

Abstract
Dysregulation of hepatic gluconeogenesis contributes to the pathogenesis of diabetes, yet the detailed molecular mechanisms remain to be fully elucidated. Here we show that FOXP1, a transcriptional repressor, plays a key role in the regulation of systemic glucose homeostasis. Hepatic expression levels of FOXP1 are decreased in diabetic mice. Modest hepatic overexpression of FOXP1 in mice inhibited the expression of gluconeogenic genes, such as peroxisome proliferators-activated receptor γ coactivator-1α (PGC-1α), phosphoenolpyruvate carboxykinase (PEPCK), and glucose-6-phosphatase (G6PC), leading to a decrease in hepatic glucose production and fasting blood glucose levels in normal mice and different mouse models of diabetes, including db/db diabetic and high-fat diet-induced obese mice. FOXP1 physically interacted with FOXO1 in vivo and competed with FOXO1 for binding to the insulin response element in the promoter region of gluconeogenic genes, thereby interfering expression of these genes. These results identify a previously unrecognized role for FOXP1 in the transcriptional control of hepatic glucose homeostasis.
Keywords: gluconeogenesis, glucose, hemostasis, transcription regulation, type 2 diabetes
Introduction
In mammals, blood glucose levels are maintained within a relatively tight range through regulation of glucose uptake by peripheral tissues and glucose secretion by the liver, protecting the body from hypoglycemia during fasting conditions and from hyperglycemia after a high-carbohydrate meal (1). In the insulin resistance state, insulin signaling is impaired, leading to dysregulation of both hepatic glucose production and glucose uptake in peripheral tissues. Abnormal elevation of hepatic glucose production contributes to fasting hyperglycemia in diabetes (2). Therefore, efforts to uncover the molecular mechanism that control hepatic gluconeogenesis are crucial to develop new therapeutic strategies.
The expression of key gluconeogenic genes, including PEPCK2 and G6PC, is controlled by hormones at the transcriptional level (3). Hormonal and nutrient regulation of hepatic glucose production primarily occurs through modulation of a complex network of transcriptional factors and cofactors, including FOXO1, cAMP response element-binding protein, cAMP response element-binding protein-regulated transcription coactivator 2 (CRTC2) and PGC-1α (1, 4–7).
The FOXO transcription factors have emerged as important targets of insulin and growth factor action. They play a central role in cell growth, differentiation, metabolism, and stress response (8). FOXO1 contains highly conserved AKT phosphorylation sites (Thr-24, Ser-253, and Ser-316) and its activity is regulated by AKT-mediated phosphorylation of these sites. Phosphorylated FOXO1 is excluded from the nucleus, thereby decreasing its transcriptional activity (9). In normal mice, the decreased blood insulin levels during fasting promote FOXO1 nuclear localization, where it collaborates with PGC-1α to increase the expression of the key gluconeogenic enzymes PEPCK and G6PC (7). FOXO1 stimulates the expression of PEPCK and G6PC via direct binding to insulin response element (IRE) mapped in the promoters of these genes (10–12). During prolonged starvation, hepatic FOXO1 stimulate glucose production to protect life-threating hypoglycemia (13). Haploinsufficiency of FOXO1 protects from insulin resistance and overexpression of an active FOXO1 mutant in liver causes insulin resistance (6).
The FOXP subfamily is defined by four members (FOXP1–FOXP4). FOXPs function as transcription factors and possess distinct transcriptional regulatory and DNA-binding domains. The N terminus of FOXPs contains a transcriptional repressor region with a zinc-finger/leucine zipper motif. The DNA-binding domain of FOXPs is uniquely positioned among forkhead proteins near the C terminus (14). FOXP1 is widely expressed in human tissues and regulates the development of many tissues, including heart, thymus, and lung (15–19). FOXP1-deficient embryos have severe defects in cardiac morphogenesis, including outflow tract septation and cushion defects, resulting in embryonic death (16). In the present study we show that FOXP1 can directly interact with FOXO1 in vivo and it also competes with FOXO1 for binding to IRE mapped in the promoter of gluconeogenic genes, preventing FOXO1 binding to these genes, thereby modulating hepatic glucose metabolism.
