

Abstract
NF-κB is best known for its pro-inflammatory and anti-apoptotic actions, but in skeletal muscle, NF-κB activation is important for atrophy upon denervation or cancer. Here, we show that also upon fasting, NF-κB becomes activated in muscle and is critical for the subsequent atrophy. Following food deprivation, the expression and acetylation of the p65 of NF-κB on lysine 310 increase markedly in muscles. NF-κB inhibition in mouse muscles by overexpression of the IκBα superrepressor (IκBα-SR) or of p65 mutated at Lys-310 prevented atrophy. Knockdown of GCN5 with shRNA or a dominant-negative GCN5 or overexpression of SIRT1 decreased p65K310 acetylation and muscle wasting upon starvation. In addition to reducing atrogene expression, surprisingly inhibiting NF-κB with IκBα-SR or by GCN5 knockdown in these muscles also enhanced AKT and mechanistic target of rapamycin (mTOR) activities, which also contributed to the reduction in atrophy. These new roles of NF-κB and GCN5 in regulating muscle proteolysis and AKT/mTOR signaling suggest novel approaches to combat muscle wasting.
Keywords: acetylation, acetyltransferase, mTOR complex (mTORC), muscle atrophy, NF-κB (NF-KB), protein degradation, GCN5, fasting
Introduction
Muscle wasting and cachexia occur in various physiological and pathophysiological conditions, including fasting, denervation, inactivity, acidosis, sepsis, certain cancers, and in aged populations (1). A common feature of these conditions is the increased rates of protein degradation in skeletal muscle, which results from activation of both the ubiquitin proteasome pathway and autophagy (1, 2). This increased proteolysis is triggered by the expression of a set of about 100 atrophy-specific genes or atrogenes that include key components of both of these degradative pathways. Their expression and resulting increase in proteolysis have been shown to be controlled by FoxO1 and FoxO3 transcription factors (3–5). Another transcription factor that appears critical for certain types of atrophy is NF-κB (6). Transgenic mice expressing the constitutively activated form of IκB kinase β (IKKβ) in muscle exhibit reduced muscle mass (7). Furthermore, the atrophy induced by denervation, cancer cachexia, and disuse (7–9) can be inhibited by knockdown of the p50 subunit of NF-κB (8) and also by overexpression of the IκBα superrepressor (IκBα-SR). This IκBα mutant inhibits NF-κB but lacks the phosphorylation sites necessary for its rapid ubiquitination and degradation (7). However, it has also been reported that overexpression of a dominant-negative mutant of the NF-κB subunit, p65, does not reduce the loss of muscle mass induced by cancer (10). Therefore, it remains unclear how NF-κB promotes atrophy, how disuse signals these changes in IKKβ or NF-κB activity, and whether NF-κB is required for other major physiological stimuli for muscle wasting, such as food deprivation.
It is well established that the NF-κB pathway is regulated exceptionally tightly by several post-translational modifications, including phosphorylation, ubiquitination, acetylation, and methylation (11–13). However, it remains to be determined whether these modifications influence muscle function, size, or atrophy. One goal of the present study is to clarify the poorly understood actions, mechanisms, and regulation of NF-κB in skeletal muscle normally and during atrophy. Among the reversible post-translational modifications that appear important for full activation of NF-κB is acetylation of its p65 subunit by p300 together with phosphorylation in non-muscle cells (11, 14). Conversely, deacetylation of NF-κB by SIRT1 inhibits its activity (15). We recently reported that overexpression of the protein deacetylase SIRT1 can block muscle wasting induced by fasting or denervation by deacetylating and decreasing the contents of FoxO1 and -3 (16). These findings led us naturally to hypothesize that a lysine acetyltransferase, perhaps p300, which has been reported to influence muscle protein balance (17), might function in an opposite fashion to SIRT1 and play an important role in controlling skeletal muscle size by acetylating p65. However, because p300 is also known to inhibit FoxO3 activity in atrophying muscle by disuse (18), the precise role of p300 in proteolysis in muscle cell remains unclear. Another lysine acetyltransferase involved in the regulation of metabolism in myocytes is GCN5, (lysine acetyltransferase 2a) (19). This enzyme has been best studied as a component of the SAGA (Spt-Ada-Gcn5 acetyltransferase) complex that acetylates histones and thus regulates transcription (20). However, in mammals, GCN5 has additional substrates including PGC-1α, Ifh1, drosha, and E2A-PBX1, whose activities are altered through acetylation by GCN5 (21–24). PGC-1α is of particular interest in this context because acetylated PGC-1α is unable to promote the expression of genes involved in activating fatty acid oxidation upon exposure of muscle cells to low glucose levels (19). Also, in budding yeast, after acetylation, Ifh1 is unable to activate transcription of ribosomal protein genes (24), and in fission yeast, GCN5 suppresses the expression of amino acid permeases and the cellular uptake of leucine (25). These studies together suggest that GCN5 may inhibit adaptive responses to low nutrient supply and reduce overall rates of protein synthesis and thus could play a role in the net loss of muscle protein triggered by nutrient deprivation.
The activation of net protein breakdown in skeletal muscle is a critical adaptation in starving animals that provides amino acids precursors for hepatic gluconeogenesis. We show here that in mouse muscles during a 2-day fast that the activity of NF-κB, the expression of both of its subunits (p50 and p65), and p65 acetylation are markedly induced, and that the NF-κB pathway is required for the muscle wasting characteristic of fasted animals. This critical acetylation of NF-κB is mediated by GCN5, and inhibition of GCN5, by lowering p65 acetylation, diminishes atrophy upon food deprivation. These findings thus provide evidence that inhibition of NF-κB by stimulating its deacetylation or by blocking GCN5 activity could be effective interventions to prevent or reverse muscle wasting in catabolic states.
Experimental Procedures
Antibodies
The following antibodies were used: anti-FLAG (F1804; Sigma), anti-acetyl-lysine (ICP0380; Immunechem), anti-GCN5 (SC-20698; Santa Cruz Biotechnology), anti-p300 (ab37143; Abcam), anti-p65 (SC-372; Santa Cruz Biotechnology), anti-p50 (number 06-886; Millipore), anti-PGC-1α (SC-13067; Santa Cruz Biotechnology), and anti-T7 (AB3790; Chemicon). The following antibodies were purchased from Cell Signaling Technology: anti-p65(K310) (number 3045), anti-phospho-p65 (Ser-536) (number 3033), anti-phospho-p65 (Ser-276) (number 3037), anti-IκBα (number 9242), anti-FoxO3 (number 9467), anti-phospho-FoxO1/3 (number 9464), anti-AKT (number 4691), anti-phospho-AKT(Thr-308) (number 9275), phospho-AKT(Ser-473) (number 4046), anti-S6K (number 9202), anti-phospho-S6K(Thr-389) (number 9205), anti-mTOR2 (number 2972), anti-phospho-mTOR(Ser-2448) (number 2971), and anti-Tubulin (number 2146).
