

Abstract
MHC class I-restricted epitopes, which carry a tumor-specific mutation resulting in improved MHC binding affinity, are preferred T cell receptor targets in innovative adoptive T cell therapies. However, T cell therapy requires efficient generation of the selected epitope. How such mutations may affect proteasome-mediated antigen processing has so far not been studied. Therefore, we analyzed by in vitro experiments the effect on antigen processing and recognition of a T210M exchange, which previously had been introduced into the melanoma gp100209–217tumor epitope to improve the HLA-A*02:01 binding and its immunogenicity. A quantitative analysis of the main steps of antigen processing shows that the T210M exchange affects proteasomal cleavage site usage within the mutgp100201–230 polypeptide, leading to the generation of an unique set of cleavage products. The T210M substitution qualitatively affects the proteasome-catalyzed generation of spliced and non-spliced peptides predicted to bind HLA-A or -B complexes. The T210M substitution also induces an enhanced production of the mutgp100209–217 epitope and its N-terminally extended peptides. The T210M exchange revealed no effect on ERAP1-mediated N-terminal trimming of the precursor peptides. However, mutant N-terminally extended peptides exhibited significantly increased HLA-A*02:01 binding affinity and elicited CD8+ T cell stimulation in vitro similar to the wtgp100209–217 epitope. Thus, our experiments demonstrate that amino acid exchanges within an epitope can result in the generation of an altered peptide pool with new antigenic peptides and in a wider CD8+ T cell response also towards N-terminally extended versions of the minimal epitope.
Keywords: antigen presentation, antigen processing, mutant, protein degradation, ubiquitin-dependent protease
Introduction
Protein degradation by the ubiquitin-proteasome system plays an important role in regulating cellular protein homeostasis. Concomitant with the degradation of proteins, the 20S proteasome, which is the catalytic core of the ubiquitin-proteasome system, generates peptides of 8–12 amino acids in length or N-terminally extended precursor peptides that, after trimming by endoplasmic reticulum (ER)3 resident amino peptidases (ERAPs), can bind MHC class I molecules in the ER to be presented at the cell surface to CD8+ T cells for immune recognition. These antigenic peptides can be produced by the proteasome by normal peptide-bond hydrolysis or by proteasome-catalyzed peptide splicing. The latter reaction generates peptides that have a different sequence than the original antigen, and it can occur by binding fragments of a single molecule (cis proteasome-catalyzed peptide splicing) or of two distinct molecules (trans proteasome-catalyzed peptide splicing) (1–4).
In vitro experiments performed with purified 20S proteasomes were shown to closely reflect the in vivo situation, making it an ideal platform to study the generation of non-spliced and spliced antigenic peptides in vitro (1, 5–11). Under ideal conditions the 20S proteasome exists in two isoforms, i.e. the standard 20S proteasome (s-proteasomes) with the active site subunits β1, β2, and β5 and the 20S immunoproteasomes (i-proteasomes) with the inducible active site subunits β1i, β2i, and β5i. Constitutive expression of true i-proteasomes appears to be restricted to a small number of mainly immune cells like B or T cells. In contrast, the expression of so-called intermediate-type proteasomes containing both standard- and immuno-active subunits appears to be more frequent. Intermediate-type proteasomes are expressed in most tumor cells and in many tissues of the human body under normal physiological nutrition and growth conditions (12). It has been recently shown that the active subunit composition of 20S proteasomes in principle does not affect the quality of proteasome-generated peptides (5, 13, 14). Nevertheless, proteasomal subunit composition can strongly affect cleavage site usage within a given substrate and hence the relative quantity of non-spliced or spliced peptides produced. Such quantitative differences in the generation of cleavage products can strongly affect cell surface presentation of MHC class I-peptide complexes and in consequence the efficacy of a peptide-specific CD8+ T cell response (5, 13–15).
Although sequence requirements for proteasomal cleavage site usage are difficult to predict, there exists frequent evidence that seemingly minor alteration in the primary sequence of a protein substrate can have an impact on proteasomal processing and thereby positively or negatively affecting the liberation of antigenic peptides and concomitantly the CD8+ T cell-dependent immune response (7, 10, 16). Mutations flanking the C-terminal residue of an antigenic peptide were shown to infer negatively as well as positively with the generation and presentation of the respective epitopes (17–20). There exist also examples of amino acid exchanges occurring within an epitope sequence that introduce a strong proteasomal cleavage site and that consequently leads to a suppression of epitope generation (16, 21). With respect of innovative adoptive T cell therapies, tumor-specific mutated epitopes with enhanced MHC class I binding affinity are of particular interest and are used for the cloning of tumor-specific T cell receptors for T cell therapy (22). Also, vaccination against the tumor with longer polypeptides requiring proteasomal processing has been shown to increase the anti-tumor immune response (23).
Although the success of T cell therapies strongly depends on efficient proteasomal processing of such mutant epitopes, almost no information exists on how such amino acid exchanges within a tumor epitope, which enhance binding affinity to the MHC class I molecules, affect proteasomal processing. We, therefore, analyzed with the help of in vitro experiments the effect on proteasome-mediated antigen processing of a T210M substitution, which was introduced into the melanoma gp100209–217 tumor epitope at the HLA-A*02:01 binding anchor position to improve its immunogenicity (24). This first comprehensive quantitative study of in vitro tumor antigen generation and presentation revealed that the T210M substitution within the mutgp100201–230 polypeptide substrate induces alteration in proteasomal cleavage site usage, epitope presentation, and recognition by CD8+ T cells. It also leads to the generation of an altered antigenic peptide pool, which may be of particular relevance when longer synthetic polypeptides are used for tumor vaccination (23).
Experimental Procedures
Cell Cultures
Lymphoblastoid cell lines are human B lymphocytes immortalized with Epstein-Barr virus. The T2 cell line is a human T cell leukemia/B cell line hybrid defective in TAP1/TAP2 (transporter associated with antigen presentation) and β1i/β5i subunits. The T2 cell line contains only standard proteasome (s-proteasomes), whereas the lymphoblastoid cell line possesses proteasomes carrying mainly immuno-subunits, and it is named here as immunoproteasome (i-proteasome) (14, 25).
Peptides and Peptide Synthesis
The sequence enumeration for the polypeptides gp100201–230 (AHSSSAFTITDQVPFSVSVSQLRALDGGNK) and mutgp100201–230 (AHSSSAFTIMDQVPFSVSVSQLRALDGGNK) is referred to the human protein gp100PMEL17. All peptides were synthesized using Fmoc (N-(9-fluorenyl)methoxycarbonyl) solid phase chemistry as previously described (26). The purity of synthetic peptides was tested by amino acid analysis (3).
