

Abstract
Sirtuin 1 (SIRT1), an NAD+-dependent histone deacetylase, plays crucial roles in various biological processes including longevity, stress response, and cell survival. Endoplasmic reticulum (ER) stress is caused by dysfunction of ER homeostasis and exacerbates various diseases including diabetes, fatty liver, and chronic obstructive pulmonary disease. Although several reports have shown that SIRT1 negatively regulates ER stress and ER stress-induced responses in vitro and in vivo, the effect of ER stress on SIRT1 is less explored. In this study, we showed that ER stress induced SIRT1 expression in vitro and in vivo. We further determined the molecular mechanisms of how ER stress induces SIRT1 expression. Surprisingly, the conventional ER stress-activated transcription factors XBP1, ATF4, and ATF6 seem to be dispensable for SIRT1 induction. Based on inhibitor screening experiments with SIRT1 promoter, we found that the PI3K-Akt-GSK3β signaling pathway is required for SIRT1 induction by ER stress. Moreover, we showed that pharmacological inhibition of SIRT1 by EX527 inhibited the ER stress-induced cellular death in vitro and severe hepatocellular injury in vivo, indicating a detrimental role of SIRT1 in ER stress-induced damage responses. Collectively, these data suggest that SIRT1 expression is up-regulated by ER stress and contributes to ER stress-induced cellular damage.
Keywords: endoplasmic reticulum stress (ER stress), gene regulation, liver injury, phosphatidylinositol signaling, sirtuin 1 (SIRT1)
Introduction
The endoplasmic reticulum (ER)2 is an important organelle functioning in protein folding, transport, processing, and storage of calcium ion. Homeostasis of ER is crucial for cellular activity and survival (1–3). Intracellular and extracellular conditions that interfere with ER function lead to the accumulation of unfolded proteins in the ER, resulting in ER stress. Cells exposed to ER stress activate the unfolded protein response (UPR), which consists of three major branches: the activating transcription factor (ATF) 6, inositol-requiring enzyme (IRE) 1-X-box binding protein (XBP) 1, and PERK-eIF2α-ATF4 pathways (4). These signaling pathways include both translational and transcriptional control mechanisms that reduce protein synthesis, increase the protein folding capacity by up-regulating the transcription of molecular chaperones, and activate the ER-associated protein degradation. By using these systems, living organisms maintain homeostasis against various stress conditions (4). Thus, malfunction of ER stress responses is associated with various diseases including diabetes, fatty liver, neurodegeneration, and chronic obstructive pulmonary disease (5–7).
Sirtuins are the mammalian orthologues of the yeast Sir2 protein that is involved in chromatin silencing and lifespan regulation. The mammalian Sirtuin 1 (SIRT1) gene encodes a nicotinamide adenosine dinucleotide (NAD+)-dependent histone deacetylase that is the closest structural ortholog of the yeast Sir2 protein (8, 9). Although the yeast Sir2 protein has been extensively characterized as a histone deacetylase, the mammalian SIRT1 protein deacetylates not only histone but also non-histone substrates including transcription factors. For example, SIRT1 expression is induced in response to acute nutritional stress leading to SIRT1 deacetylation of FOXO1, a cell metabolism regulator, thereby contributing to the activation of gluconeogenesis (10). The deacetylation of E2F1 by SIRT1 inhibits its transcriptional activity that contributes to the suppression of apoptosis induced by DNA damage (11).
Recent reports have shown that SIRT1 deacetylates XBP1 and inhibits its transcriptional activity to promote ER stress-induced apoptosis, and SIRT1 suppresses the PERK-eIF2α-dependent translational inhibition in mammals (12–14). Thus, SIRT1 has been known to be a negative regulator for ER stress response. However, compared with the effect of SIRT1 on ER stress response, the effect of ER stress on SIRT1 is less studied. In addition, the role of SIRT1 in hepatic steatosis is controversial in high fat diet-induced obese mice whose livers are exposed to ER stress (15). Chen et al. (16) showed that SIRT1 liver-specific knock-out mice are protected from liver weight gain, whereas Purushotham et al. (17) demonstrated that liver-specific SIRT1 deficiency enhances liver steatosis. Moreover, Li et al. (18) reported that hepatic overexpression of SIRT1 ameliorates hepatic steatosis. However, because mouse models of high fat diet-induced obesity are affected and complicated by multiple factors, the effect of SIRT1 on ER stress-induced hepatic injury is still largely unclear.
In this study, we examined the effect of ER stress on the expression of SIRT1. We show that ER stress increases the expression of SIRT1 in vitro and in vivo. We also provide evidence that the PI3K-Akt-GSK3β signaling pathway is required for the induction of SIRT1 expression by ER stress. Furthermore, we show that ER stress-induced cell death and hepatic injury are positively regulated by SIRT1. These data support the growing evidence of a direct link between ER stress and SIRT1.
Experimental Procedures
Reagents, Plasmids, and Antibodies
Thapsigargin, SB203580,SP600125, and wortmannin were purchased from Calbiochem. Tunicamycin was purchased from Wako (Osaka, Japan). EX527 and LiCl were purchased from Sigma. CHIR99021 was purchased from Cayman Chemicals. pcDNA3.1 plasmid was obtained from Invitrogen. The plasmids for ATF4; ATF6α(1–373), which corresponds to the active form of ATF6 (amino acids 1–373); and spliced XBP1 (XBP1s) were described previously (19–21). Antibodies for SIRT1 (H-300), γ-tubulin (C-20), β-actin (AC-15), and phospho-GSK3β (Tyr-216) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-KDEL antibody for glucose-related protein 78 kDa (GRP78) (SPA-827) was purchased from Stressgen (San Diego, CA). Antibodies against phospho-Akt (Ser-473), Akt, phospho-GSK3β (Ser-9), and GSK3β were purchased from Cell Signaling Technology. Anti-p53 acetyl-lysine 379 rabbit polyclonal antibody (SAB4503018) was from Sigma. Anti-p65 acetyl-lysine 310 rabbit polyclonal antibody (ab19870) was from Abcam (UK). Horseradish peroxidase-conjugated anti-rabbit, anti-goat, and anti-mouse antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA).