Animal Studies and Electroporation of Adult Muscle
In all experiments, adult male CD1 mice (28–30 g) were used. Tibialis anterior (TA) muscles were transfected by electroporation as described previously (26). After electroporation at the time of analysis, no microscopic evidence for inflammation in the muscle was observed. For fasting studies, 4 days after electroporation, the mice were given free access to food or deprived of food at 10 a.m. Animals were killed, and tissues removed and analyzed 2 days later. Muscles were denervated by sectioning the sciatic nerve at the same time as the electroporation, and muscles were collected 12 days later. To monitor NF-κB activity in skeletal muscles, 10 μg of a firefly luciferase construct containing multiple binding sites of NF-κB (kind gift from Dr. Tom Maniatis) and 5 μg of Renilla luciferase plasmid (Promega) were co-electroporated together with either FLAG-SIRT1 (5 μg) or FLAG-SIRT1H355A (5 μg) (kindly provided by Dr. Pere Puigserver). Constructs for IκBα-SR (number 15264), FLAG-GCN5 (number 14106), and FLAG-GCN5 dominant-negative (number 14425) were purchased from Addgene. All mouse experiments were performed with the approval of the Institutional Animal Care and Use Committee and are in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Measurement of Cross-sectional Area
Electroporated muscles were cross-sectioned (10-μm thick) with a cryostat (Leica CM3050) and fixed with 4% paraformaldehyde as described previously (26, 27). Fibers stained with a T7 antibody (Chemicon) or expressing GFP and an equal number of non-transfected fibers from 5 to 7 muscles for each frequency histogram were used for measurement of cross-sectional areas using Metamorph (Molecular Devices). Due to day-to-day variations in animal size, cross-sectional areas of fibers in tibialis anterior were compared between experimental and control mice handled and sacrificed in parallel. Differences in fiber size are expressed as the means of the cross-sectional area of each group and are statistically significant (p < 0.01). No evidence for inflammation in muscles as a result of the in vivo transfection procedure was noted.
Immunoprecipitation
The muscle lysates precleared by incubating mouse IgG and Protein A-agarose for 1 h at 4 °C were incubated with the indicated antibodies for overnight. After extensive washing with buffer containing 500 mm KCl, co-immunoprecipitated proteins were subject to SDS-PAGE and examined by Western blot.
RNA Extraction and Quantitative Real-time PCR
Total RNA extraction with TRIzol (Invitrogen) and reverse transcription reaction were performed as described previously. Quantitative real-time PCR was performed with mouse gene-specific primers (see below) and DyNAmo HS SYBR Green qPCR kit (Finnzymes) using C100 Thermal Cycler (Bio-Rad). The following primers were used: Atrogin-1 (5′-CAGTGTCATGGTTCCTTTGC-3′ and 5′-TCAGCCTCTGCATGATGTTC-3′), MuRF1 (5′-AGGACAACCTCGTGCCTACAAG-3′ and 5′-ACAACCTGTGCCGCAAGTG-3′), p65 (5′-TCTGCTTCCAGGTGACAGTG-3′ and 5′-ATCTTGTCGGCAGTGTT-3′), p105 (5′-ACCTGGCCGTGGAGTACGAC-3′ and 5′-GACCCTCTTCCGGCCGCTAT-3′), Gabarapl1 (5′-CATCGTGGAGAAGGCTCCTA-3′ and 5′-ATACAGCTGGCCCATGGTAG-3′), Bnip3 (5′-TTCCACTAGCACCTTCTGATGA-3′ and 5′-GAACACCGCATTTACAGAACAA-3′), and Tubulin (5′-GATCATTGACCTTGTCCTGGACA-3′ and 5′-GAGCCGCTCCATCAGCAG-3′).
Statistical Analysis
The analysis of all biochemical data were performed using Student's t test, and significant differences (p < 0.05) are demonstrated with an asterisk. Data are presented as mean ± S.E.
Results
Fasting Enhances NF-κB Activity, Expression, and Acetylation in Muscles
Although NF-κB has been shown to be important for the induction of muscle wasting upon disuse (7, 8) and cancer cachexia (7, 9), nutrient deprivation involves quite different extracellular and intracellular signaling mechanisms. To determine whether NF-κB may also trigger muscle wasting during starvation, we tested if NF-κB is activated in skeletal muscle upon food deprivation. TA muscles were electroporated with reporter plasmids for the luciferase gene under control of a promoter containing multiple NF-κB binding sites (Fig. 1A). To our surprise, NF-κB activity increased severalfold 2 days after food deprivation (Fig. 1A). Thus, NF-κB activation coincided with the marked induction of fiber atrophy and loss of muscle mass.
FIGURE 1.