20S Proteasome Purification
20S proteasomes were purified from T2 and the lymphoblastoid cell lines as previously described (26). Purity of the preparation is depicted in Fig. 8.
FIGURE 8.
Purity of 20S proteasome preparation. 20S proteasomes purified from T2 and lymphoblastoid (LcL) cell lines (1 μg) were separated by SDS-PAGE and stained by Coomassie Blue to detect possible contaminations. Only the characteristic bands of 20S proteasome, with sizes between 21 and 31 kDa, are visible, confirming the high purity of the proteasome preparation. For instance, no contamination with Hsp molecules at high molecular mass (above 60 kDa), which are often present in less pure proteasome preparations, could be detected.
In Vitro Digestion of Synthetic Peptides by 20S Proteasomes and Recombinant ERAP1
Synthetic polypeptides (40 μm) were digested by 3 μg of 20S proteasomes in 100 μl of TEAD buffer (20 mm Tris, 1 mm EDTA, 1 mm NaN3, 1 mm DTT, pH 7.2) over time (0–4 h) at 37 °C.
50 μm concentrations of the peptides wtgp100209–217, wtgp100208–217, wtgp100207–217, wtgp100206–217, wtgp100205–217,mutgp100209–217, mutgp100208–217, and mutgp100207–217 were digested in vitro by 2.5 ng of recombinant ERAP1 (recombinant human aminopeptidase PILS/ALAP/ERAP1; R&D Systems) in 20 μl of assay buffer (25 mm Tris, pH 7.5, 150 mm NaCl, 0.5 μg/ml albumin) at 37 °C over time. To test the ERAP1 dependence of the reaction, 3 ng of recombinant ERAP1 were incubated in buffer in the presence of 30 μm leucinethiol (27) at room temperature for 20 min before co-incubation with the gp100 synthetic peptides. A reduction of 60% of ERAP1 activity was observed (data not shown).
Quantification of Peptides and Substrate Site-specific Cleavage Strength (SCS) Computation
Liquid chromatography mass spectrometry (LC-MS) analyses of polypeptide digestion products were performed as previously described (28) by the ESI-ion trap instrument DECA XP MAX (ThermoFisher Scientific). Database searching was performed using SpliceMet's ProteaJ, which allowed the identification of spliced and non-spliced peptide products (28). Quantification of proteasome-generated non-spliced and spliced peptides and computation of the SCS was carried out by applying the QME (Quantification with Minimal Effort) method to the LC-MS analyses (3). QME estimates the absolute content of spliced and non-spliced peptide products based on their MS ion strength measured in the digestion probe. QME is an optimization tool that makes use of the law of mass conservation and MS instrument features. For example it considers the dependence of the MS ion strength of a given peptide on the peptide sequence and length. The QME algorithm parameters were empirically computed in our previous study (3) and were applied here. SCS describes the relative frequencies of proteasome cleavage after any given residue of the synthetic polypeptide substrates (3). The SCS value showed in this study are the average of SCS measured over time (3).
In the in vitro digestion of the gp100-derived peptides by recombinant ERAP1 we did not apply QME because the low molecular N-terminal trimmed peptides cannot be detected by LC-ESI-MS. Therefore, we performed a canonical titration of the digestion products using synthetic peptides as previously described (28). In particular, synthetic peptides were mixed with 15 ng of inactive recombinant ERAP1 in 100 μl of digestion buffer, their precursor ion intensity was measured in triplicate by LC-MS, and the derived linear titration curves were used to calculate the pmol of trimmed fragments in the in vitro ERAP1 digestion over time.
MHC Class I-Peptide Complex Binding Affinity and Stability
Stability of the HLA-A*02:01-peptide complex was determined by pulsing of 105 T2 cells (HLA-A*02:01+) for 16 h by synthetic peptides at concentrations exhibiting the largest binding efficiency, i.e. 100 μm (data not shown). Afterward, cells were washed twice, and the expression of the HLA-A*02:01 at the cell surface was measured over time by FACS analysis using FITC mouse anti-human HLA-A*02 antibody (1:100, BD Bioscience). The stability of the complex MHC-epitope is expressed by its half-life (t½) at the cell surface and was calculated as t½ = ln(2)/k. k was estimated by minimization of the equation C = C0 exp(−kt), where C is the signal over time, C0 is the signal measured at time 0, and t is the time. Therefore, k describes the rate of the exponential decay of the complex MHC-epitope.
The binding affinity of the epitope wtgp100213–218/220–222 to the HLA-B*07:02 complex was measured in classical competition binding assays utilizing purified MHC and a high affinity radiolabeled ligand, as previously described (29). Under the conditions utilized for these assays, where [label] < [MHC] and IC50 ≥ [MHC], the measured IC50 values represent reasonable approximations of true Kd values (30). Each competitor peptide was tested in 3 or more independent assays at 6 different concentrations covering a 100,000-fold dose range.
The binding affinity of the whole pool of 8–12-mer spliced or non-spliced peptides identified in the proteasome digestions were evaluated for their predicted binding affinity to the most frequent HLA-A and -B haplotypes (i.e. HLA-A*01:01, -A*02:01, -A*03:01, -A*24:02, -A*26:01, -B*07:02, -B*08:01, -B*27:05, -B*39:01, -B*40:01, -B*58:01, -B*15:01) by using the ANN-NetMHC prediction software version 3.4 (31). In the analysis we included all 8–14-mer peptides produced by proteasome and allowed only N-terminal trimming of the peptides to generate the binder peptides. Because the high inter-experimental variability of the MHC class I- peptide stability assay depended on technical factors (the same antigen presenting cells (APCs) and peptides are used in all assays), we reported in Fig. 6A the S.E. instead of the S.D.
FIGURE 6.

Presentation to and recognition by CD8+ T cells of the gp100209–217 epitope and its N-terminally extended peptides is affected by the T210M substitution. A, binding to the HLA-A*02:01 molecule; the stability of the complex MHC class I bound to the wt and mut gp100207–217, gp100208–217, and gp100209–217 peptides is shown. The MHC class I-peptide stability was measured by pulsing the synthetic peptides onto T2 cells (expressing the HLA- A*02:01 molecule) over time and by assessing the HLA-A*02:01 expression on the cell surface by FACS analysis. The t½ of the HLA-A*02:01-peptide is reported in the panel. Values are the mean, and bars are the S.E. of two independent experiments measured in duplicate. FU, fluorescence unit. B, CTL response; IFN-γ release by gp100209–217-specific PBLs triggered by HLA-A*02:01-expressing K652 cells pulsed with synthetic peptides. The EC50 (concentration of peptide triggering half of the measured maximal PBL response) is reported in the right panel. Values are the means, and bars are the S.E. of independent experiments (n = 5–16) measured in duplicate.