Cell Culture, Treatment, and Transfection
Human lung epithelial A549 cells, human embryonic kidney (HEK) 293 cells, and mouse embryonic fibroblast cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Wako) supplemented with 10% FBS and antibiotics. Human monocytic THP-1 cells were cultured in RPMI 1640 medium supplemented with 10% of heat-inactivated FBS and antibiotics. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Treatment of cells with 5 μg/ml tunicamycin or 1 μm thapsigargin was carried out at the indicated time and concentration. Transient transfections of plasmids were performed using Trans-IT LT1 (Mirus, Madison, WI) following the manufacturer's recommendations. Specifically, Trans-IT LT1 reagent diluted with Opti-MEM (Gibco) was mixed with total DNA in a ratio of 1:4 (DNA:LT1) and applied to subconfluent cells in medium. TransIT-TKO reagent was used for knockdown experiments by siRNA. siRNA for luciferase was used as control siRNA. Forty-eight hours after siRNA transfection, cells were collected for assay.
Quantitative RT-PCR (QPCR) Analysis
Total RNA was isolated from cells with RNAiso Plus (TaKaRa, Japan) according to the manufacturer's instructions. Real time quantitative RT-PCR analysis for SIRT1, CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP), GRP78, ER DnaJ homologue 4 (ERdj4), XBP1, ATF4, mIL-6, mCTGF, and 18S ribosomal RNA (18S rRNA) was carried out using PrimeScript® RT reagent kit (TaKaRa) and SYBR Premix Ex TaqTM II (TaKaRa). PCR amplifications were performed as described previously (22). The threshold cycle values for each gene amplification were normalized by subtracting the threshold cycle value calculated for 18S rRNA (internal control). The normalized gene expression values were expressed as the relative quantity of gene-specific mRNA (SIRT1, CHOP, GRP78, XBP1, ATF4, mIL-6, mCTGF, and ERdj4). The oligonucleotide primers used in real time quantitative PCR amplifications are shown in Table 1.
TABLE 1.
Sequences of oligonucleotides used as primer for cloning of human SIRT1 promoter and real time QPCR
| Primer | Sequence |
|---|---|
| For SIRT1 promoter | |
| PF-1 (−1200) | 5′-CTCGAGAACCCATACTAGGCTTAAGGG-3′ |
| PF-2 (−691) | 5′-CTCGAGATCGGCTGATCTCCAAAC-3′ |
| PF-3 (−457) | 5′-CTCGAGAGAACGACTATCCAAC-3′ |
| PF-4 (−47) | 5′-CTCGAGGTTTAAATCTCCCGCAG-3′ |
| PR-1 (+53) | 5′-AAGCTTCTTCCAACTGCCTCTCTG-3′ |
| For QPCR | |
| SIRT1 | Forward, 5′-TGGCAAAGGAGCAGATTAGTAGG-3′ |
| Reverse, 5′-CTGCCACAAGAACTAGAGGATAAGA-3′ | |
| CHOP | Forward, 5′-ATGGCAGCTGAGTCATTGCCTTTC-3′ |
| Reverse, 5′-AGAAGCAGGGTCAAGAGTGGTGAA-3′ | |
| ERdj4 | Forward, 5′-AGTAGACAAAGGCATCATTTCCAA-3′ |
| Reverse, 5′-CTGTATGCTGATTGGTAGAGTCAA-3′ | |
| GRP78 | Forward, 5′-ACCAATTATCAGCAAACTCTATGGAA-3′ |
| Reverse, 5′-CATCTTTTTCTGCTGTATCCTCTTCA-3′ | |
| XBP1 | Forward, 5′-CCGCAGCAGGTGCAGG-3′ |
| Reverse, 5′-GAGTCAATACCGCCAGAATCC-3′ | |
| ATF4 | Forward, 5′-AGTGGCATCTGTATGAGCCCA-3′ |
| Reverse, 5′-GCTCCTATTTGGAGAGCCCCT-3′ | |
| 18S rRNA | Forward, 5′-CGGCTACCACATCCAAGGAA-3′ |
| Reverse, 5′-GCTGGAATTACCGCGGCT-3′ | |
| mSIRT1 | Forward, 5′-TTGCAAAGGAGCAGATTAGTAAGC-3′ |
| Reverse, 5′-TGCCACAGGAACTAGAGGACAA-3′ | |
| mIL-6 | Forward, 5′-TAGTCCTTCCTACCCCAATTTCC-3′ |
| Reverse, 5′-TTGGTCCTTAGCCACTCCTTC-3′ | |
| mCTGF | Forward, 5′-GGGCCTCTTCTGCGATTTC-3′ |
| Reverse, 5′-ATCCAGGCAAGTGCATTGGTA-3′ | |
| m18S rRNA | Forward, 5′-CCATCCAATCGGTAGTAGCG-3′ |
| Reverse, 5′-GTAACCCGTTGAACCCCATT-3′ | |
Western Blotting
Western blotting analysis was performed as described previously (23). Briefly, whole cell lysates were prepared with radioimmunoprecipitation assay buffer (50 mm Tris-HCl, 150 mm NaCl, 1 mg/ml sodium deoxycholate, 1% Nonidet P-40) containing 1% protease inhibitor mixture and rotated overnight at 4 °C. Equal amounts of samples were fractionated by 8% SDS-PAGE and transferred to polyvinylidene difluoride membrane. After blocking, the membrane was probed with the appropriate antibodies, and blots were visualized using SuperSignal (Pierce).