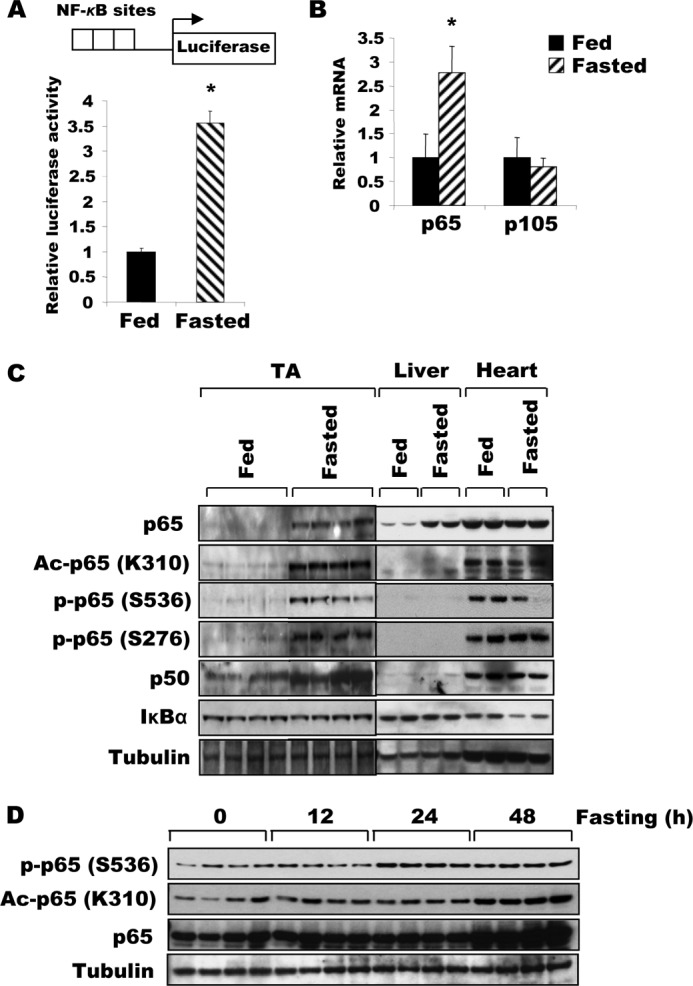
Fasting increases the expression, acetylation, and activity of NF-κB in skeletal muscle. A, NF-κB transcriptional activity in TA muscle increases after food deprivation. Four days after electroporation of plasmids encoding a reporter gene under the control of a promoter containing a NF-κB-responsive element, animals were either deprived of food or maintained on the standard diet for 2 days. Muscle lysates were prepared for the reporter assay. n = 3; *, p < 0.05. B, expression of p65 mRNA increased in muscles during fasting. TA muscles from fed mice or mice deprived of food for 2 days were used for RNA extraction. RT-PCR was performed to analyze the expression of p65 or p105 (p50). n = 3; *, p < 0.05. C, upon fasting, the expression of both NF-κB subunits and p65 acetylation all increase in TA muscles, but not in the liver or heart. The expression of p65, p50, and IκBα, and the acetylation of the p65 subunit of NF-κB in different tissues in fed or fasted mice were determined by Western blot. D, the levels of p65 amount, its phosphorylation and Lys-310 acetylation gradually increase after 24 h of food deprivation in TA muscles.
To define the mechanism for this enhancement of NF-κB activity upon fasting, we initially measured the content and expression of the NF-κB subunits, p65 and p50. Both levels of the p65 protein and its corresponding mRNA were increased (Fig. 1, B and C) in TA muscles of mice fasted for 2 days. The level of the p50 protein, which is generated by proteolytic processing of its precursor, p105 (28), is also increased markedly in these muscles, even though the amount of mRNA encoding p105 was not significantly elevated (Fig. 1, B and C). Thus, p65 rises in fasting through increased transcription, whereas p50 increases by accelerated proteasomal processing of the precursor or reduced degradation of mature p50. It is noteworthy that the content of both NF-κB subunits increased in fasting in skeletal muscles, but not in liver or heart (Fig. 1C).
In the muscles atrophying due to fasting, there was not only an increase in the phosphorylation of p65 at serines 276 and 536, but there was also a marked increase in the acetylation of p65 at Lys-310 (Fig. 1C). The induction of acetylation and phosphorylation of p65 indicated that activation of the NF-κB pathway gradually begins 24 h after fasting (Fig. 1D). No such change was observed in the liver despite the marked induction of hepatic p65 protein during fasting (Fig. 1C). Therefore, the enhancement of p65 acetylation in muscle is not simply due to the increase in p65 levels. Together, these data demonstrate that fasting increases the expression, phosphorylation, and, importantly, the acetylation of NF-κB, which presumably contributes to its increased transcriptional activity as shown below.
Blocking p65K310 Acetylation in Muscle Inhibited Atrophy
We recently reported that SIRT1 overexpression inhibits muscle wasting upon fasting and denervation by causing the deacetylation and inactivation of FoxO1, FoxO3, and PGC-1α (16). To determine whether SIRT1 also inactivates NF-κB by deacetylation of p65 in muscle, as it does in other tissues (15, 29), TA muscles were electroporated with plasmids encoding the NF-κB reporter gene either alone or together with constructs encoding FLAG-SIRT1 or FLAG-SIRT1H355A, which lacks deacetylase activity (Fig. 2A). Overexpression of SIRT1 completely abolished the stimulation of NF-κB activity by food deprivation in muscle, and concomitantly decreased p65 acetylation without reduction in the p65 content (Fig. 2A, lanes 1–4). However, no such inhibition was observed after electroporation of the inactive mutant SIRT1H355A, even though this mutant protein interacted with p65 and could be coimmunoprecipitated with p65 (Fig. 2B). Thus, the inhibition of NF-κB activity by SIRT1 was not due to its physical interaction with p65, but to its deacetylation of p65. It is noteworthy that acetylation of p65 on Lys-310 was only observed in muscle on fasting for 2 days (Fig. 2A, lane 2), even though the total p65 levels in muscles of the fed and fasted mice were increased similarly by electroporation (see below).
FIGURE 2.


Inhibition of NF-κB activity in adult muscle-reduced muscle wasting upon fasting. A, the increase in NF-κB activity in muscles upon fasting was blocked by SIRT1 overexpression, but was enhanced by overexpression of the inactive mutant SIRT1H355A. NF-κB reporter constructs were co-electroporated into TA muscles with F-SIRT1, F-SIRT1H355A, or a control vector. After food deprivation for 2 days, luciferase activity was measured. n = 3; *, p < 0.05 versus Fed; **, p < 0.05 versus Control vector in Fasted. Lower panel, fasting induced acetylation of the p65 subunit of NF-κB, especially after electroporation of the H355A mutant, but WT SIRT1 decreased acetylation of p65 (but not total p65). B, SIRT1 associates with p65 in fasted TA muscle. Constructs for F-SIRT1 or F-SIRT1H355A genes were electroporated, and 2 days later, muscle lysates were prepared for an immunoprecipitation assay with an antibody against FLAG. After extensive washing with a buffer containing 500 mm KCl, proteins bound to FLAG antibodies were eluted with FLAG peptides and subjected to SDS-PAGE for Western blot (WB). α-Actin was used as a nonspecific control protein. Input corresponds to 10% of the lysates used for immunoprecipitation. One-fourth of the eluate was used for FLAG-SIRT1 and one-half for p65. C, increase in p65 mRNA level upon electroporation. TA muscles of fed mice that are not electroporated (non elec) or electroporated with a control vector (ctrl. v) or FLAG-SIRT1 (F-SIRT1) were used to measure the levels of p65 mRNA (upper) and protein (lower, 10 μg of lysate per lane) (A). n = 4; *, p < 0.05 versus non electroporated muscle. D, the constructs encoding the T7-p65K310A mutant were electroporated into TA muscles. Four days later, mice were fed or deprived of food for 2 days. Fibers expressing p65K310A were stained with a T7 antibody (marked by asterisks), and cross-sectional areas were measured. Scale bar, 20 μm. E, IκBα-SR overexpression protected muscle fibers from atrophy upon fasting. Bicistronic DNA constructs encoding IκBα-SR and GFP were electroporated into TA muscles, and mice were deprived of food for 2 days and cross-sectional areas were measured. Typical image showing the electroporated fibers (GFP-labeled and marked by asterisks) by fluorescence microscopy (right image) is shown. Scale bar, 15 μm. F, GFP overexpression (marked by asterisks) does not influence the size of muscle fibers in either fasted animals. Electroporation of TA muscle with DNA constructs encoding GFP, food deprivation procedures, and measurement of fiber sizes were performed as described in the legend to Fig. 2B. Scale bar, 20 μm. G, IκBα-SR electroporation does not induce any increase in muscle size in fed mice. Six days after electroporation of IκBα-SR, cross-sectional areas were measured. Scale bar, 20 μm. H, GFP overexpression (marked by asterisks) does not alter fiber sizes in fed mice. Scale bar, 20 μm. I, overexpression of IκBα-SR inhibits the expression in fasting of atrogin1 and MuRF1 but not autophagy genes. TA muscles were electroporated with constructs for either a control vector or IκBα-SR. Four days later, mice were fed or deprived of food for 2 days. n = 3; *, p < 0.05 versus Fasted + Control vector. J, after IκBα-SR overexpression, the levels of phosphorylated AKT and FoxO3 in muscles of fasted mice. TA muscles were electroporated with IκBα-SR or a control vector, and the animals were fed or deprived of food as in D. Muscle extracts were analyzed for different components of AKT-FoxO pathway by Western blot.
