 in complex with a retinoid mimetic indicates that the polyene chain of carotenoids binds at a position distant from the iron center, which excludes formation of an Fe-O2-retinoid complex (26). Thus, the commonality in reaction mechanisms between alkene-cleaving CCOs and RPE65 appears unrelated to their O2 requirements but rather relate to their use of iron to bind oxygen: for O2 activation in the former and as an ester-polarizing Lewis acid in the latter.
Author Contributions
X. S., M. G., K. P., and P. D. K. conceived and designed the study. X. S., M. G., J. Z., K. A. K., J. v. L., K. P., and P. D. K. performed experiments and analyzed the data. X. S. and P. D. K. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Leslie T. Webster, Jr., and all members of the Palczewski laboratory (Case Western Reserve University) for valuable comments on this manuscript. We also thank Randall B. Clapp for assistance in protein expression and characterization.
This work was supported by Department of Veterans Affairs Career Development Award IK2BX002683 (to P. D. K.) and National Institutes of Health Grants EY009339 (to K. P.), EY023948 (to M. G.), and EY020551 (to K. P. and J. v. L.). A portion of this work is based upon research conducted at the Advanced Photon National Institutes of Health Grant GM103403 from the NCRR. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.
The atomic coordinates and structure factors (code 5E47) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CCO
- carotenoid cleavage oxygenase
- ACO
- apocarotenoid oxygenase
- 4-HBA
- 4-hydroxybenzaldehyde
- 3,5-DHBA
- 3,5-dihydroxybenzaldehyde
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- BCO
- β-carotene oxygenase
- PDB
- Protein Data Bank
- RAL
- all-trans-retinal
- C10-apocarotenal
- 8′-hydroxy-15′-apocarotenal
- DFT
- density functional theory
- RPE
- retinal pigment epithelium
- BisTris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane.
References
- 1.Kloer D. P., and Schulz G. E. (2006) Structural and biological aspects of carotenoid cleavage. Cell. Mol. Life Sci. 63, 2291–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giuliano G., Al-Babili S., and von Lintig J. (2003) Carotenoid oxygenases: cleave it or leave it. Trends Plant Sci. 8, 145–149 [DOI] [PubMed] [Google Scholar]
- 3.Auldridge M. E., McCarty D. R., and Klee H. J. (2006) Plant carotenoid cleavage oxygenases and their apocarotenoid products. Curr. Opin. Plant Biol. 9, 315–321 [DOI] [PubMed] [Google Scholar]
- 4.Ernst O. P., Lodowski D. T., Elstner M., Hegemann P., Brown L. S., and Kandori H. (2014) Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 114, 126–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spudich J. L., Yang C. S., Jung K. H., and Spudich E. N. (2000) Retinylidene proteins: structures and functions from archaea to humans. Annu. Rev. Cell Dev. Biol. 16, 365–392 [DOI] [PubMed] [Google Scholar]
- 6.Kamoda S., and Saburi Y. (1993) Cloning, expression, and sequence analysis of a lignostilbene-α,β-dioxygenase gene from Pseudomonas paucimobilis TMY1009. Biosci. Biotechnol. Biochem. 57, 926–930 [DOI] [PubMed] [Google Scholar]
- 7.Brefort T., Scherzinger D., Limón M. C., Estrada A. F., Trautmann D., Mengel C., Avalos J., and Al-Babili S. (2011) Cleavage of resveratrol in fungi: characterization of the enzyme Rco1 from Ustilago maydis. Fungal Genet. Biol. 48, 132–143 [DOI] [PubMed] [Google Scholar]
- 8.Poliakov E., Gubin A. N., Stearn O., Li Y., Campos M. M., Gentleman S., Rogozin I. B., and Redmond T. M. (2012) Origin and evolution of retinoid isomerization machinery in vertebrate visual cycle: hint from jawless vertebrates. PLoS ONE 7, e49975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albalat R. (2012) Evolution of the genetic machinery of the visual cycle: a novelty of the vertebrate eye? Mol. Biol. Evol. 29, 1461–1469 [DOI] [PubMed] [Google Scholar]