Cloning of SIRT1 Promoter and Reporter Assay
Genomic DNA was isolated from A549 cells as described previously (24). Human SIRT1 promoter sequence was amplified by PfuUltra HS with primers indicated in Table 1. Amplified products (−1200 to +53, −691 to +53, −457 to +53, and −47 to +53) were subcloned into pGL3-Basic vector by using XhoI and HindIII sites. A549 cells were transfected with promoter plasmids and phRG-TK plasmids for the normalization of transfection efficiency and subjected to a dual reporter assay 48 h after transfection with the PicaGene Dual Sea Pansy Luminescence kit (Toyo Inki, Tokyo).
Lactate Dehydrogenase Assay
Cells were assayed for lactate dehydrogenase release according to a protocol described previously (25). Lactate dehydrogenase release was expressed as the percentage of lactate dehydrogenase in the medium over the total lactate dehydrogenase (medium and lysate). Values are means ± S.E. of triplicate testing for a representative experiment. At least three independent experiments were performed.
Animal Experiments and Tissue Sample Analysis
Male C57BL/6 J mice (Charles River Laboratories, Inc., Kanagawa, Japan) were housed in a vivarium in accordance with the guidelines of the animal facility center of Kumamoto University (number 25-230E). The mice were maintained on food and water ad libitum. For the tunicamycin injection experiments, 6–8-week-old mice were injected with tunicamycin intraperitoneally at 1 μg/g of body weight for 24 h (26). Livers were harvested, and total RNA was isolated by using RNAiso Plus with recombinant DNase I (TaKaRa) according to the manufacturer's instructions. Liver samples were also subjected to immunohistochemical analysis. Briefly, livers were fixed with 4% paraformaldehyde and embedded into paraffin. Paraffin sections were subjected to antigen retrieval and blocking with goat serum. Primary antibody against SIRT1 was used to detect mouse SIRT1 protein. Rabbit IgG was used as a negative control. In some experiments, mice were co-injected intraperitoneally with tunicamycin (1 μg/g) and EX527 (2 μg/g) on Day 0 and Day 1. Livers were harvested 72 h after first injection and subjected to H&E staining, QPCR analysis, and immunohistochemistry (for acetylated p65 and acetylated p53).
Statistical Analysis
Data are presented as mean ± S.E. For statistical analysis, the data were analyzed by one-way analysis of variance with either a Bonferroni or Dunnett multiple comparison test or Student's t test (JMP software, SAS Institute, Cary, NC) as indicated in each figure legend. The differences were considered statistically significant when the p value was less than 0.05.
Results
ER Stress Increases the Protein Expression of SIRT1 in Vitro
We first examined whether ER stress influences the expression of SIRT1 protein. Human epithelial A549 cells were treated with thapsigargin (TG) for the indicated time to induce ER stress (Fig. 1A). SIRT1 protein was increased at 8–24 h of treatment with TG (Fig. 1, A and B). The expression of GRP78, a molecular chaperone induced by ER stress, was also increased in response to TG (Fig. 1, A and C). TG increased the SIRT1 expression at 0.1–2 μm (Fig. 1D). We treated A549 cells with another ER stress inducer, tunicamycin (TM). TM dose-dependently increased SIRT1 protein expression at 12 h of treatment (Fig. 1E). To further investigate the effect of ER stress on SIRT1 expression in other cell types, we treated THP-1 cells, HEK293 cells, and mouse embryonic fibroblasts with TG. The expression of SIRT1 was also up-regulated in these cell lines by TG treatment, suggesting a general effect of ER stress on the induction of SIRT1 protein in vitro (Fig. 1, F–H).
FIGURE 1.

ER stress increases the protein expression of SIRT1 in vitro. A, A549 cells were treated with 1 μm TG for the indicated time. B and C, relative protein quantity of SIRT1 (B) and GRP78 (C) was analyzed by Image Gauge software and normalized with γ-tubulin. Values are the mean ± S.E. (error bars) (n = 3 for B and C). *, p < 0.05 versus control as assessed by one-way analysis of variance with Dunnett procedure. D and E, A549 cells were treated with the indicated concentrations of TG for 24 h (D) or with TM for 12 h (E). THP-1 cells (F) and HEK293 cells (G) were treated for 12 h with TG (1 μm). H, mouse embryonic fibroblast (MEF) cells were treated for 24 h with TG (1 μm). Expression of SIRT1 and GRP78 protein was determined by Western blotting analysis of whole cell lysate. γ-Tubulin was used as a loading control. CON, control.
ER Stress Increases the Protein Expression of SIRT1 in Vivo
We next determined whether SIRT1 is induced by ER stress in vivo. C57BL/6J mice were intraperitoneally injected with TM at a dose of 1 μg/g of body weight. Liver tissues were harvested at 4, 8, and 24 h after injection. Mouse SIRT1 (mSIRT1) protein expression was assessed by Western blotting. Consistent with in vitro results, the expression of SIRT1 protein was up-regulated at 8 and 24 h after TM injection compared with saline-injected control (Fig. 2, A and B). ER stress in livers of TM-treated mice was confirmed by mouse GRP78 (mGRP78) up-regulation (Fig. 2, A and C). Immunohistochemical experiments showed an increase of nuclear SIRT1 expression in hepatocytes in TM-injected mouse livers (Fig. 2D, bottom panels) compared with saline-injected control (Fig. 2D, upper panels). These data indicated that ER stress increases the protein expression of SIRT1 in vivo.
FIGURE 2.