CTL Assay
CTL assays were performed with peripheral blood lymphocytes (PBLs) of a healthy donor transduced with gp100mel209–217 epitope-specific T cell receptor, which was generated as described elsewhere.4 As APCs, we used the K652 cell line transduced with HLA-A*02:01, which does not express other HLA complexes. APCs were pulsed for 2 h at 4 °C with the synthetic peptides, washed twice, and co-cultivated with the transduced PBLs for 16 h. The specific response of transduced PBLs against APCs pulsed with different concentrations of the epitopes were measured by IFN-γ release, which was determined using a commercially available human ELISA kit (BD Biosciences) according to the manufacturer's instructions. The EC50 (concentration of peptide triggering half of the measured maximal PBL response) was computed by linear interpolation of the values IFN-γ versus peptide amount (Fig. 6B) in proximity of half of the measured maximal PBL response. Because the high inter-experimental variability of the IFN-γ release absolute amount in the CTL assay depended on technical factors (the same CTL clone and APCs are used in all assays), we reported in Fig. 6B the S.E. instead of the S.D.
Results
A Single Thr/Met Substitution in the Polypeptide gp100201–230Affects Cleavage Site Usage
Assessment of the impact of a mutation within an antigen on its cleavage-site usage by 20S proteasomes requires a method permitting comparison of the quality and quantity of the digestion products derived from both wild type (wt) and mutated (mut) synthetic gp100201–230 polypeptides. Previous approaches considered only a portion of the entire product pool and used the MS fragment ionization intensity as a surrogate marker for quantity (16). However, this approach is not applicable when peptides of different lengths are analyzed (3). To solve these caveats and limitations we applied the QME method for quantitative analysis (3). QME allows quantification of the amount of each product of a proteasome digestion identified by MS analysis and computation of the cleavage site usage, i.e. the SCS. Importantly, QME features also allow minimizing the impact of the difference in MS ionization of wt and mut peptides in determining their amount in the digestion sample (see “Experimental Procedures”). A mutation introduced in an antigen sequence might change the quality of the peptide pool generated upon proteasome digestion of the antigen. In addition, the digestion products generated by cleavage at the same sites of the wt and mut substrates may differ in their sequence if the mutation is within the substrate cleavage sites (Fig. 1). Both aspects might temper the correct quantitation of the SCS and of the product's amount by QME and were thus tested.
FIGURE 1.

gp100 T210M substitution alters the product peptide pool. Schematic representation of the number of common or specific peptides generated by 20S proteasomes by cleaving in vitro the synthetic substrates wtgp100201–230 or mutgp100201–230.
In fact, upon processing of the synthetic wtgp100201–230 or mutgp100201–230 polypeptides by 20S s- and i-proteasomes, we identified 94 peptide products (10 spliced peptides) and 93 products (5 spliced peptides), respectively (Fig. 1). In agreement with our previous results (14), the peptide pools produced by both s- or i-proteasomes perfectly overlapped with respect to numbers and sequences of the products (data not shown). 56 products (2 spliced peptides, i.e. gp100201–204/201–209 and gp100205–210/214–217) were generated from both wt and mut substrates. Hence, the T210M substitution led to a variation of the pool of in vitro generated peptides by 20S proteasomes of ∼40% (Fig. 1).
For initial validation of our quantitative approach we applied QME to the MS dataset containing only the 56 peptides common to wt and mut substrate digestions. We then compared the resulting SCS with the SCS computed by applying QME to the full peptide datasets of either the wtgp100201–230 and mutgp100201–230 substrate digestion. By adopting this strategy we could understand whether the difference in the substrate cleavage-site usage introduced by the T210M substitution depended only on the peptides specific to wt and mut substrates. The wt and mut substrate SCSs computed by applying QME to full peptide (i.e. 94 and 93 peptides) or to the common peptide (i.e. 56 peptides) datasets did not dramatically differ (Fig. 2, A and B). This indicates that the peptides common to wtgp100201–230 and mutgp100201–230 substrate digestions were the most frequently generated peptides and represent the core of the peptide pools. The analysis of the quantitative kinetics of representative peptides verified that the QME calculation was remarkably unbiased by the size of the MS datasets compared in this study. These peptides, one long product (26-mer) and two 9-mers located at the N and C termini of the substrates, were chosen to test potential bias of QME calculation for peptides of different length or position within the wt and mut gp100 substrates (Fig. 2C). Thus, all controls supported the use of QME on the wt and mut peptide datasets to evaluate the effect of the substitution.
FIGURE 2.

Product peptides specific for wt or mut gp100201–230 substrate degradation only marginally influence the substrate cleavage-site usage by proteasome. The SCSs of the wt or mut gp100201–230 substrate computed by QME considering the full products pool or only the peptides common to both substrates is shown. The analysis is performed for both s- (A) and i- (B) proteasome digestions. Histograms represent the mean of four time points, and bars represent the S.D. of three independent experiments measured in duplicate. C, quantitative kinetics of the representative peptide products gp100201–209, gp100201–226, and gp100221–230 calculated by applying QME to the full or the common peptide pools of the wt and mut gp100201–230 substrate digestion. No major difference in the absolute amount of each peptide was observed between the analysis done with full or the common peptide pools. It proves that the QME method can be applied to this study and does not introduce artifacts due to the difference in the database size. The peptides represent the N- and C-terminal area of the substrates (gp100201–209 and gp100221–230) and two different peptide sizes (gp100201–226 versus gp100201–209 and gp100221–230). We chose these peptides as controls because both the location and the length of the peptide products are relevant in the QME computation of the peptide absolute amount (3). Only the results obtained from s-proteasome-mediated degradation are shown.
When we compared the SCSs based on the common peptide datasets of the wtgp100201–230 and mutgp100201–230 substrate digestions, we observed that the T210M substitution strongly altered the frequency of cleavage after several substrate residues even distant to the substitution site such as the sites His-202, Val-217, Leu-222, and Leu-225 (Fig. 3). Of note, the T210M substitution did not significantly altered the preference of s- or i-proteasomes for specific substrate sites (Fig. 4) previously identified (14).
FIGURE 3.

gp100 T210M substitution alters the substrate cleavage-site usage by 20S proteasomes. The SCS by s- (A) and i- (B) proteasomes of the synthetic substrates wtgp100201–230 or mutgp100201–230. SCS was computed by QME from kinetics experiments. Histograms represent the mean of four time points, and bars represent the S.D. of three independent experiments measured in duplicate.
FIGURE 4.