Surprisingly, in these electroporated muscles, we also observed an increase in the levels of p65 mRNA and protein (Fig. 2, A and C) above the levels in untreated animals. This induction of p65 must be a nonspecific response to surgery or electroporation, because it was seen even upon electroporation of a control plasmid in fed or fasted mice (Fig. 2A, lanes 1 and 2). However, the control plasmid did not cause p65 acetylation, which only increased in the starving mice (Fig. 2A, lanes 1 and 2). Thus, the inhibition of NF-κB activity by SIRT1 was not due to a reduction in the p65 content (Fig. 2A), but to its deacetylation.
To further establish the importance of p65 acetylation in triggering the loss of muscle mass upon fasting, TA muscles were electroporated with plasmids encoding p65 mutated at lysine 310 to alanine (p65K310A). Those fibers overexpressing p65K310A and thus unable to be acetylated, were markedly protected from wasting upon fasting (Fig. 2D). Like wild type p65, p65K310A was localized in the nucleus of the muscle. Therefore, the inhibition of atrophy resulted from blocking the acetylation of p65 and not from its sequestration in the cytoplasm.
Inhibition of NF-κB by IκBα-SR Reduces Atrophy and Maintains AKT Activity
To determine whether the activation of NF-κB is required for the loss of muscle mass upon starvation, we electroporated into the TA of fed mice bicistronic constructs encoding GFP to mark the electroporated fibers and a mutant form of IκBα, the IκBα super-repressor (IκBα-SR), which cannot be phosphorylated by IKKs, and thus, is a potent inhibitor of NF-κB activation. After food deprivation for 2 days, those fibers overexpressing IκBα-SR were much larger than non-electroporated fibers in the same muscles (Fig. 2E) and control fibers overexpressing GFP alone (Fig. 2F). In fact, the cross-sectional areas of the TA fibers expressing IκBα-SR in fasting resembled those of TA fibers in the fed controls (Fig. 2, E and F). By contrast, in fed mice, overexpression of IκBα-SR did not significantly alter the cross-sectional areas of the fibers (Fig. 2, G and H). Thus, NF-κB function is essential for muscle wasting upon food deprivation, but does not appear to influence muscle mass normally.
To determine whether IκBα-SR prevents the atrophy process by inhibiting the expression of atrogenes, we electroporated IκBα-SR and then measured atrogin1 and MuRF1 mRNA after food deprivation. Indeed, overexpression of IκBα-SR consistently reduced the induction of atrogin1 and MuRF1 (by about 50%) (Fig. 2I). Interestingly, induction of some atrogenes, two autophagy genes (Gabarapl1 and Bnip3) were not affected by IκBα-SR overexpression (Fig. 2I). These findings, together with our prior observations on SIRT1 (16) indicate that NF-κB influences the expression of these ubiquitin ligases; however, the induction of the autophagy genes appears to be more critically dependent on transcription factors other than NF-κB and FoxO3 as both are inhibited by IκBα-SR (Fig. 2J).
Because IκBα-SR reduces the induction of genes for increased proteolysis, some other mechanism must be more directly contributing to its capacity to block fiber atrophy. Decreased FoxO3 and AKT phosphorylation are characteristic responses of muscle and many other cells to fasting and are critical for the loss of muscle mass (27). Surprisingly, the expression of IκBα-SR in muscles during fasting blocked most of the expected fall in the phosphorylation of FoxO3 as well as that of its upstream kinase AKT (Fig. 2J). In fact, the levels of phospho-FoxO3 and phospho-AKT in muscles expressing IκBα-SR approached those in fed controls, which can help account in part for the dramatic suppression of atrophy by IκBα-SR (Fig. 2E) and the smaller induction of two atrophy-related ubiquitin ligases (Fig. 2I) in the muscles of starving mice (Fig. 2J). Perhaps more importantly, the maintenance of phospho-AKT levels, and its downstream kinase, mTOR (based on phospho-S6K), should prevent the decrease in muscle protein synthesis normally seen in fasting.
GCN5 Catalyzes p65 Acetylation and Is Essential for Muscle Wasting in Fasting
Because p65 activation by acetylation appeared to be essential for the induction of muscle atrophy during starvation, we attempted to identify the responsible acetyltransferase. GCN5 was a likely candidate for this role, because GCN5 (but not its paralog, P300/CBP-associated factor) is known to acetylate and inhibit PGC-1α in myocytes in low-glucose media, and one key role of PGC-1α is to inhibit muscle wasting upon fasting and denervation (30). To investigate whether GCN5 acetylates p65 upon fasting and is important in signaling muscle wasting, we decreased the level of GCN5 in the TA muscle of adult mice by electroporation of a plasmid encoding GFP and shRNA against GCN5. Upon fasting, atrophy was greatly attenuated in the fibers with reduced levels of GCN5 (Fig. 3A). Moreover, the reduction in GCN5 by shRNA also markedly decreased the content of acetylated p65 in the muscle (Fig. 3B), as well as acetylated PGC-1α (Fig. 3C). In U2OS and HEK293 cells, GCN5 has also been reported to stimulate p65 degradation by promoting the association of p65 with a Cul2-containing ubiquitin ligase (31). However, we were not able to demonstrate GCN5-facilitated degradation of p65 in skeletal muscles, and neither knockdown nor inhibition of GCN5 increased p65 levels in muscles of fed or starved mice (Figs. 3B and 4E).