ER stress increases the protein expression of SIRT1 in vivo. A–C, C57BL/6J mice were intraperitoneally injected with TM (1 μg/g of body weight), and liver tissues were harvested at 4, 8, or 24 h after injection. Expression of mSIRT1 and mGRP78 protein was determined by Western blotting analysis of whole cell lysate. Mouse γ-tubulin (mγ-tubulin) was used as a loading control. The relative quantity of mSIRT1 (B) and mGRP78 (C) protein was analyzed by Image Gauge software. Values are the mean ± S.E. (error bars) (n = 3 for B and C). *, p < 0.05 versus control; ***, p < 0.001 versus control; †, p < 0.1 versus control as assessed by Student's t test. n.s., not significant. D, immunohistochemical analysis of SIRT1 in TM-injected mouse liver tissue. Livers were collected 24 h after intraperitoneal injection with TM. Rabbit IgG was used as a negative control. Scale bars indicate 20 μm. CON, control.
ER Stress Increases the Expression of SIRT1 at the Transcriptional Level
We next investigated whether SIRT1 is up-regulated at the transcriptional level during ER stress. A549 cells were treated with TG or TM for the indicated times (Fig. 3, A and B, respectively). As shown in Fig. 3A, the expression of SIRT1 mRNA was significantly increased at 3, 6, and 12 h of treatment with TG. TM also significantly increased the expression of SIRT1 mRNA in vitro at 3 h of treatment (Fig. 3B). Mice were administered intraperitoneally with TM, and liver tissue was harvested at 4, 8, and 24 h after injection. The expression of SIRT1 mRNA was up-regulated at 4 h after TM injection (Fig. 3C). These results showed that ER stress increases the expression of SIRT1 at the transcriptional level in vitro and in vivo.
FIGURE 3.

ER stress increases the expression of SIRT1 at transcriptional level. A and B, A549 cells were treated with TG (1 μm) (A) or with TM (5 μg/ml) (B) for the indicated time. The relative quantity of SIRT1 mRNA was analyzed by real time quantitative RT-PCR. 18S rRNA was used as an internal control. C, mice were administered intraperitoneally with TM (1 μg/g of body weight), and liver tissues were harvested at the indicated time after injection. The relative quantity of mouse SIRT1 mRNA was analyzed by real time quantitative RT-PCR using mouse 18S rRNA as an internal control. Values are the mean ± S.E. (error bars) (n = 3 for A and B). *, p < 0.05; **, p < 0.01 as assessed by Student's t test. n.s., not significant; CON, control.
XBP1, ATF4, and ATF6 Are Likely Dispensable for ER Stress-induced SIRT1 Up-regulation
In silico analysis of the SIRT1 promoter using the Transcription Element Search System revealed that two putative binding sites for the ATF/cAMP-responsive element-binding protein family and four putative binding sites for XBP1 are contained within 1 kbp upstream from the transcription initiation site. To determine which UPR signaling pathway is responsible for SIRT1 up-regulation by ER stress, we cloned human SIRT1 promoter (−1200 to +53 bp) and investigated SIRT1 promoter activity in response to TG treatment. As shown in Fig. 4A, TG treatment significantly increased SIRT1 promoter activity. To further specify the ER stress-responsive region, we generated three reporter plasmids containing shorter lengths of SIRT1 promoter (−691 to +53, −457 to +53, and −47 to +53). A reporter assay showed that the region containing four XBP1 putative binding sites (−457 to −47) is required for TG-induced up-regulation of SIRT1 promoter activity (Fig. 4A). We next examined the effect of XBP1s, ATF4, or ATF6α(1–373) on the expression of SIRT1 mRNA. Overexpression of XBP1s significantly increased the expression of SIRT1 and ERdj4, a known XBP1s target gene (Fig. 4B). Overexpression of ATF4 slightly increased SIRT1 expression along with CHOP, a specific ATF4 target gene (Fig. 4C). Conversely, overexpression of ATF6 failed to induce SIRT1 expression, although GRP78, a specific ATF6 target gene, was up-regulated (Fig. 4D). To clarify the role of XBP1 and ATF4 in ER stress-induced SIRT1 up-regulation, we used siRNA to knock down XBP1 and ATF4. Although expressions of XBP1 and ATF4 and their target genes ERdj4 and CHOP, respectively, were significantly reduced by siRNA, knockdown of XBP1 or ATF4 had no effect on TG-induced SIRT1 mRNA expression (Fig. 4, E–G). Moreover, knockdown of both ATF4 and XBP1 did not suppress the TG-induced SIRT1 mRNA expression (Fig. 4E). Together, these data indicated that although overexpression of XBP1s or ATF4 has a positive effect on SIRT1 expression both transcription factors do not seem to be the main mediators for ER stress-induced SIRT1 up-regulation.
FIGURE 4.

XBP1, ATF4, and ATF6 are likely dispensable for ER stress-induced SIRT1 up-regulation. A, A549 cells were transfected with SIRT1 promoter plasmids (−1200, −691, −457, or −47 bp) and treated with 1 μm TG for 24 h. Luciferase activity was determined 48 h after plasmid transfection and is expressed as -fold activation over untreated samples. B–D, A549 cells were transfected with 0.1 or 1.0 μg XBP1s (B), ATF4 (C), or ATF6α(1–373) (D). E–G, A549 cells were transfected with siRNA for XBP1 and/or ATF4. Forty-eight hours post-transfection, cells were treated with TG (1 μm) for 3 h. The relative quantity of the indicated gene was analyzed by real time quantitative RT-PCR. 18S rRNA was used as an internal control. Values are the mean ± S.E. (error bars) (n = 3 for A–G). *, p < 0.05; **, p < 0.01; ***, p < 0.001 as assessed by Student's t test. n.s., not significant; CON, control.