gp100 T210M mutation does not impinge upon the s- and i-proteasome usage of the preferential substrate cleavage sites. The SCSs of the synthetic wtgp100201–230 (A) and mutgp100201–230 (B) substrates by s- and i- proteasomes is compared. The cleavage sites that we previously described as significantly more used by i-proteasome (i.e. gp100 Ile-209, Val-219, and Leu-222) or s-proteasome (i.e. gp100 Val-213, Ser-220, and Asp-226) (14) are consistently preferred during the digestion of wt or mut gp100201–230 substrates. SCSs are computed by applying QME on kinetics experiments. Histograms represent the mean of four time points, and bars represent the S.D. of three independent experiments measured in duplicate.
gp100201–230 T210M Substitution Affects the Generation of Non-spliced Antigenic Peptides
Our study also showed that the T210M substitution altered not only the substrate cleavage-site usage but also the quality and quantity of the potential MHC class I-restricted pool of antigenic peptides by affecting their kinetics and/or their overall generation (Table 1). For instance, the synthetic substrate wtgp100201–230 contains the well known HLA-A*02:01-restricted wtgp100209–217 epitope (24). As observed before for the wtgp100209–217 epitope (14, 32), the mutgp100209–217 epitope was also not generated by the 20S proteasome as such but was generated as N-terminally extended precursor peptides (data not shown). In fact, from the wtgp100201–230 substrate we obtained the N-terminally extended peptides wtgp100208–217, wtgp100207–217, wtgp100206–217,and wtgp100205–217, whereas from the mutgp100201–230 substrate the elongated peptides mutgp100208–217 and mutgp100207–217 were produced by the 20S proteasome (Fig. 5A). Only the generation kinetics of the wtgp100206–217 peptide was influenced by the proteasome isoform used in the assay (Fig. 5A).
TABLE 1.
8–12-mer peptides that potentially bind the most frequent HLA-A and -B haplotypes and are produced by proteasome-catalyzed digestion of the wt and/or mut gp100201–230 polypeptides
The non-spliced and spliced 8–12-mer peptides shown were produced by proteasomal processing of the wt and/or mut gp100201–230 polypeptides as such or as N-terminally extended precursors. Binding affinities of the peptides were predicted in silico by using ANN-NetMHC software version 3.4 (31). We included in the analysis the most frequent HLA-A and -B haplotypes (i.e., HLA-A*01:01, -A*02:01, -A*03:01, -A*24:02, -A*26:01, -B*07:02, -B*08:01, -B*27:05, -B*39:01, -B*40:01, -B*58:01, -B*15:01). Only 8–14-mer peptides were used for the in silico analysis, and only N-terminally extended peptides were considered as MHC class I binder precursors (i.e. the binder and the precursor peptides must have the same C terminus). Only 8–12-mer peptides with predicted IC50 values <200 nm were considered as efficient binders. In the peptide sequence the T210M is marked in bold. When both wt and mut peptides were produced by proteasome and predicted to be MHC class I binders, we reported the wt sequence and the substitution as following T(M). When both wt and mut peptides had a IC50 < 200 nm we stated whether the IC50 was affected by the T210M substitution.
| Precursors (gp100_) | Binder (gp100_) | Sequence | HLA- | IC50 influenced by T210M substitution? |
|---|---|---|---|---|
| 8–12-Mer peptide binders common to wt and mut gp100201–230-produced peptide pool | ||||
| 201–209 | 202–209 | HSSSAFTI | B*58:01 | No |
| 208–217 | 209–217 | IT(M)DQVPFSV | A*02:01 | Yes |
| 201–209 | 201–209 | AHSSSAFTI | B*39:01 | No |
| 205–215 | 207–215 | FTIT(M)DQVPF | B*15:01, B58:01 | Yes |
| 208–217 | 208–217 | TIT(M)DQVPFSV | A*02:01 | Yes |
| 207–217 | 207–217 | FTIT(M)DQVPFSV | A*02:01 | Yes |
| 205–215 | 205–215 | SAFTIT(M)DQVPF | B*15:01 | Yes |
| 8–12-Mer peptide binders unique for the wtgp100201–230-produced peptide pool | ||||
| 208–219 | 208–219 | TITDQVPFSVSV | A*02:01 | |
| 210–218/220–222 | 213–218/220–222 | VPFSVSSQL | B*07:02 | |
| 201–207/201–207 | 207/201–207 | FAHSSSAF | B*15:01 | |
| 201–207/201–207 | 206–207/201–207 | AFAHSSSAF | B*15:01 | |
| 201–207/201–207 | 205–207/201–207 | SAFAHSSSAF | B*15:01 | |
| 201–207/201–207 | 204–207/201–207 | SSAFAHSSSAF | B*15:01 | |
| 201–207/201–207 | 203–207/201–207 | SSSAFAHSSSAF | B*58:01 | |
| 210–212/214–222 | 212/214–222 | QPFSVSVSQL | B*39:01 | |
| 8–12-Mer peptide binders unique for the mutgp100201–230-produced peptide pool | ||||
| 205–215 | 208–215 | TIMDQVPF | B*15:01 | |
| 201–204/201–207 | 204/201–207 | SAHSSSAF | B*15:01 | |
| 201–204/201–207 | 203–204/201–207 | SSAHSSSAF | B*15:01 | |
| 201–204/201–207 | 202–204/201–207 | HSSAHSSSAF | B*15:01 | |
FIGURE 5.

gp100 T210M substitution alters the generation of the gp100209–217 epitope and its N-terminally extended peptides. A, proteasome-mediated generation; quantitative kinetics of the wtgp100201–230 and mutgp100201–230 substrate degradation and of the production of the peptides wt gp100205–217 and gp100206–217 as well as the wt and mut gp100207–217 and gp100208–217 peptides from the carried out by 20S s- and i-proteasomes. Peptide amount values are the means, and bars are the S.D. of three independent in vitro experiments. B, ERAP1-mediated trimming; quantitative kinetics of the substrates wtgp100205–217 and wtgp100206–217 (upper panels) or wt and mut gp100207–217 and gp100208–217 (lower panels) trimmed by recombinant ERAP1. Peptide amount values are the means, and bars are the S.D. of three independent in vitro experiments.
Although the substrate degradation rate was not affected by the T210M substitution (Fig. 5A), the T210M substitution affected the production of the N-terminally extended precursors of the epitope mutgp100209–217. In comparison to the wild type counterparts the Thr/Met residue exchange abolished the production of the peptides mutgp100206–217 and mutgp100205–217and improved the generation of the peptides mutgp100208–217and mutgp100207–217 regardless of which proteasome isoform was used in the assay (Fig. 5A). We can exclude that the peptides mutgp100206–217 and mutgp100205–217 were not detected due to a sequence-dependent lower MS ion signal of the mutated peptides because the intensity of the MS ion signals of the synthetic wt and mut peptides were comparable (data not shown).