FIGURE 3.

GCN5 is required for the acetylation of p65K310 in skeletal muscle. A, bicistronic DNA constructs encoding shRNA against GCN5 and GFP were electroporated into the TA muscles, and mice were deprived of food for 2 days and fiber cross-sectional areas were measured. Representative images showing the electroporated (GFP-labeled) and non-electroporated fibers (marked by asterisks) (right image). Scale bar, 15 μm. B, GCN5 knockdown reduced p65K310 acetylation without a change in p300 content. Muscle lysates were used to determine the levels of the indicated proteins by Western blot. C, after immunoprecipitation of PGC-1α using the same muscle lysates of Fig. 3B, levels of acetylation were examined by Western blots. IP, immunoprecipitation. D, GCN5 overexpression in skeletal muscles stimulated by p65K310 acetylation. Wild type GCN5 was overexpressed in TA muscles, and mice were fed or fasted for 2 days. The same amounts (20 μg) of muscle extracts from each animal were analyzed for p65K310 acetylation and total p65 by Western blot (WB). Intensity of dots was quantified using ImageJ (National Institutes of Health). n = 4; *, p < 0.05. E, HEK293 cells were transiently transfected with the constructs in combinations as indicated above the each panel. Acetylation of p65 was determined by antibody to acetylated p65K310.
FIGURE 4.

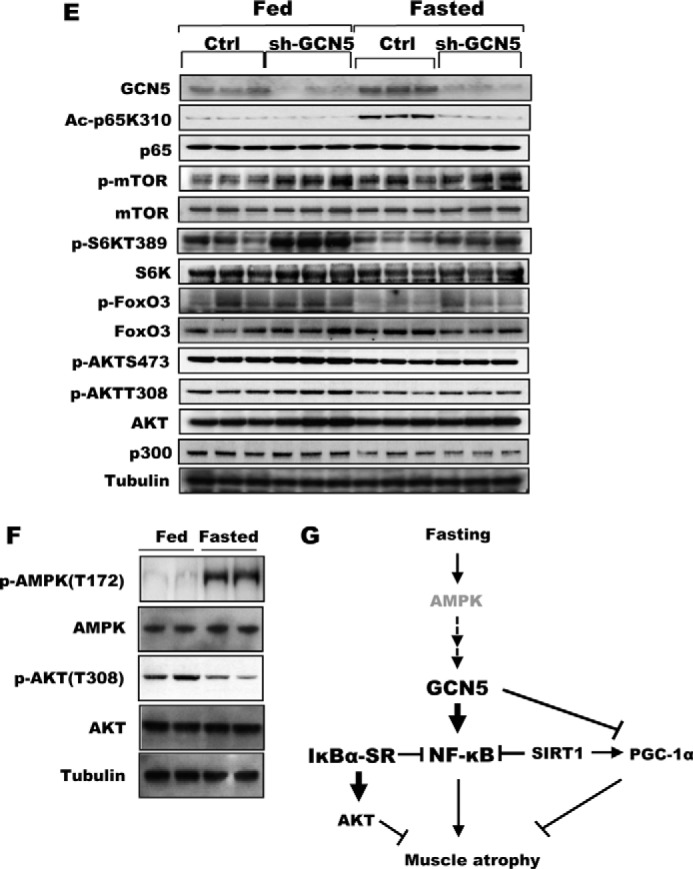
Inhibition of GCN5 by a dominant-negative mutant attenuated wasting and sustained mTOR/S6K activity. A, bicistronic plasmids encoding a GCN5 mutant that lacks acetyltransferase activity and GFP were electroporated into TA muscles. Four days later, mice were deprived of food for 2 days, and cross-sectional areas were measured. A representative image showing the electroporated (GFP) and non-electroporated fibers (marked by asterisks) (right image). Scale bar, 20 μm. B, GCN5 inhibition by overexpression of its dominant-negative mutant (GCN5DN) does not alter size of muscle fibers in fed mice. Frequency histograms show the distribution of the cross-sectional areas of muscle fibers. Scale bar, 20 μm. C, inhibition of GCN5 activity repressed induction of atrogenes upon fasting. TA muscles were electroporated with constructs for either a control vector or FLAG-GCN5DN. Four days later, mice were fed or deprived of food for 2 days. n = 3; *, p < 0.05 versus Fasted + Control vector. D, transient transfection of GCN5 into C2C12 cells elevated the activity of the atrogin1 promoter. C2C12 myoblasts were transiently transfected with plasmids encoding a reporter gene together with FLAG-GCN5, FLAG-GCN5DN, or a control vector, and cell lysates were prepared for the reporter assay. n = 3; *, p < 0.05. E, knockdown of GCN5 maintained mTOR/S6K activity at a high level in fasting. TA muscles were electroporated with sh-GCN5, and the animals were deprived of food for 2 days. Muscle extracts were analyzed for several different components of the mTOR/S6K pathway by Western blot. F, AMPK activation in skeletal muscle upon fasting for 2 days. The levels of phospho-AMPK and phospho-AKT were examined with the muscles lysates of fed or fasted mice. G, summary of actions of NF-κB and GCN5 in the muscle identified here. New findings about the regulation of muscle wasting in this study are highlighted in bold.
Another protein transacetylase, p300, has been shown to acetylate p65 in cultured cells (11), and it was therefore possible that the inhibitory effect of GCN5 knockdown was mediated indirectly through down-regulation of p300. However, p300 is known to inhibit the activity of FoxOs and atrogin1 and MuRF1 transcription with disuse in rodents, and dexamethasone and starvation in C2C2 cells (18). Thus, p300 could not be causing the activation of FoxO and atrogin1 induction during fasting. Moreover, the content of p300 was not affected by GCN5 knockdown (Fig. 3B). Therefore, the decrease in p65 acetylation and the inhibition of fiber atrophy upon fasting resulted specifically from the decrease in GCN5.
Because knockdown of GCN5 decreased p65 acetylation, we determined whether overexpression of GCN5 enhances p65 acetylation. In TA muscle electroporated with FLAG-tagged GCN5, p65K310 acetylation was increased in muscles from both fed and fasted mice (Fig. 3D). Similarly, transient transfection of HEK293 cells with GCN5 also increased p65 acetylation but co-transfection of SIRT1 deacetylase prevented this increase (Fig. 3E). Together, these findings indicate that GCN5 and SIRT1 play opposing roles in the regulation of NF-κB in skeletal muscle.