ER Stress-induced SIRT1 Up-regulation Is Mediated by the PI3K-Akt-GSK3β Signaling Pathway
To clarify the molecular mechanism of how ER stress regulates SIRT1 expression, we screened a variety of inhibitors for the intracellular signaling pathways NF-κB, ERK, JNK, p38 MAPK, PI3K, GSK3β, calcineurin, and sterol regulatory element-binding protein (Fig. 5A and data not shown). TG significantly induced SIRT1 promoter (−457 to +53) activity, which was suppressed by CHIR99021, a specific inhibitor for GSK3β, whereas TG-induced SIRT1 up-regulation was greatly enhanced by wortmannin, a specific inhibitor for PI3K. SP600125, a JNK inhibitor, enhanced basal SIRT1 promoter activity but suppressed TG-induced activity. p38 MAPK inhibitor SB203580 showed no effect on SIRT1 expression. The PI3K-Akt signaling pathway is known as an important regulator of GSK3β activity (27). PI3K activates Akt via Ser-473 phosphorylation. Activated Akt phosphorylates GSK3β at Ser-9 to inactivate it, whereas phosphorylation of GSK3β at Tyr-216 leads to its activation. The phosphorylation of GSK3β at Tyr-216 is mediated by several kinases including PYK-2, FYN, and GSK3β itself (27, 28). Therefore, we investigated the effect of ER stress on phosphorylation of Akt and GSK3β. TG treatment transiently suppressed phosphorylation of Akt (Ser-473) in A549 cells (Fig. 5B). Maximum suppression was observed at 30 min after TG treatment. Consistently, TG treatment inhibited the phosphorylation of GSK3β at Ser-9 at 60–120 min. In contrast, phosphorylation of GSK3β at Tyr-216 was increased at 60–120 min by TG treatment (Fig. 5B). We next investigated the effects of these signaling pathways on ER stress-induced SIRT1 mRNA expression. Unlike in the SIRT1 promoter assay (Fig. 5A), JNK inhibitor SP600125 showed no effect on ER stress-induced SIRT1 mRNA up-regulation (Fig. 5C). In line with Fig. 5, A and B, wortmannin enhanced but CHIR99021 suppressed SIRT1 induction by ER stress in a dose-dependent manner (Fig. 5, D and E). Moreover, up-regulation of SIRT1 was also inhibited by LiCl, another inhibitor of GSK3β (Fig. 5F). The importance of GSK3β for SIRT1 induction by ER stress was confirmed by knockdown of GSK3β (Fig. 5, G and H). These results suggested that ER stress suppresses the PI3K-Akt pathway and subsequently activates GSK3β to induce SIRT1 expression.
FIGURE 5.

ER stress-induced SIRT1 up-regulation is mediated by the PI3K-Akt-GSK3β signaling pathway. A, A549 cells were transfected with SIRT1 promoter plasmid (−457 bp). Cells were pretreated with inhibitors SB203580 (SB; 20 μm), CHIR99021 (CHIR; 2 μm), SP600125 (SP; 20 μm), and wortmannin (WM; 1 μm) for 1 h and co-treated with TG (1 μm) for 24 h. Luciferase activity was determined 48 h after plasmid transfection and is expressed as -fold activation over TG-untreated samples. B, A549 cells were treated with 1 μm TG for the indicated time and subjected to Western blotting analysis. β-Actin was used as a loading control. C–F, A549 cells were pretreated with SP600125 (C), wortmannin (D), CHIR99021 (E), and LiCl (F) for 1 h and treated with TG for 3 h. G and H, A549 cells were transfected with siRNA for GSK3β. Forty-eight hours after transfection, cell lysates were subjected to Western blotting (G) or TG treatment for QPCR analysis (H). Relative SIRT1 mRNA expression was analyzed by QPCR analysis. 18S rRNA was used as an internal control. *, p < 0.05; **, p < 0.01; ***, p < 0.001 as assessed by Student's t test (A) and by one-way analysis of variance with Bonferroni test (C–F and H). Values are the mean ± S.E. (error bars) (n = 3 for A and C–F). n.s., not significant; CON, control; NT, no treatment; p-Akt, phospho-Akt; p-GSK3β, phospho-GSK3β.
Pharmacological Inhibition of SIRT1 Ameliorates ER Stress-induced Cell Death and Hepatic Injury
To examine the function of SIRT1 up-regulation during ER stress, we evaluated the effect of SIRT1-specific inhibitor EX527. A549 cells were treated with TG in the presence or absence of 10 μm EX527 for 48 h, and cell death was measured by lactate dehydrogenase release. TG treatment induced cell death, but intriguingly, co-treatment with TG and EX527 significantly suppressed cell death compared with TG alone (Fig. 6A). We next investigated the effect of SIRT1 induction by ER stress in vivo using a multiple TM-injected mouse model. TM induced severe hepatic injury, and vacuoles were detected in hepatocytes in a high magnification field (Fig. 6B, upper right panels). Hepatic injury was ameliorated by EX527 co-treatment (Fig. 6B, bottom panels). Moreover, the hepatic injury score was also improved in EX527-treated mice (Fig. 6C). Consistently, TM induced IL-6 and CTGF mRNA expression in mouse liver that was reduced by EX527 treatment (Fig. 6, D and E). Because SIRT1 deacetylates various substrates including p65 and p53, we stained for acetylated p65 and p53 in mouse livers treated with TM or TM +EX527. Although basal acetylation status of both p65 and p53 is low, TM treatment further decreased the signals. Conversely, compared with samples treated with TM only, the addition of EX527 increased the acetylation of p65 and p53 as determined by immunohistochemical analysis (Fig. 6F), indicating that TM-induced deacetylation of both p65 and p53 is dependent on SIRT1. Collectively, these data suggested that ER stress-induced cellular death and hepatic injury are positively regulated by SIRT1.