Theoretically, all the identified N-terminally extended precursors peptides may be further trimmed by ERAPs to produce the epitope. As shown in Fig. 5B, in vitro the trimming of the N-terminally extended peptides wtgp100208–217, wtgp100207–217,wtgp100206–217, and wtgp100205–217 as well as mutgp100208–217 and mutgp100207–217 was catalyzed by recombinant ERAP1. Trimming by ERAP1 stopped when the minimal antigenic peptides wtgp100209–217 and mutgp100209–217 were generated (data not shown). No effects of the T210M substitution were observed in the ERAP1-dependent kinetics of peptide generation (Fig. 5B).
Previous studies focused on MHC class I-restricted epitopes documented antigenic peptides longer than the canonical 8–9-mers presented onto MHC class I complexes and able to elicit CD8+ T cell response (33–36). Thus, the N-terminally extended peptides of the epitopes wtgp100209–217 and mutgp100209–217 may not only be precursors of the 9-mer epitopes but also exhibit sufficient affinity by themselves to bind the HLA-A*02:01 complex. To test this hypothesis, we measured in vitro the stability of the HLA-A*02:01-peptide complex of the two wt and mut gp100207–217 (11-mer) and gp100208–217 (10-mer) precursor peptides generated by the 20S proteasome and of the wt and mut gp100209–217 9-mer epitopes produced by ERAP1-catalyzed trimming. Both wt and mut gp100207–217 and gp100208–217 peptides exhibited sufficient affinity for binding the HLA-A*02:01 molecules, although the formed MHC class I-peptide complexes were less stable than those formed with the 9-mer peptides (Fig. 6A). As expected, the stability of the HLA-A*02:01-peptide complex was considerably improved by the T210M substitution. In fact, theThr/Met exchange enhanced the binding of the 11-mer mutgp100207–217 and 10-mer mutgp100208–217 peptides that was more stable than the ones formed with the wtgp100209–217 epitope (Fig. 6A).
These data suggest that the proteasome-generated N-terminally extended peptides may not only be a source for the production of the known epitopes wtgp100209–217 and mutgp100209–217 but might also be able to elicit a CTL response. To test this hypothesis, we performed CTL assays with gp100209–217-specific CD8+ T cells exposed to HLA-A*02:01+ APCs pulsed with different concentrations of the synthetic wt and mut gp100207–217 11-mer, gp100208–217 10-mer, and gp100209–217 9-mer peptides. In agreement with the MHC class I-peptide stability assays, the wtgp100209–217 and mutgp100209–217epitopes triggered a stronger CTL response than their N-terminally extended versions, although the mutgp100208–217 and mutgp100207–217 epitopes elicited a CTL response similar or even stronger than the wtgp100209–217 epitope (Fig. 6B).
gp100201–230 T210M Impinges upon the Generation of Spliced Epitope Precursors
The second group of peptides of interest are the spliced peptides, which are generated by proteasome-catalyzed peptide splicing, whose immunological relevance has been emerging in the past years. We found that the T210M substitution strongly affected the generation of such peptides; 8 out of 10 spliced peptides were generated only from the wtgp100201–230 substrate, whereas 3 out of 5 spliced peptides were generated exclusively from the mutgp100201–230 substrates (Fig. 1). Among the spliced peptides only generated from the wtgp100201–230 substrate, five were potential precursors of predicted HLA-A or -B binders (predicted IC50 < 200 nm). For instance, the peptides gp100210–218/220–222 and gp100210–212/214–222 were produced by cis proteasome-catalyzed peptide splicing; the peptide gp100210–218/220–222 was trimmed in vitro by ERAP1, thereby generating the antigenic peptide gp100213–218/220–222[VPFSVS][SQL] (Fig. 7, A and B), which efficiently binds the HLA-B*07:02 complex (IC50, 28 nm; Fig. 7C); the peptide gp100210–212/214–222 might be trimmed up to produce the peptide gp100212/214–222, which was predicted to efficiently bind the HLA-B*39:01 complex (Table 1). In contrast, the spliced peptides gp100201–207/201–207 and gp100201–209/201–207, both representing potential precursors of the HLA-B*15:01-binding antigenic peptides gp100207/201–207 (and N-extended variants) and gp100209/201–207 (and N-extended variants), respectively, were produced by trans proteasome-catalyzed peptide splicing (Table 1 and data not shown). However, because trans proteasome-catalyzed peptide splicing seems to occur with low efficiency in cells (1), these spliced peptides appear to be less relevant from the immunological point of view. The same applies for the trans spliced peptide gp100201–204/201–207,i.e. the precursor of the antigenic peptide gp100203–204/201–207,which is specific for the substrate mutgp100201–230 (Table 1). The purity of the 20S proteasome preparation (Fig. 8) excluded that the peptide splicing reaction observed in our assays could be catalyzed by other proteases.
FIGURE 7.
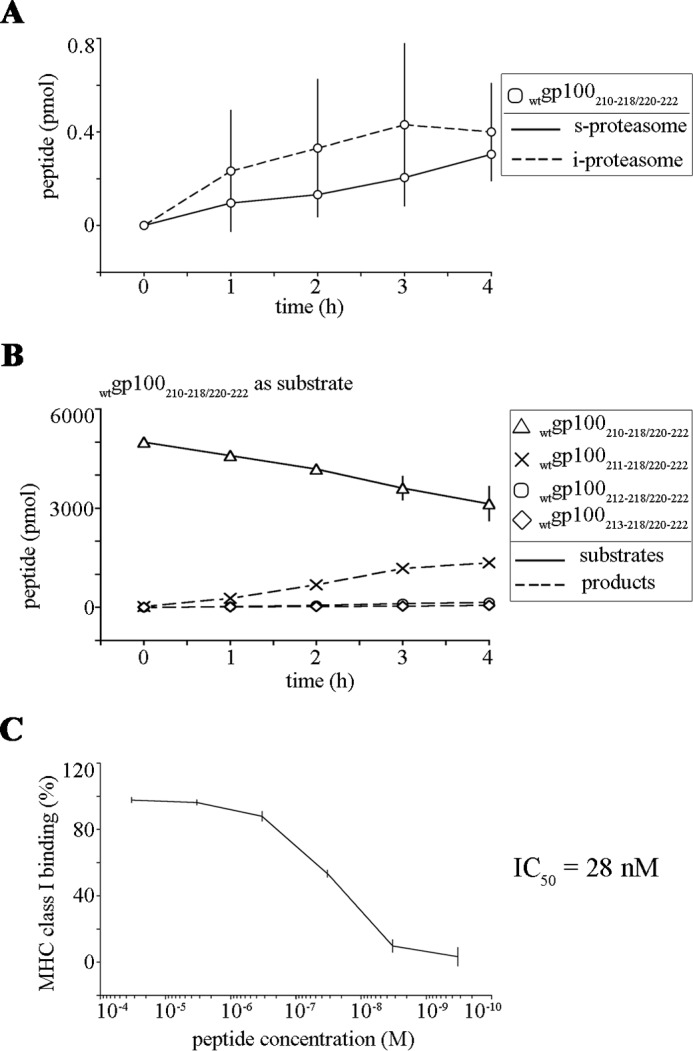
Quantitative kinetics displaying the processing of the spliced peptide gp100210–218/220–222 by 20S proteasomes and ERAP1. A, proteasome-mediated generation kinetics of the peptide gp100210–218/220–222 from the wt and mut gp100201–230 substrate digestions carried out by 20S s- and i-proteasomes. Peptide amount values are the means, and bars are the S.D. of three independent in vitro experiments. B, ERAP1 trimming kinetics of the substrate wtgp100210–218/220–222. No products shorter than the HLA-B*07:02-restricted epitope wtgp100213–218/220–222 were detected in the assay. Peptide amount values are the means, and bars are the S.D. of three independent in vitro experiments. C, binding to the HLA-B07:02 molecule by the epitope wtgp100213–218/220–222 (IC50 = 28 nm).