Inhibition of GCN5 Diminishes Loss of Muscle Mass and Attenuates Induction of Atrogenes
To determine whether the inhibition of atrophy by GCN5 knockdown resulted from the reduction in its acetylase activity, we inhibited endogenous GCN5 activity by electroporating a dominant-negative GCN5 mutant (GCN5DN) that lacks acetyltransferase activity. Like knockdown of GCN5, inhibition of its enzymatic activity diminished muscle wasting induced by fasting (Fig. 4A). By contrast, in fed animals, overexpression of the dominant-negative did not alter fiber size (Fig. 4B), which is consistent with the lack of effect of NF-κB in the fed state (Fig. 2G). The attenuation of muscle loss by knockdown or GCN5DN and the reduction in p65 acetylation on fasting suggested that the expression of atrogenes might be regulated by GCN5. In fact, upon fasting, the content of mRNAs for several atrogenes critical for muscle wasting (i.e. atrogin1, MuRF1, Gabarapl1, and Bnip3) was significantly reduced by overexpression of GCN5DN (Fig. 4C). Conversely, overexpression of WT GCN5 could by itself induce atrogin1 expression. Indeed, when C2C12 myoblast cells were transfected with the luciferase gene under control of the atrogin1 promoter (3.5 kb) together with a plasmid-encoding GCN5, overexpression of GCN5 stimulated atrogin1 transcription in the acetylase-dependent manner (Fig. 4D). These observations clearly demonstrate a critical role for this protein acetylase in activating the transcriptional program for muscle atrophy in starvation.
GCN5 Influences Activity of the PI3K-AKT Pathway
Whether a muscle undergoes hypertrophy or atrophy is determined by the balance between overall protein synthesis and degradation (32). Thus, in fasting and other systemic forms of atrophy, the acceleration of proteolysis is accompanied by an overall decrease in rates of protein synthesis due to reduced signaling by the PI3K-AKT-mTOR pathway (33). We therefore determined whether the inhibition of wasting upon down-regulation of GCN5 was also in part due to maintenance of PI3K-AKT-mTOR signaling and protein synthesis, as was observed in muscles overexpressing IκBα-SR (Fig. 2J). In fact, knockdown of GCN5 prevented the marked decreases in phosphorylation of S6K in muscles of fasted mice, although this preventive effect was less pronounced but reproducible in AKT and mTOR (Fig. 4E). Thus, GCN5 is not only important in activation of the NF-κB pathway during starvation by acetylating p65, but it also inhibits the major anabolic pathway of the cell and thus protein synthesis.
Discussion
NF-κB Activation in Muscle during Starvation
In most cells, especially cancer cells, NF-κB promotes cell survival and thus favors cell proliferation, and it plays a critical role in triggering inflammatory responses especially in immune cells (12). However, activation of this transcription factor also occurs in disused skeletal muscle and contributes to the decrease in muscle size in part by up-regulating muscle-specific ubiquitin ligases (6). In this study, we demonstrated a new mode of NF-κB regulation and an important new function of NF-κB in fasting in mediating muscle protein breakdown, which supplies precursors for hepatic gluconeogenesis or direct oxidation by the starving organism. This activation of NF-κB occurs through increased expression of p65 and p50 and acetylation of p65 in atrophying muscle upon fasting was unexpected (Fig. 1), because this pleiotropic transcription factor had not been implicated in metabolic adaptations to reduced nutrient supply. However, a recent study showed that IKKβ inhibits PI3K upon leucine depletion or exposure to amino acid-deficient medium (Hanks' balanced salt solution) in cultured cell lines and short term fasting (6–12 h fasting) in mouse liver (34). Surprisingly, this inhibition of PI3K by IKKβ appears to be independent of p65. It is noteworthy that in skeletal muscle and heart, the inhibition of PI3K by IKKβ was only observed with food deprivation for 6 h, but was not seen upon fasting for 24 h (34). By contrast, the increase in expression of both p65 and p50 subunits was found here at 48 h, which is exactly when fiber atrophy becomes clearly evident. Furthermore, this loss of muscle mass between 24 and 48 h required the activity of NF-κB. Thus, electroporation of IκBα-SR caused a marked inhibition of muscle wasting, presumably because it reduced the expression of critical atrogenes (Atrogin-1 and MuRF1).
A surprising observation during these studies was that electroporation of even a control vector induced the expression of p65 mRNA and protein (Fig. 2, A and C). This nonspecific response must be a consequence of either the brief local surgery or the electric stimulus, although there was no sign of muscle damage, inflammation, or discomfort. Most importantly, this induction of p65 did not lead to an increase in p65 acetylation (see below) and did not influence muscle size. These findings thus provide further evidence of the critical importance of p65 acetylation for NF-κB activation upon fasting.
Acetylation of p65K310 by GCN5 in Atrophying Muscles during Starvation
In addition to the increased expression of NF-κB subunits, the stimulation of NF-κB transcriptional activity during fasting required p65 acetylation at Lys-310 (Fig. 1). This modification of Lys-310 could be reversed by overexpression of the SIRT1 deacetylase, which in turn completely prevented NF-κB activation (Fig. 2A). These findings and our recent observations on the effect of SIRT1 (16) on FoxO1 and -3 indicate that inhibition of muscle wasting in fasting by SIRT1 resulted from the inactivation of both FoxOs and NF-κB. Moreover, the critical role of p65K310 acetylation in triggering fiber atrophy was confirmed by electroporation of p65K310A. Those fibers expressing p65K310A showed little or no atrophy despite food deprivation for 2 days (Fig. 2D). An important question raised by these results is whether p65K310 acetylation is involved in other types of muscle wasting, such as cancer cachexia and denervation atrophy. Another major question concerns what genes are regulated by NF-κB specifically when p65K310 is acetylated and promote fiber atrophy. In light of our finding of functional links between NF-κB and FoxO3 (Fig. 2J and see below), future studies of which genes are controlled by either or both of these transcription factors could provide valuable insights into the mechanisms that activate the atrophy-related transcriptional program.