FIGURE 6.
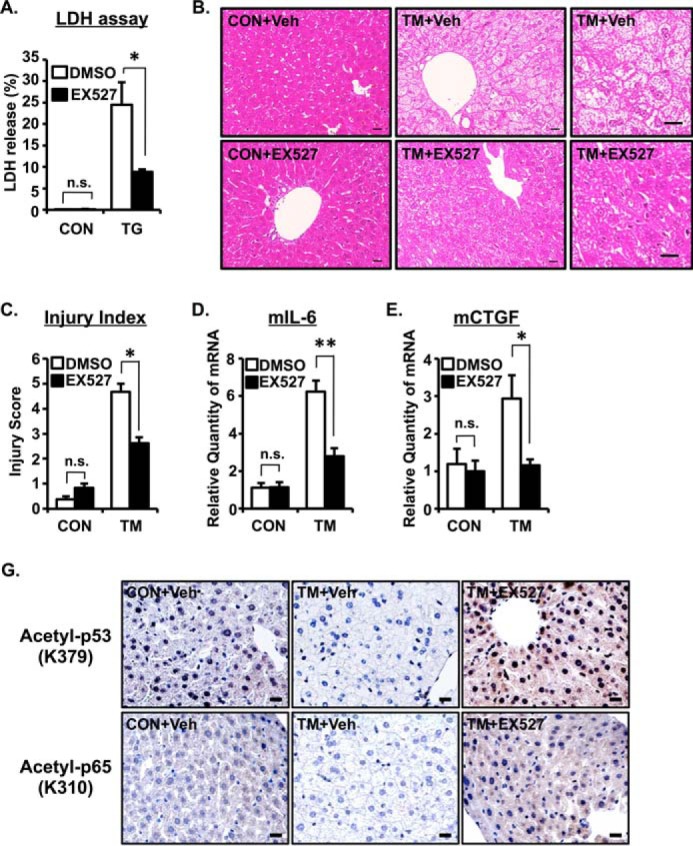
ER stress-induced cell death is positively regulated by SIRT1. A, A549 cells were treated with TG (1 μm) for 24 h with or without EX527, a SIRT1 inhibitor (10 μm). The relative -fold increase of cell mortality was assessed by lactate dehydrogenase (LDH) assay. B–E, mice were injected with TM (1 μg/g) and EX527 (2 μg/g) on Day 0 and Day 1. Livers were collected 72 h after the first injection and subjected to H&E staining (B), hepatic injury quantification (C), and real time quantitative PCR analysis (D and E). 18S rRNA was used as an internal control. Values are the mean ± S.E. (error bars) (n = 3–4 for A, C, and D). Scale bars indicate 20 μm (B). Right panels are high magnification photos of middle panels (B). *, p < 0.05; **, p < 0.01 as assessed by analysis of variance with Dunnett procedure. n.s., not significant. F, TM- and TM + EX527-treated mouse livers were subjected to immunohistochemistry for acetylated p53 (upper panels) and acetylated p65 (bottom panels). Scale bars indicate 20 μm. CON, control; Veh, vehicle.
Discussion
Several reports have shown that SIRT1 acts as a negative regulator for ER stress response. Viswanathan et al. (29) recently reported that Sir-2.1, the Caenorhabditis elegans Sir2 ortholog, is a negative regulator of UPR genes. In mammals, Wang et al. (13) showed that SIRT1 negatively regulates XBP1 transcriptional activity by deacetylating XBP1 protein and promotes cellular apoptosis. Ghosh et al. (12) have demonstrated that SIRT1 associates with eIF2-α and negatively regulates cellular stress responses. Moreover, it was also reported that SIRT1 attenuates ER stress in vitro and in vivo (18, 30). Therefore, the role of SIRT1 as a negative regulator for ER stress response is conserved from C. elegans to mammals, indicating a crucial link between SIRT1 and ER stress. However, the impact of ER stress on SIRT1 remained unclear. In this study, we showed that ER stress induces SIRT1 expression in vitro and in vivo at the mRNA and protein levels (Figs. 1–3). We also provide evidence that the PI3K-Akt-GSK3β signaling pathway is required for induction of SIRT1 expression by ER stress. Furthermore, ER stress-induced cell death and hepatic injury are positively regulated by SIRT1. These data add to the growing evidence of a direct link between ER stress and SIRT1 (Fig. 7) and suggest that an autoregulatory negative feedback mechanism exists between ER stress and SIRT1.
FIGURE 7.

Schematic diagram of how ER stress regulates SIRT1 expression. The up-regulation of SIRT1 expression by ER stress is not through the conventional ER stress pathways but rather through the PI3K-Akt-GSK3β signaling pathway. The ER stress-induced SIRT1 leads to hepatocellular damage.