Discussion
Increasing evidence suggests that tumor-specific somatic mutations enhancing the MHC class I binding affinity of an antigenic peptide are potentially the best targets for adoptive T cell therapy of established tumors (22). However, factors like processing of a tumor antigen by proteasomes, trimming of antigenic precursor peptides by ERAPs, and binding of peptides to MHC class I with sufficient affinity will determine whether a given peptide may be a suitable therapeutic target. Very little is known about how such mutations within a tumor epitope sequence will affect proteasomal processing of the respective antigen and in consequence the generation of the mutated epitope. In the present communication we, therefore, analyzed the effect of the experimentally introduced T210M amino acid exchange within the melanoma antigen-derived HLA-A*02:01-restricted gp100209–217 epitope on proteasomal processing of the gp100201–230 polypeptide. This antigenic peptide was chosen as a suitable model antigen because the T201M exchange at the anchor position of the epitope increased its MHC binding affinity as well as its immunogenicity (24) thereby exhibiting two features that are mandatory for peptide targets suited for T cell therapy.
To study the impact of the T201M exchange within the gp100209–217 epitope, we developed a novel biochemical method for the quantitative evaluation of the effect of a mutation on the processing of a given substrate and connected it with the generation and presentation of antigenic peptides on the MHC class I molecules. The previous pioneering study by Tenzer et al. (16), which challenged a similar issue and carried out a quantitative evaluation of the effect of HIV mutations on antigen presentation, had some caveats and limitations regarding the quantification of the in vitro products of proteasomal digestions and the computation of the substrate cleavage site usage. Such restrictions have been solved using the recently developed QME method (3). By applying this method we assessed how the T210M substitution influenced the wt and mut gp100201–230 substrate cleavage site usage by proteasomes. Our quantitative analysis shows that the substitution not only affects the cleavage site usage by the proteasome close to the substitution but also major cleavage sites as distant as 15 residues away from the substitution. The influence of a given mutation on cleavages that are four-five residues apart from the site of the mutation might be explained by a mutation-induced alteration of substrate binding to the S4-S1 and S1′-S4′ substrate-binding sites of the proteasome's catalytic pockets (37). On the contrary, the effects on the substrate cleavage site usage that are 15 residues apart form the T210M exchange are more difficult to assess and may be due to alterations of substrate transport along the proteasome cavities or the binding to non-catalytic modifier sites (38–41).
As observed in the in vitro experiments, the gp100 T210M substitution also resulted in a surprising and substantial alteration of the peptide pools produced by the 20S proteasome. The fact that the gp100 T210M substitution results in the generation of 37 unique non-spliced and 3 unique spliced peptides and that this pool contains potential antigenic peptides, which are not generated from the wt substrate (Table 1) indicates that tumor-specific somatic mutations may also induce the generation of new epitopes located not too far from the mutation. Although such epitopes do not carry somatic mutations, they would be tumor-specific and thus represent potential targets for specific adoptive T cell transfer therapy.
Our quantitative analysis of the post-proteasomal antigen presentation steps also revealed some interesting aspects of the potential immune relevance of N-terminally extended epitope-precursor peptides. Probably the most relevant result of these analyses is that mutations at the anchor position of an epitope that increase the MHC class I binding affinity of the minimal epitope peptide can also enhance the MHC class I binding affinity of N-terminally elongated variants of the minimal epitopes. In consequence, mutant N-terminally extended variants of the minimal epitopes may be presented on the cell surface as efficiently as the wt minimal epitope (e.g. wtgp100209–217) and trigger a specific CD8+ T cell response. These results also indicate that even when proteasomes generate the 9-mer epitope, ERAPs can trim proteasome-generated N-terminally extended precursor peptides (>12-mers) thereby producing 10–12-mers (or longer) that will bind efficiently the MHC class I complex and thus may contribute directly to the CD8+ T cell response.
In summary, our quantitative analyses of the main steps in antigen processing suggest that tumor-specific somatic mutations increasing the MHC class I binding affinity of an epitope may also broaden the specific T cell response by enhancing the presentation of immune reactive N-terminally extended precursor peptides. Furthermore, mutations may affect the generation of new unpredicted non-spliced and spliced epitopes, which will be of particular relevance when mutant polypeptides are used as tumor vaccines to improve the immune response. The impact of a tumor-specific somatic mutation could interest several immunogenic peptides located in proximity of the mutation, and only a systemic and quantitative biochemical study could estimate the outcome from an immunological point of view.
Author Contributions
P. M. K. coordinated the study and wrote the paper. M. M. conceived and coordinated the study, wrote the paper, and designed, performed, and analyzed the experiments shown in Figs. 1–8. K. T.-T. designed, performed, and analyzed the experiments shown in Figs. 1–7. C. K. performed the experiments shown in Figs. 6–8. J. L. analyzed the experiments shown in Fig. 6. P. H. provided technical assistance. J. S. and A. S. performed and analyzed the experiments shown in Fig. 7. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank F. K. M. Lorenz and W. Uckert for technical assistance and critical reading of the manuscript and P. Kunert and B. Brecht-Jachan for peptide synthesis.
This work was supported by a grant of the Berlin Institute of Health (CRG1-TP1) and the Einstein Stiftung Berlin (A2013-174) (to P. M. K.) and by NC3Rs (National Center for the Replacement, Refinement, and Reduction of Animal in Research) through a David Sainsbury Fellowship (to J. L.) (NC/K001949/1). The authors declare that they have no conflict of interest.