- 10.Neidig M. L., and Solomon E. I. (2005) Structure-function correlations in oxygen activating non-heme iron enzymes. Chem. Commun. 2005 47, 5843–5863 [DOI] [PubMed] [Google Scholar]
- 11.Leuenberger M. G., Engeloch-Jarret C., and Woggon W. D. (2001) The reaction mechanism of the enzyme-catalyzed central cleavage of β-carotene to retinal. Angew. Chem. Int. Ed. Engl. 40, 2613–2617 [DOI] [PubMed] [Google Scholar]
- 12.dela Seña C., Riedl K. M., Narayanasamy S., Curley R. W. Jr., Schwartz S. J., and Harrison E. H. (2014) The human enzyme that converts dietary provitamin A carotenoids to vitamin A is a dioxygenase. J. Biol. Chem. 289, 13661–13666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marasco E. K., and Schmidt-Dannert C. (2008) Identification of bacterial carotenoid cleavage dioxygenase homologues that cleave the interphenyl α,β double bond of stilbene derivatives via a monooxygenase reaction. Chembiochem 9, 1450–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt H., Kurtzer R., Eisenreich W., and Schwab W. (2006) The carotenase AtCCD1 from Arabidopsis thaliana is a dioxygenase. J. Biol. Chem. 281, 9845–9851 [DOI] [PubMed] [Google Scholar]
- 15.Ryu J. Y., Seo J., Park S., Ahn J. H., Chong Y., Sadowsky M. J., and Hur H. G. (2013) Characterization of an isoeugenol monooxygenase (Iem) from Pseudomonas nitroreducens Jin1 that transforms isoeugenol to vanillin. Biosci. Biotechnol. Biochem. 77, 289–294 [DOI] [PubMed] [Google Scholar]
- 16.Byrn M., and Calvin M. (1966) Oxygen-18 exchange reactions of aldehydes and ketones. J. Am. Chem. Soc. 88, 1916–1922 [Google Scholar]
- 17.During A., and Harrison E. H. (2004) Intestinal absorption and metabolism of carotenoids: insights from cell culture. Arch. Biochem. Biophys. 430, 77–88 [DOI] [PubMed] [Google Scholar]
- 18.Mutti F. G. (2012) Alkene cleavage catalysed by heme and nonheme enzymes: reaction mechanisms and biocatalytic applications. Bioinorg. Chem. Appl. 2012, 626909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borowski T., Blomberg M. R., and Siegbahn P. E. (2008) Reaction mechanism of apocarotenoid oxygenase (ACO): a DFT study. Chemistry 14, 2264–2276 [DOI] [PubMed] [Google Scholar]
- 20.Kiser P. D., Golczak M., Lodowski D. T., Chance M. R., and Palczewski K. (2009) Crystal structure of native RPE65, the retinoid isomerase of the visual cycle. Proc. Natl. Acad. Sci. U.S.A. 106, 17325–17330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiser P. D., and Palczewski K. (2010) Membrane-binding and enzymatic properties of RPE65. Prog. Retin. Eye Res. 29, 428–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Redmond T. M., Poliakov E., Kuo S., Chander P., and Gentleman S. (2010) RPE65, visual cycle retinol isomerase, is not inherently 11-cis-specific: support for a carbocation mechanism of retinol isomerization. J. Biol. Chem. 285, 1919–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McBee J. K., Kuksa V., Alvarez R., de Lera A. R., Prezhdo O., Haeseleer F., Sokal I., and Palczewski K. (2000) Isomerization of all-trans-retinol to cis-retinols in bovine retinal pigment epithelial cells: dependence on the specificity of retinoid-binding proteins. Biochemistry 39, 11370–11380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sui X., Kiser P. D., Lintig Jv., and Palczewski K. (2013) Structural basis of carotenoid cleavage: from bacteria to mammals. Arch. Biochem. Biophys. 539, 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison P. J., and Bugg T. D. (2014) Enzymology of the carotenoid cleavage dioxygenases: reaction mechanisms, inhibition and biochemical roles. Arch. Biochem. Biophys. 544, 105–111 [DOI] [PubMed] [Google Scholar]