GCN5 Is Required in Muscle Wasting for Acetylation of p65
In certain cell lines (e.g. 239T and COS-7), p300 has been shown to acetylate p65. However, in skeletal muscle, this enzyme appears to serve other regulatory functions. For example, in myotubes, p300 is important for the acceleration of protein degradation by dexamethasone, specifically for the acetylation of C/EBPβ, C/EBPδ, FoxO1, FoxO3, and NF-κB/p65 (17). By contrast, in atrophying muscle triggered by disuse, p300 acetylates FoxOs 1, 3, and 4 and inhibits their activity. p300 thus represses the induction of key atrogenes (e.g. atrogin1 and MuRF1), and a dominant-negative form of p300 increases the expression of these genes (18). In addition, the reduction in p65K310 acetylation by knockdown of GCN5 (Fig. 3B) indicated that p300 is not the primary enzyme for p65 acetylation in muscle upon fasting. Also, preventing GCN5 activity by overexpressing a dominant-negative mutant (GCN5DN) spared muscle mass, and suppressed the induction of critical atrogenes (Fig. 4, A and C). Conversely, p65K310 acetylation and the activity of the atrogin1 promoter were increased by overexpression of GCN5 in muscle and C2C12 myoblasts (Figs. 3D and 4D). In addition to activating NF-κB and promoting atrophy by increasing the induction of key atrogenes, GCN5 also inhibits PGC-1α/β (Fig. 3C), which normally inhibits muscle wasting by blocking both FoxO and NF-κB function (19, 26).
Because GCN5 expression was not induced by fasting despite the enhanced p65 acetylation by GCN5 (Figs. 3B and 4E), its acetyltransferase activity must be posttranslationally enhanced upon food deprivation. An important goal for future work will be to determine how GCN5 senses the lack of nutrients and becomes activated. In budding yeast, GCN5 is phosphorylated by AMPK, which in turn promotes expression of GCN5 target genes (35). In the atrophying muscles where p65K310 acetylation is induced, phosphorylated AMPK was elevated (Fig. 4F). Therefore, it is likely that a similar regulatory mechanism involving AMPK is conserved in skeletal muscle, but we have been unable thus far to demonstrate direct phosphorylation and activation of GCN5 by AMPK.
NF-κB Also Decreases AKT/mTOR/S6K Signaling in Fasting
One completely unexpected finding in this study was that the inhibition of NF-κB by IκBα-SR (Fig. 2J) or GCN5 knockdown (Fig. 4E) involved not only an attenuation of the induction of key atrogenes and muscle wasting, but also prevented the decrease in the activity of AKT and mTOR (Fig. 4G). Because the content of AKT, mTOR, or S6K proteins did not increase under these conditions, their expression is not inhibited by NF-κB. One possibility is that NF-κB induces the expression of a negative regulator of AKT or mTOR (e.g. a phosphatase). In this way, IκBα-SR or GCN5KD would indirectly enhance AKT and mTOR activities. Another possibility is that the increase in proteolysis induced by FoxO3 and NF-κB are suppressed by IκBα-SR or GCN5KD, because they decrease the reduction in AKT/mTOR signaling upon fasting. It is also noteworthy that in the muscles of fed mice, NF-κB inhibition by IκBα-SR or GCN5KD did not induce hypertrophy (Figs. 2G and 4B), even though activity in the AKT/mTOR pathway was enhanced. Activation of AKT/mTOR signaling would be expected to increase the overall rates of protein synthesis and promote muscle growth. Therefore, the decrease in muscle wasting in starvation must be primarily due to blocking the excessive proteolysis and the atrophy program, rather than to a general anabolic effect.
Interestingly, in knock-out mice lacking all three FoxOs (1, 3, and 4) in their muscles, the activities of AKT and mTOR were reduced in both fed and fasted conditions, whereas AMPK activity was stimulated (36). In light of our findings upon inhibition of NF-κB (Figs. 2J and 4E), this inhibition of the major growth-promoting kinases (AKT and mTOR) and activation of the growth inhibitory AMPK raise several intriguing questions for future study, whether loss of FoxOs also suppresses NF-κB activity and whether our approaches (i.e. IκBα-SR overexpression or GCN5 knockdown) would also enhance AKT/mTOR in the triple KO mice.
These findings also imply that the two critical transcription factors in muscle atrophy, FoxO3 and NF-κB, do not function independently. The activation of NF-κB in fasting, by decreasing AKT activity, should enhance dephosphorylation of FoxO3, which would promote the induction of some atrogenes. Indeed, IKKβ is known to phosphorylate and inhibit the PI3K subunit, p85 (34), which should favor dephosphorylation of FoxO3 and thus increase its capacity to stimulate expression of atrogenes (e.g. atrogin1 and MuRF1). Such effects probably also account in part for the ability of IκBα-SR expression to decrease muscle atrophy upon denervation (7) and cancer cachexia (7, 37), as well as in fasting (Fig. 2E). This new role for NF-κB in skeletal muscle in promoting protein loss and reducing AKT/mTOR signaling may also function in other differentiated or nutrient-limited cells and contrasts with its best characterized roles in promoting proliferation and survival of many cell types (e.g. cancer).
Author Contributions
D. L. and A. L. G. conceived and designed the study. D. L. performed the experiments. D. L. and A. L. G. analyzed the data, and wrote and approved the final version of the manuscript.
Acknowledgments
We are grateful to Galen Collins and Zhe Sha for helpful comments. We have received valuable assistance from the Nikon Imaging Center at Harvard Medical School.
This work was supported, in whole or in part, by National Institutes of Health Grant 2 R01 AR055255-06A1 from the NIA and the Muscular Dystrophy Association. The authors declare no conflict of interest.
- mTOR
- mechanistic target of rapamycin
- TA
- tibialis anterior
- AMPK
- AMP-activated protein kinase.