F. Ebstein et al., personal communication.
- ER
- endoplasmic reticulum
- ERAP
- ER amino peptidase
- i-proteasome
- immunoproteasome
- s-proteasome
- standard proteasome
- SCS
- substrate site-specific cleavage strength
- ESI
- electrospray ionization
- QME
- quantification with minimal effort
- APC
- antigen presenting cell
- PBL
- peripheral blood lymphocyte
- mut
- mutant.
References
- 1.Dalet A., Vigneron N., Stroobant V., Hanada K., and Van den Eynde B. J. (2010) Splicing of distant Peptide fragments occurs in the proteasome by transpeptidation and produces the spliced antigenic peptide derived from fibroblast growth factor-5. J. Immunol. 184, 3016–3024 [DOI] [PubMed] [Google Scholar]
- 2.Vigneron N., Stroobant V., Chapiro J., Ooms A., Degiovanni G., Morel S., van der Bruggen P., Boon T., and Van den Eynde B. J. (2004) An antigenic peptide produced by peptide splicing in the proteasome. Science 304, 587–590 [DOI] [PubMed] [Google Scholar]
- 3.Mishto M., Goede A., Taube K. T., Keller C., Janek K., Henklein P., Niewienda A., Kloss A., Gohlke S., Dahlmann B., Enenkel C., and Kloetzel P. M. (2012) Driving forces of proteasome-catalyzed peptide splicing in yeast and humans. Mol. Cell Proteomics 11, 1008–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanada K., Yewdell J. W., and Yang J. C. (2004) Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature 427, 252–256 [DOI] [PubMed] [Google Scholar]
- 5.Zanker D., Waithman J., Yewdell J. W., and Chen W. (2013) Mixed proteasomes function to increase viral peptide diversity and broaden antiviral CD8+ T Cell Responses. J. Immunol. 191, 52–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalet A., Stroobant V., Vigneron N., and Van den Eynde B. J. (2011) Differences in the production of spliced antigenic peptides by the standard proteasome and the immunoproteasome. Eur. J. Immunol. 41, 39–46 [DOI] [PubMed] [Google Scholar]
- 7.Beekman N. J., van Veelen P. A., van Hall T., Neisig A., Sijts A., Camps M., Kloetzel P. M., Neefjes J. J., Melief C. J., and Ossendorp F. (2000) Abrogation of CTL epitope processing by single amino acid substitution flanking the C-terminal proteasome cleavage site. J. Immunol. 164, 1898–1905 [DOI] [PubMed] [Google Scholar]
- 8.Sijts A. J., Standera S., Toes R. E., Ruppert T., Beekman N. J., van Veelen P. A., Ossendorp F. A., Melief C. J., and Kloetzel P. M. (2000) MHC class I antigen processing of an adenovirus CTL epitope is linked to the levels of immunoproteasomes in infected cells. J. Immunol. 164, 4500–4506 [DOI] [PubMed] [Google Scholar]
- 9.Deol P., Zaiss D. M., Monaco J. J., and Sijts A. J. (2007) Rates of processing determine the immunogenicity of immunoproteasome-generated epitopes. J. Immunol. 178, 7557–7562 [DOI] [PubMed] [Google Scholar]
- 10.Seifert U., Liermann H., Racanelli V., Halenius A., Wiese M., Wedemeyer H., Ruppert T., Rispeter K., Henklein P., Sijts A., Hengel H., Kloetzel P. M., and Rehermann B. (2004) Hepatitis C virus mutation affects proteasomal epitope processing. J. Clin. Invest. 114, 250–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strehl B., Joeris T., Rieger M., Visekruna A., Textoris-Taube K., Kaufmann S. H., Kloetzel P. M., Kuckelkorn U., and Steinhoff U. (2006) Immunoproteasomes are essential for clearance of Listeria monocytogenes in nonlymphoid tissues but not for induction of bacteria-specific CD8+ T cells. J. Immunol. 177, 6238–6244 [DOI] [PubMed] [Google Scholar]
- 12.Vigneron N., and Van den Eynde B. J. (2012) Proteasome subtypes and the processing of tumor antigens: increasing antigenic diversity. Curr. Opin. Immunol. 24, 84–91 [DOI] [PubMed] [Google Scholar]
- 13.Zanker D., and Chen W. (2014) Standard and immunoproteasomes show similar peptide degradation specificities. Eur. J. Immunol. 44, 3500–3503 [DOI] [PubMed] [Google Scholar]
- 14.Mishto M., Liepe J., Textoris-Taube K., Keller C., Henklein P., Weberruss M., Dahlmann B., Enenkel C., Voigt A., Kuckelkorn U., Stumpf M. P., and Kloetzel P. M. (2014) Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur. J. Immunol. 44, 3508–3521 [DOI] [PubMed] [Google Scholar]
- 15.Kincaid E. Z., Che J. W., York I., Escobar H., Reyes-Vargas E., Delgado J. C., Welsh R. M., Karow M. L., Murphy A. J., Valenzuela D. M., Yancopoulos G. D., and Rock K. L. (2012) Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol. 13, 129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tenzer S., Wee E., Burgevin A., Stewart-Jones G., Friis L., Lamberth K., Chang C. H., Harndahl M., Weimershaus M., Gerstoft J., Akkad N., Klenerman P., Fugger L., Jones E. Y., McMichael A. J., Buus S., Schild H., van Endert P., and Iversen A. K. (2009) Antigen processing influences HIV-specific cytotoxic T lymphocyte immunodominance. Nat. Immunol. 10, 636–646 [DOI] [PubMed] [Google Scholar]
- 17.Del Val M., Schlicht H. J., Ruppert T., Reddehase M. J., and Koszinowski U. H. (1991) Efficient processing of an antigenic sequence for presentation by MHC class I molecules depends on its neighboring residues in the protein. Cell 66, 1145–1153 [DOI] [PubMed] [Google Scholar]
- 18.Theobald M., Ruppert T., Kuckelkorn U., Hernandez J., Häussler A., Ferreira E. A., Liewer U., Biggs J., Levine A. J., Huber C., Koszinowski U. H., Kloetzel P. M., and Sherman L. A. (1998) The sequence alteration associated with a mutational hotspot in p53 protects cells from lysis by cytotoxic T lymphocytes specific for a flanking peptide epitope. J. Exp. Med. 188, 1017–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eggers M., Boes-Fabian B., Ruppert T., Kloetzel P. M., and Koszinowski U. H. (1995) The cleavage preference of the proteasome governs the yield of antigenic peptides. J. Exp. Med. 182, 1865–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Velders M. P., Weijzen S., Eiben G. L., Elmishad A. G., Kloetzel P. M., Higgins T., Ciccarelli R. B., Evans M., Man S., Smith L., and Kast W. M. (2001) Defined flanking spacers and enhanced proteolysis is essential for eradication of established tumors by an epitope string DNA vaccine. J. Immunol. 166, 5366–5373 [DOI] [PubMed] [Google Scholar]