ER stress-exposed cells activate the UPR that consists of ATF6, IRE1-XBP1, and PERK-eIF2α-ATF4 pathways. However, our data showed that these pathways are dispensable. Although we cannot totally eliminate the possible involvement of XBP1 and ATF4, knockdown experiments revealed that these factors have no significant impact on ER stress-induced SIRT1 up-regulation (Fig. 4). Winnay et al. (31, 32) reported the inhibitory role of PI3K on ER stress response through direct interaction with XBP1. In our study, we showed that XBP1 is not involved in ER stress-induced SIRT1 induction (Fig. 4), suggesting a regulatory involvement of PI3K in an XBP1-independent, Akt-GSK3β-dependent manner. But how ER stress suppresses the PI3K-Akt pathway is unknown. Several reports have shown that ER stress suppresses Akt phosphorylation (33). Zhang et al. (35) reported the involvement of PERK-dependent nuclear transport of PTEN in ER stress-mediated Akt dephosphorylation, whereas Qin et al. (34) showed that PTEN, a phosphatase for phosphatidylinositol 1,4,5-trisphosphate, is not involved in ER stress-mediated Akt dephosphorylation. In addition, a recent report showed that ER stress-induced GRP78-Akt interaction leads to inhibition of Akt phosphorylation at Ser-473 (36). Because Akt dephosphorylation was transiently observed at 30 min after TG treatment in our study (Fig. 5B), it might be possible that PTEN contributes to SIRT1 induction by ER stress through the suppression of Akt. It is also still unclear how GSK3β up-regulates SIRT1 expression. In silico analysis of human, mouse, and rat SIRT1 promoter using VISTA software revealed that the specific region (−157 to +150 bp) is highly conserved among species. There are two E-boxes (CACGTG) in the −157 to −47 promoter region. However, our preliminary investigation revealed that USF1, a protein that binds the E-box sequence, is dispensable in the up-regulation of SIRT1 by ER stress (data not shown). Future studies will focus on the role of other E-box-binding transcription factors in ER stress-induced SIRT1 expression.
SIRT1 is considered to accelerate cell survival because its expression in some cancers is increased and it inhibits the function of tumor suppressor p53 (9). However, recent reports revealed that SIRT1 expression is reduced in some types of cancer and that tumorigenesis is increased by SIRT1 deficiency. In addition, ectopic SIRT1 overexpression inhibits cancer formation in p53 or adenomatous polyposis coli mutant mice. Because SIRT1 inactivates tumor promoter NF-κB, survivin, and β-catenin, SIRT1 attenuates cell survival (8). Thus, the role of SIRT1 in cell survival and death is controversial and might depend on intracellular and extracellular conditions. In the present study, we showed that pharmacological inhibition of SIRT1 by EX527 inhibited TG-induced cell death (Fig. 6A). Wang et al. (13) reported that SIRT1 promotes TM-induced cellular apoptosis. Consistent with this finding, we also found that SIRT1 promotes ER stress-induced hepatic injury. Pharmacological inhibition of SIRT1 ameliorated cell death in vitro and hepatocellular injury in vivo (Fig. 6, A–E). Based on these data, we propose that SIRT1 acts as a negative regulator of the ER stress-induced cell-protective pathway. This hypothesis is also supported by a previous report showing that ER stress activates XBP1-dependent UPR target genes Edem1, Ero1α, and Sec61α to protect from cellular death; their expressions are enhanced by SIRT1 deficiency (13). In addition, SIRT1 deficiency prolonged global translational attenuation through the enhanced phosphorylation of eIF2-α that allows the induction of molecular chaperones and subsequently accelerates protein folding (12). Future studies will reveal the detailed mechanisms of how SIRT1 contributes to ER stress-induced hepatocellular injury. Although the deacetylating activity of SIRT1 on various proteins is relatively well known, its transcriptional regulation is less studied. In summary, our findings provide insight into the transcriptional regulation mechanism of SIRT1 in the context of ER stress response, confirm the regulatory link of ER stress and SIRT1, and contribute to our understanding on the role of SIRT1 in ER stress response.
Author Contributions
T. K., M. A. S., S. S., and H. K. designed the research. T. K., M. A. S., S. S., E. W., Y. K., K. K., and K. O. performed the experiments. K. M., S. H., and M. N. contributed new reagents and analytic tools. T. K., M. A. S., S. S., E. W., Y. K., S. M.-K., T. Sa., T. Sh., and H. K. analyzed the data. T. K,, M. A. S., and S. S, wrote the paper.
This work was supported by Ministry of Education, Science, Sports and Culture of Japan Grant 19390045, the Global Centers of Excellence Program (Cell Fate Regulation Research and Education Unit) (to H. K.), and Japan Society for the Promotion of Science Research Fellowship Grant-in-aid 20-8140 (to T. K.). The authors declare no conflict of interest.
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- ATF
- activating transcription factor
- IRE
- inositol-requiring enzyme
- XBP
- X-box-binding protein
- XBP1s
- spliced XBP1
- SIRT1
- Sirtuin 1
- QPCR
- quantitative RT-PCR
- CHOP
- CCAAT/enhancer-binding protein (C/EBP) homologous protein
- GRP78
- glucose-related protein 78 kDa
- ERdj4
- ER DnaJ homologue 4
- m
- mouse
- mCTGF
- mouse connective tissue growth factor
- TG
- thapsigargin
- TM
- tunicamycin
- PERK
- protein kinase RNA-like endoplasmic reticulum kinase
- PTEN
- phosphatase and tensin homologue.