References
- 1.Sandri M. (2013) Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 45, 2121–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schiaffino S., Dyar K. A., Ciciliot S., Blaauw B., and Sandri M. (2013) Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 280, 4294–4314 [DOI] [PubMed] [Google Scholar]
- 3.Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S. J., Di Lisi R., Sandri C., Zhao J., Goldberg A. L., Schiaffino S., and Sandri M. (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471 [DOI] [PubMed] [Google Scholar]
- 4.Sandri M. (2008) Signaling in muscle atrophy and hypertrophy. Physiology 23, 160–170 [DOI] [PubMed] [Google Scholar]
- 5.Zhao J., Brault J. J., Schild A., Cao P., Sandri M., Schiaffino S., Lecker S. H., and Goldberg A. L. (2007) FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 6, 472–483 [DOI] [PubMed] [Google Scholar]
- 6.Mourkioti F., and Rosenthal N. (2008) NF-κB signaling in skeletal muscle: prospects for intervention in muscle diseases. J. Mol. Med. 86, 747–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai D., Frantz J. D., Tawa N. E. Jr., Melendez P. A., Oh B. C., Lidov H. G., Hasselgren P. O., Frontera W. R., Lee J., Glass D. J., and Shoelson S. E. (2004) IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell 119, 285–298 [DOI] [PubMed] [Google Scholar]
- 8.Hunter R. B., and Kandarian S. C. (2004) Disruption of either the NFκB1 or the Bcl3 gene inhibits skeletal muscle atrophy. J. Clin. Invest. 114, 1504–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mourkioti F., Kratsios P., Luedde T., Song Y. H., Delafontaine P., Adami R., Parente V., Bottinelli R., Pasparakis M., and Rosenthal N. (2006) Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J. Clin. Invest. 116, 2945–2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cornwell E. W., Mirbod A., Wu C. L., Kandarian S. C., and Jackman R. W. (2014) C26 cancer-induced muscle wasting is IKKβ-dependent and NF-κB-independent. PloS One 9, e87776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen L. F., Williams S. A., Mu Y., Nakano H., Duerr J. M., Buckbinder L., and Greene W. C. (2005) NF-κB RelA phosphorylation regulates RelA acetylation. Mol. Cell. Biol. 25, 7966–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker R. G., Hayden M. S., and Ghosh S. (2011) NF-κB, inflammation, and metabolic disease. Cell Metab. 13, 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy D., Kuo A. J., Chang Y., Schaefer U., Kitson C., Cheung P., Espejo A., Zee B. M., Liu C. L., Tangsombatvisit S., Tennen R. I., Kuo A. Y., Tanjing S., Cheung R., Chua K. F., Utz P. J., Shi X., Prinjha R. K., Lee K., Garcia B. A., Bedford M. T., Tarakhovsky A., Cheng X., and Gozani O. (2011) Lysine methylation of the NF-kappaB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-κB signaling. Nat. Immunol. 12, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L. F., Fischle W., Verdin E., and Greene W. C. (2001) Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293, 1653–1657 [DOI] [PubMed] [Google Scholar]
- 15.Yeung F., Hoberg J. E., Ramsey C. S., Keller M. D., Jones D. R., Frye R. A., and Mayo M. W. (2004) Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee D., and Goldberg A. L. (2013) SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J. Biol. Chem. 288, 30515–30526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamberlain W., Gonnella P., Alamdari N., Aversa Z., and Hasselgren P. O. (2012) Multiple muscle wasting-related transcription factors are acetylated in dexamethasone-treated muscle cells. Biochem. Cell Biol. 90, 200–208 [DOI] [PubMed] [Google Scholar]
- 18.Senf S. M., Sandesara P. B., Reed S. A., and Judge A. R. (2011) p300 acetyltransferase activity differentially regulates the localization and activity of the FOXO homologues in skeletal muscle. Am. J. Physiol. Cell Physiol. 300, C1490–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerhart-Hines Z., Rodgers J. T., Bare O., Lerin C., Kim S. H., Mostoslavsky R., Alt F. W., Wu Z., and Puigserver P. (2007) Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 26, 1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagy Z., and Tora L. (2007) Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 26, 5341–5357 [DOI] [PubMed] [Google Scholar]
- 21.Tang X., Wen S., Zheng D., Tucker L., Cao L., Pantazatos D., Moss S. F., and Ramratnam B. (2013) Acetylation of drosha on the N-terminus inhibits its degradation by ubiquitination. PLoS ONE 8, e72503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lerin C., Rodgers J. T., Kalume D. E., Kim S. H., Pandey A., and Puigserver P. (2006) GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1α. Cell Metab. 3, 429–438 [DOI] [PubMed] [Google Scholar]
- 23.Holmlund T., Lindberg M. J., Grander D., and Wallberg A. E. (2013) GCN5 acetylates and regulates the stability of the oncoprotein E2A-PBX1 in acute lymphoblastic leukemia. Leukemia 27, 578–585 [DOI] [PubMed] [Google Scholar]
- 24.Downey M., Knight B., Vashisht A. A., Seller C. A., Wohlschlegel J. A., Shore D., and Toczyski D. P. (2013) Gcn5 and sirtuins regulate acetylation of the ribosomal protein transcription factor Ifh1. Curr. Biol. 23, 1638–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi H., Sun X., Hamamoto M., Yashiroda Y., and Yoshida M. (2012) The SAGA histone acetyltransferase complex regulates leucine uptake through the Agp3 permease in fission yeast. J. Biol. Chem. 287, 38158–38167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brault J. J., Jespersen J. G., and Goldberg A. L. (2010) Peroxisome proliferator-activated receptor γ coactivator 1α or 1β overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J. Biol. Chem. 285, 19460–19471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S. H., and Goldberg A. L. (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palombella V. J., Rando O. J., Goldberg A. L., and Maniatis T. (1994) The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell 78, 773–785 [DOI] [PubMed] [Google Scholar]
- 29.Lee J. H., Song M. Y., Song E. K., Kim E. K., Moon W. S., Han M. K., Park J. W., Kwon K. B., and Park B. H. (2009) Overexpression of SIRT1 protects pancreatic β-cells against cytokine toxicity by suppressing the nuclear factor-κB signaling pathway. Diabetes 58, 344–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sandri M., Lin J., Handschin C., Yang W., Arany Z. P., Lecker S. H., Goldberg A. L., and Spiegelman B. M. (2006) PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. U.S.A. 103, 16260–16265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao X., Gluck N., Li D., Maine G. N., Li H., Zaidi I. W., Repaka A., Mayo M. W., and Burstein E. (2009) GCN5 is a required cofactor for a ubiquitin ligase that targets NF-κB/RelA. Genes Dev. 23, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitch W. E., and Goldberg A. L. (1996) Mechanisms of muscle wasting: the role of the ubiquitin-proteasome pathway. N. Engl. J. Med. 335, 1897–1905 [DOI] [PubMed] [Google Scholar]
- 33.Egerman M. A., and Glass D. J. (2014) Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 49, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Comb W. C., Hutti J. E., Cogswell P., Cantley L. C., and Baldwin A. S. (2012) p85α SH2 domain phosphorylation by IKK promotes feedback inhibition of PI3K and Akt in response to cellular starvation. Mol. Cell 45, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y., Xu X., and Kuo M. H. (2010) Snf1p regulates Gcn5p transcriptional activity by antagonizing Spt3p. Genetics 184, 91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milan G., Romanello V., Pescatore F., Armani A., Paik J. H., Frasson L., Seydel A., Zhao J., Abraham R., Goldberg A. L., Blaauw B., DePinho R. A., and Sandri M. (2015) Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 6, 6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Acharyya S., Butchbach M. E., Sahenk Z., Wang H., Saji M., Carathers M., Ringel M. D., Skipworth R. J., Fearon K. C., Hollingsworth M. A., Muscarella P., Burghes A. H., Rafael-Fortney J. A., and Guttridge D. C. (2005) Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 8, 421–432 [DOI] [PubMed] [Google Scholar]