- 21.Ossendorp F., Eggers M., Neisig A., Ruppert T., Groettrup M., Sijts A., Mengedë E., Kloetzel P. M., Neefjes J., Koszinowski U., and Melief C. (1996) A single residue exchange within a viral CTL epitope alters proteasome-mediated degradation resulting in lack of antigen presentation. Immunity 5, 115–124 [DOI] [PubMed] [Google Scholar]
- 22.Blankenstein T., Leisegang M., Uckert W., and Schreiber H. (2015) Targeting cancer-specific mutations by T cell receptor gene therapy. Curr. Opin. Immunol. 33, 112–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melief C. J., and van der Burg S. H. (2008) Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat. Rev. Cancer 8, 351–360 [DOI] [PubMed] [Google Scholar]
- 24.Parkhurst M. R., Salgaller M. L., Southwood S., Robbins P. F., Sette A., Rosenberg S. A., and Kawakami Y. (1996) Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J. Immunol. 157, 2539–2548 [PubMed] [Google Scholar]
- 25.Mishto M., Santoro A., Bellavista E., Sessions R., Textoris-Taube K., Dal Piaz F., Carrard G., Forti K., Salvioli S., Friguet B., Kloetzel P. M., Rivett A. J., and Franceschi C. (2006) A structural model of 20S immunoproteasomes: effect of LMP2 codon 60 polymorphism on expression, activity, intracellular localisation and insight into the regulatory mechanisms. Biol. Chem. 387, 417–429 [DOI] [PubMed] [Google Scholar]
- 26.Mishto M., Luciani F., Holzhütter H. G., Bellavista E., Santoro A., Textoris-Taube K., Franceschi C., Kloetzel P. M., and Zaikin A. (2008) Modeling the in vitro 20S proteasome activity: the effect of PA28-αβ and of the sequence and length of polypeptides on the degradation kinetics. J. Mol. Biol. 377, 1607–1617 [DOI] [PubMed] [Google Scholar]
- 27.Saric T., Chang S. C., Hattori A., York I. A., Markant S., Rock K. L., Tsujimoto M., and Goldberg A. L. (2002) An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat. Immunol. 3, 1169–1176 [DOI] [PubMed] [Google Scholar]
- 28.Liepe J., Mishto M., Textoris-Taube K., Janek K., Keller C., Henklein P., Kloetzel P. M., and Zaikin A. (2010) The 20S proteasome splicing activity discovered by SpliceMet. PLoS Comput. Biol. 6, e1000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sidney J., Southwood S., Moore C., Oseroff C., Pinilla C., Grey H. M., and Sette A. (2013) Measurement of MHC/peptide interactions by gel filtration or monoclonal antibody capture. Curr. Protoc. Immunol. Chapter 18, Unit 18.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gulukota K., Sidney J., Sette A., and DeLisi C. (1997) Two complementary methods for predicting peptides binding major histocompatibility complex molecules. J. Mol. Biol. 267, 1258–1267 [DOI] [PubMed] [Google Scholar]
- 31.Lundegaard C., Lamberth K., Harndahl M., Buus S., Lund O., and Nielsen M. (2008) NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 36, W509–W512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chapiro J., Claverol S., Piette F., Ma W., Stroobant V., Guillaume B., Gairin J. E., Morel S., Burlet-Schiltz O., Monsarrat B., Boon T., and Van den Eynde B. J. (2006) Destructive cleavage of antigenic peptides either by the immunoproteasome or by the standard proteasome results in differential antigen presentation. J. Immunol. 176, 1053–1061 [DOI] [PubMed] [Google Scholar]
- 33.Tynan F. E., Burrows S. R., Buckle A. M., Clements C. S., Borg N. A., Miles J. J., Beddoe T., Whisstock J. C., Wilce M. C., Silins S. L., Burrows J. M., Kjer-Nielsen L., Kostenko L., Purcell A. W., McCluskey J., and Rossjohn J. (2005) T cell receptor recognition of a “super-bulged” major histocompatibility complex class I-bound peptide. Nat. Immunol. 6, 1114–1122 [DOI] [PubMed] [Google Scholar]
- 34.Liu Y. C., Miles J. J., Neller M. A., Gostick E., Price D. A., Purcell A. W., McCluskey J., Burrows S. R., Rossjohn J., and Gras S. (2013) Highly divergent T-cell receptor binding modes underlie specific recognition of a bulged viral peptide bound to a human leukocyte antigen class I molecule. J. Biol. Chem. 288, 15442–15454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hassan C., Chabrol E., Jahn L., Kester M. G., de Ru A. H., Drijfhout J. W., Rossjohn J., Falkenburg J. H., Heemskerk M. H., Gras S., and van Veelen P. A. (2015) Naturally processed non-canonical HLA-A*02:01 presented peptides. J. Biol. Chem. 290, 2593–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mommen G. P., Frese C. K., Meiring H. D., van Gaans-van den Brink J., de Jong A. P., van Els C. A., and Heck A. J. (2014) Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD). Proc. Natl. Acad. Sci. U.S.A. 111, 4507–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huber E. M., Basler M., Schwab R., Heinemeyer W., Kirk C. J., Groettrup M., and Groll M. (2012) Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148, 727–738 [DOI] [PubMed] [Google Scholar]
- 38.Liepe J., Holzhutter H. G., Bellavista E., Kloetzel P. M., Stumpf M. P., and Mishto M. (2015) Quantitative time-resolved analysis reveals intricate, differential regulation of standard- and immuno-proteasomes. Elife 10.7554/eLife.07545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidtke G., Emch S., Groettrup M., and Holzhutter H. G. (2000) Evidence for the existence of a non-catalytic modifier site of peptide hydrolysis by the 20 S proteasome. J. Biol. Chem. 275, 22056–22063 [DOI] [PubMed] [Google Scholar]
- 40.Kisselev A. F., Kaganovich D., and Goldberg A. L. (2002) Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20 S proteasomes. Evidence for peptide-induced channel opening in the α-rings. J. Biol. Chem. 277, 22260–22270 [DOI] [PubMed] [Google Scholar]
- 41.Kisselev A. F., Garcia-Calvo M., Overkleeft H. S., Peterson E., Pennington M. W., Ploegh H. L., Thornberry N. A., and Goldberg A. L. (2003) The caspase-like sites of proteasomes, their substrate specificity, new inhibitors and substrates, and allosteric interactions with the trypsin-like sites. J. Biol. Chem. 278, 35869–35877 [DOI] [PubMed] [Google Scholar]