Experimental Procedures
Preparation of Recombinant Adenoviruses and Constructs
Recombinant adenovirus expressing FOXP1 were generated as previously described (20). The adenovirus was amplified in HEK293 cells and purified by cesium chloride density centrifugation (21). The full-length mouse FOXP1 gene was amplified from C57BL/6J mouse kidney cDNA by PCR, then FLAG-tagged FOXP1 was cloned into pcDNA4. Truncated FOXP1 (FOXP1 amino acids 145 to 653) was generated by PCR, then cloned into pcDNA4 and pGEX-4T-1 (Amersham Biosciences), respectively. A series of truncated G6PC promoters and PEPCK promoter were amplified by PCR and cloned into pGL3-Basic Luciferase reporter vector.
Animal Treatments
Male db/db and C57BL/6J mice at 6–8 weeks of age were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China) and housed in a controlled environment with a 12-h light/dark photoperiod with unrestricted water and food. Wild type C57BL/6J mice were fed ad libitum either a high fat diet (D12492; Research Diets, New Brunswick, NJ) to establish a diet-induced obese model or a control chow diet. For fasting experiments, food was removed for the indicated amount of time before mice were sacrificed. Mice received tail vein injection with Ad-GFP or Ad-FOXP1 at a dose of 1–1.5 × 109 active viral particles in 150 μl of saline. 5–7 days later, mice were fasted for the indicated hours and subsequently sacrificed. Plasma and livers were harvested for further analysis.
Glucose Tolerance Test and Pyruvate Tolerance Test
Glucose tolerance test (GTT) and pyruvate tolerance test (PTT) were performed as previously described (22). For GTT, we mice were fasted for 16 h (5 p.m. to 9 a.m.) and intraperitoneally injected with d-glucose solution. Blood glucose levels were measured at 0, 15, 30, 45, 60, 90, and 120 min after glucose injection. The dose of glucose injection was 1 g/kg−1 for db/db and DIO mice, 2 g/kg−1 for C57BL/6J mice. For PTT, mice were fasted for 16 h (5 p.m. to 9 a.m.) and intraperitoneally injected with sodium pyruvate solution. Blood glucose levels were measured at 0, 15, 30, 45, 60, 90, and 120 min after pyruvate injection. The dose of pyruvic acid sodium was 0.5 kg−1 for db/db and DIO mice, 1.5 kg−1 for C57BL/6J mice. Mouse blood glucose levels were measured from tail vein with a glucose monitor (OneTouch; LifeScan, Inc., Milpitas, CA).
Analytical Procedures and Chemicals
The hepatic TG and total cholesterol content were measured using a colorimetric diagnostic kit (Applygen Technologies, Beijing, China). The serum concentrations of glucose, TG, cholesterol, free fatty acids, alanine aminotransferase, and aspartate aminotransferase were determined using an automated Monarch device (Peking Union Medical College Hospital, Beijing, China). Insulin, forskolin, dexamethasone, pyruvic acid, and sodium were purchased from Sigma.
Cell Culture
Mouse primary hepatocytes were isolated as previously reported (23), and maintained in RPMI 1640 (GIBCO/BRL, 37 °C, 5% CO2) containing 10% FBS, 100 units/ml of penicillin, and 0.1 mg/ml of streptomycin.
RNA Extraction and Quantitative RT-PCR
Total RNA was extracted from cells and liver tissues with TriZOL reagent (Roche Applied Science), and reverse transcribed to cDNA using high capacity reverse transcription kit (Applied Biosystem, Grand Island, NY). Quantitative real-time PCR was performed using GoTaq qPCR Master Mix (Promega). Relative mRNA levels were normalized to β-actin expression levels. All primers used for the PCR are listed in Table 1.
TABLE 1.
Primers used in PCR
F, forward; R, reverse.