References
- 1.Ellgaard L., and Helenius A. (2003) Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191 [DOI] [PubMed] [Google Scholar]
- 2.Hetz C., and Mollereau B. (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249 [DOI] [PubMed] [Google Scholar]
- 3.Xu C., Bailly-Maitre B., and Reed J. C. (2005) Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Investig. 115, 2656–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hetz C. (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 [DOI] [PubMed] [Google Scholar]
- 5.Hetz C., Chevet E., and Harding H. P. (2013) Targeting the unfolded protein response in disease. Nat. Rev. Drug. Discov. 12, 703–719 [DOI] [PubMed] [Google Scholar]
- 6.Jorgensen E., Stinson A., Shan L., Yang J., Gietl D., and Albino A. P. (2008) Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer 8, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Min T., Bodas M., Mazur S., and Vij N. (2011) Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J. Mol. Med. 89, 577–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng C. X. (2009) SIRT1, is it a tumor promoter or tumor suppressor? Int. J. Biol. Sci. 5, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raynes R., Brunquell J., and Westerheide S. D. (2013) Stress inducibility of SIRT1 and its role in cytoprotection and cancer. Genes Cancer 4, 172–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Houtkooper R. H., Pirinen E., and Auwerx J. (2012) Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang C., Chen L., Hou X., Li Z., Kabra N., Ma Y., Nemoto S., Finkel T., Gu W., Cress W. D., and Chen J. (2006) Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 8, 1025–1031 [DOI] [PubMed] [Google Scholar]
- 12.Ghosh H. S., Reizis B., and Robbins P. D. (2011) SIRT1 associates with eIF2-α and regulates the cellular stress response. Sci. Rep. 1, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F. M., Chen Y. J., and Ouyang H. J. (2011) Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation. Biochem. J. 433, 245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang F. M., Galson D. L., Roodman G. D., and Ouyang H. (2011) Resveratrol triggers the pro-apoptotic endoplasmic reticulum stress response and represses pro-survival XBP1 signaling in human multiple myeloma cells. Exp. Hematol. 39, 999–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herranz D., and Serrano M. (2010) SIRT1: recent lessons from mouse models. Nat. Rev. Cancer 10, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen D., Bruno J., Easlon E., Lin S. J., Cheng H. L., Alt F. W., and Guarente L. (2008) Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 22, 1753–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purushotham A., Schug T. T., Xu Q., Surapureddi S., Guo X., and Li X. (2009) Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9, 327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y., Xu S., Giles A., Nakamura K., Lee J. W., Hou X., Donmez G., Li J., Luo Z., Walsh K., Guarente L., and Zang M. (2011) Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 25, 1664–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haze K., Yoshida H., Yanagi H., Yura T., and Mori K. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida H., Matsui T., Yamamoto A., Okada T., and Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 21.Yoshida H., Uemura A., and Mori K. (2009) pXBP1(U), a negative regulator of the unfolded protein response activator pXBP1(S), targets ATF6 but not ATF4 in proteasome-mediated degradation. Cell Struct. Funct. 34, 1–10 [DOI] [PubMed] [Google Scholar]
- 22.Shuto T., Furuta T., Oba M., Xu H., Li J. D., Cheung J., Gruenert D. C., Uehara A., Suico M. A., Okiyoneda T., and Kai H. (2006) Promoter hypomethylation of Toll-like receptor-2 gene is associated with increased proinflammatory response toward bacterial peptidoglycan in cystic fibrosis bronchial epithelial cells. FASEB J. 20, 782–784 [DOI] [PubMed] [Google Scholar]
- 23.Koga T., Lim J. H., Jono H., Ha U. H., Xu H., Ishinaga H., Morino S., Xu X., Yan C., Kai H., and Li J. D. (2008) Tumor suppressor cylindromatosis acts as a negative regulator for Streptococcus pneumoniae-induced NFAT signaling. J. Biol. Chem. 283, 12546–12554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koga T., Suico M. A., Nakamura H., Taura M., Lu Z., Shuto T., Okiyoneda T., and Kai H. (2005) Sp1-dependent regulation of myeloid Elf-1 like factor in human epithelial cells. FEBS Lett. 579, 2811–2816 [DOI] [PubMed] [Google Scholar]
- 25.Taura M., Fukuda R., Suico M. A., Eguma A., Koga T., Shuto T., Sato T., Morino-Koga S., and Kai H. (2010) TLR3 induction by anticancer drugs potentiates poly I:C-induced tumor cell apoptosis. Cancer Sci. 101, 1610–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto K., Takahara K., Oyadomari S., Okada T., Sato T., Harada A., and Mori K. (2010) Induction of liver steatosis and lipid droplet formation in ATF6α-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 21, 2975–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medina M., and Wandosell F. (2011) Deconstructing GSK-3: the fine regulation of its activity. Int. J. Alzheimers Dis. 2011, 479249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cole A. R., Knebel A., Morrice N. A., Robertson L. A., Irving A. J., Connolly C. N., and Sutherland C. (2004) GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 279, 50176–50180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viswanathan M., Kim S. K., Berdichevsky A., and Guarente L. (2005) A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev. Cell 9, 605–615 [DOI] [PubMed] [Google Scholar]
- 30.Jung T. W., Lee K. T., Lee M. W., and Ka K. H. (2012) SIRT1 attenuates palmitate-induced endoplasmic reticulum stress and insulin resistance in HepG2 cells via induction of oxygen-regulated protein 150. Biochem. Biophys. Res. Commun. 422, 229–232 [DOI] [PubMed] [Google Scholar]
- 31.Winnay J. N., Boucher J., Mori M. A., Ueki K., and Kahn C. R. (2010) A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 16, 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winnay J. N., Dirice E., Liew C. W., Kulkarni R. N., and Kahn C. R. (2014) p85α deficiency protects β-cells from endoplasmic reticulum stress-induced apoptosis. Proc. Natl. Acad. Sci. U.S.A. 111, 1192–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hosoi T., Hyoda K., Okuma Y., Nomura Y., and Ozawa K. (2007) Akt up- and down-regulation in response to endoplasmic reticulum stress. Brain Res. 1152, 27–31 [DOI] [PubMed] [Google Scholar]
- 34.Qin L., Wang Z., Tao L., and Wang Y. (2010) ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 6, 239–247 [DOI] [PubMed] [Google Scholar]
- 35.Zhang W., Neo S. P., Gunaratne J., Poulsen A., Boping L., Ong E. H., Sangthongpitag K., Pendharkar V., Hill J., and Cohen S. M. (2015) Feedback regulation on PTEN/AKT pathway by the ER stress kinase PERK mediated by interaction with the Vault complex. Cell Signal 27, 436–442 [DOI] [PubMed] [Google Scholar]
- 36.Yung H. W., Charnock-Jones D. S., and Burton G. J. (2011) Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS One 6, e17894. [DOI] [PMC free article] [PubMed] [Google Scholar]