- 26.Kiser P. D., Zhang J., Badiee M., Li Q., Shi W., Sui X., Golczak M., Tochtrop G. P., and Palczewski K. (2015) Catalytic mechanism of a retinoid isomerase essential for vertebrate vision. Nat. Chem. Biol. 11, 409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konagurthu A. S., Whisstock J. C., Stuckey P. J., and Lesk A. M. (2006) MUSTANG: a multiple structural alignment algorithm. Proteins 64, 559–574 [DOI] [PubMed] [Google Scholar]
- 28.Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guindon S., Lethiec F., Duroux P., and Gascuel O. (2005) PHYML Online–a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33, W557–W559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Darriba D., Taboada G. L., Doallo R., and Posada D. (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sui X., Kiser P. D., Che T., Carey P. R., Golczak M., Shi W., von Lintig J., and Palczewski K. (2014) Analysis of carotenoid isomerase activity in a prototypical carotenoid cleavage enzyme, apocarotenoid oxygenase (ACO). J. Biol. Chem. 289, 12286–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golczak M., Kiser P. D., Lodowski D. T., Maeda A., and Palczewski K. (2010) Importance of membrane structural integrity for RPE65 retinoid isomerization activity. J. Biol. Chem. 285, 9667–9682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Read R. J., Adams P. D., Arendall W. B. 3rd, Brunger A. T., Emsley P., Joosten R. P., Kleywegt G. J., Krissinel E. B., Lütteke T., Otwinowski Z., Perrakis A., Richardson J. S., Sheffler W. H., Smith J. L., Tickle I. J., Vriend G., and Zwart P. H. (2011) A new generation of crystallographic validation tools for the Protein Data Bank. Structure 19, 1395–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jastrzebska B., Palczewski K., and Golczak M. (2011) Role of bulk water in hydrolysis of the rhodopsin chromophore. J. Biol. Chem. 286, 18930–18937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.dela Seña C., Narayanasamy S., Riedl K. M., Curley R. W. Jr., Schwartz S. J., and Harrison E. H. (2013) Substrate specificity of purified recombinant human β-carotene 15,15′-oxygenase (BCO1). J. Biol. Chem. 288, 37094–37103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kloer D. P., Ruch S., Al-Babili S., Beyer P., and Schulz G. E. (2005) The structure of a retinal-forming carotenoid oxygenase. Science 308, 267–269 [DOI] [PubMed] [Google Scholar]
- 41.Messing S. A., Gabelli S. B., Echeverria I., Vogel J. T., Guan J. C., Tan B. C., Klee H. J., McCarty D. R., and Amzel L. M. (2010) Structural insights into maize viviparous14, a key enzyme in the biosynthesis of the phytohormone abscisic acid. Plant Cell 22, 2970–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Redmond T. M., Poliakov E., Yu S., Tsai J. Y., Lu Z., and Gentleman S. (2005) Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc. Natl. Acad. Sci. U.S.A. 102, 13658–13663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moiseyev G., Takahashi Y., Chen Y., Gentleman S., Redmond T. M., Crouch R. K., and Ma J. X. (2006) RPE65 is an iron(II)-dependent isomerohydrolase in the retinoid visual cycle. J. Biol. Chem. 281, 2835–2840 [DOI] [PubMed] [Google Scholar]
- 44.Kiser P. D., Farquhar E. R., Shi W., Sui X., Chance M. R., and Palczewski K. (2012) Structure of RPE65 isomerase in a lipidic matrix reveals roles for phospholipids and iron in catalysis. Proc. Natl. Acad. Sci. U.S.A. 109, E2747–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pei J., Tang M., and Grishin N. V. (2008) PROMALS3D web server for accurate multiple protein sequence and structure alignments. Nucleic Acids Res. 36, W30–W34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gouet P., Courcelle E., Stuart D. I., and Metoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]