| Forward | Reverse | |
|---|---|---|
| Real-time PCR primers | ||
| PGC-1α | F, 5′-CATTTGATGCACTGACAGATGGA-3′ | R, 5′-GTCAGGCATGGAGGAAGGAC-3′ |
| G6PC | F, 5′-TGCAAGGGAGAACTCAGCAA-3′ | R, 5′-GGACCAAGGAAGCCACAATG-3′ |
| PEPCK | F, 5′-ACACACACACATGCTCACAC-3′ | R, 5′-ATCACCGCATAGTCTCTGAA-3′ |
| β-Actin | F, 5′-CCAGCCTTCCTTCTTGGGTAT-3′ | R, 5′-TGCTGGAAGGTGGACAGTGAG-3′ |
| FOXP1 | F, 5′-CCTCGCTCAAGGCATGATTC-3′ | R, 5′-AGGACTTGGAAGGTGCCGAG-3′ |
| SREBP-1c | F, 5′-GGAGCCATGGATTGCACATT-3′ | R, 5′-GGCCCGGGAAGTCACTGT-3′ |
| FAS | F, 5′-GTAAGTTCTGTGGCTCCAGAG-3′ | R, 5′-GCCCTCCCGTACACTCACTC-3′ |
| ACC | F, 5′-AGGAAGATGGCGTCCGCTCTG-3′ | R, 5′-GGTGAGATGTGCTGGGTCAT-3′R |
| CPT1a | F, 5′-GAACCCCAACATCCCCAAAC-3′ | R, 5′-TCCTGGCATTCTCCTGGAAT-3′ |
| MCAD | F, 5′-AACACTTACTATGCCTCGATTGCA-3′ | R, 5′-CCATAGCCTCCGAAAATCTGAA-3′ |
| PPARα | F, 5′-ACAAGGCCTCAGGGTACCA-3′ | R, 5′-GCCGAAAGAAGCCCTTACAG-3′ |
| PCR primers used for gene and promoter amplification | ||
| FOXP1 gene | F, 5′-ATGATGCAAGAATCTGGGTCTGAG-3′; F, 5′-AGATCTCCACCATGATGCAAGAATCTG-3′ | R, 5′-CCATGTCCTCATTTACTGGTTCGTC-3′; R, 5′-CTCGAGTCACTTGTCATCGTCATCCTTGTAGTCCTCCATGTCCTCAT-3′ |
| G6PC-922 | F, 5′-ACGCGTAGACATGAGGCCAATACCAGG-3′ | |
| G6PC-300 | F, 5′-ACGCGTACTCTGTCCTGTGTCTCTGG-3′ | |
| G6PC-133 | F, 5′-ACGCGTCATCAACCTACTGGTGATGC-3′ | R, 5′-AGATCTTTCCTTGGCACCTCAGGAAG-3′ |
| PEPCK promoter | F, 5′-GATCCGGAGAAATCCCTGCCCTCA-3′ | R, 5′-GATCCGAAGGGAGATCCACAGGTT-3′ |
Western Blot Analysis
Liver tissues or cultured hepatocytes were lysed in RIPA lysis buffer, 60–100 μg of proteins were resolved by SDS-PAGE and eletrotransferred to PVDF membranes. Western blot assays were performed using antibodies specific for FOXP1 (CST, Abcolonal), FOXO1 (Abcam, Cambridge, UK), PCK1 (R&D Systems), α-Tubulin (Beijing ComWin Biotech Co.), FLAG (Sigma), and HA (Sigma).
Glucose Output Assay
Mouse primary hepatocytes were seeded in 6-well plates, and then infected with Ad-GFP, Ad-FOXP1. Twenty-four hours after infection, cells were washed 2 times with PBS, and maintained in 2 ml/well of DMEM without glucose, l-glutamine, and phenol red (MACGENE Biotech) for 2 h. Medium was collected and subjected to glucose measurement using the Amplex Red Glucose/Glucose Oxidase Assay Kit (Applygen Technologies). Cells were lysed, and the protein concentration was determined for each lysate. The glucose output rate was normalized by cellular protein content.
Co-immunoprecipitation (Co-IP) Experiments
HEK293A cells or liver tissues were lysed in RIPA lysis buffer. The cellular extract was incubated with primary antibodies overnight at 4 °C followed by treatment with protein G-agarose beads for 2 h. The immunoprecipitates were washed with lysis buffer 5 times, and boiled for 5 min in SDS-PAGE sample buffer. The eluted proteins were immunoblotted with indicated antibodies. The antibodies include FLAG (Sigma), HA (Sigma), FOXO1A (Abcam), and FOXP1 (Abcolonal).
GST Pulldown Assay
GST-FOXP1 fusion protein was expressed in BL21(DE3) strain of Escherichia coli using the pGEX-4T-1 vector. GST fusion protein was purified with glutathione-Sepharose 4B (GE Healthcare Life Sciences). HA-tagged FOXO1 plasmid was transfected into HEK293A cells using VigoFect (Vigorous Biotechnology Beijing Co.), whole cell lysate was collected and mixed with purified glutathione Sepharose-bound GST-FOXP1 overnight at 4 °C with gentle rocking. The glutathione-Sepharose beads were washed five times with lysis buffer, and subjected to SDS-PAGE, and immunoblotted using antibodies against HA tag.
ChIP Assay
Hepatocytes isolated from C57BL/6J mice were infected with adenoviruses for 24 h, then the cells were treated with RPMI1640 medium plus Dex and Fsk for 2 h. Liver tissues isolated from C57BL/6J and db/db mice were snipped into small pieces. Cells and tissue pieces were lysed and sonicated as previously described (23). The protein-DNA complexes were co-immunoprecipitated with antibodies: anti-FOXO1A (Abcam), anti-FLAG (Sigma), anti-FOXP1 (Abcam), or control IgG. The DNA fragments were subjected to Real-time PCR with the following primers: PEPCK, 5′-GAGGCCTCCCAACATTCAT-3′, and 5′-CGCTGAGCGCCTTGCCGGA-3′; G6PC, 5′-GTCAAGCAGTGTGCCCAAGTTA-3′ and 5′-TGTGCCTTGCCCCTGTTTTATATG-3′.
EMSA
The EMSAs were performed as previously described (23). The biotin-labeled oligonucleotides corresponding to FOXO1 binding sites of the G6PC promoter were synthesized: 5′-CGATCAGGCTGTTTTTGTGTGCCTGTTTTTCTATTTTACGTA-3′.
Statistical Analysis
Data represent the mean ± S.D. values of three independent duplicate experiments. Statistical analysis was performed with Student's t test (*, p < 0.05; **, p < 0.01).
Study Approval
All animal experiments were conducted according to procedures approved by the Institutional Animal Care and Use Committee, Chinese Academy of Medical Science, and Peking Union Medical College.
Results
Hepatic FOXP1 Is Dysregulated in Diabetic Mice
FOXO1 is a well known transcription factor regulating hepatic gluconeogenesis. However, whether and how other members of FOX family regulate hepatic gluconeogenesis is largely unclear. To better understand the mechanisms regulating hepatic gluconeogenesis, we systematically investigated the expression of the FOX family under pathophysiological status. We found that the mRNA levels of FOXP1, a transcriptional repressor, are significantly decreased in the livers of db/db and ob/ob mice compared with wild type control mice. Correspondingly, its protein levels are also lower in diabetic mice (Fig. 1A). Consistent with a previous report (1), expression levels of PGC-1α and gluconeogenic genes, including PEPCK and G6PC, are up-regulated in db/db mouse liver (Fig. 1A). Taken together, these findings suggest a correlation between hepatic FOXP1 levels and gluconeogenesis.
FIGURE 1.
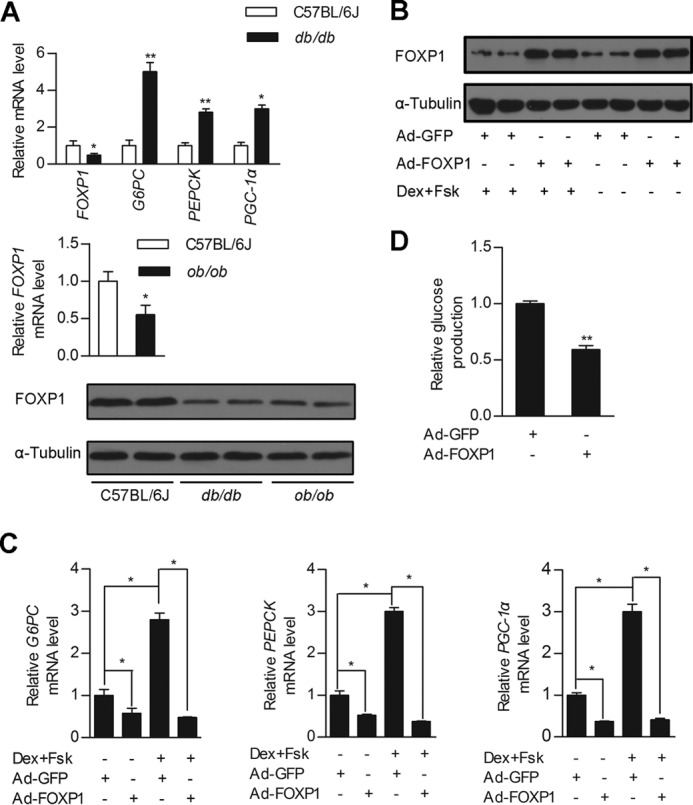
FOXP1 regulates cellular glucose production in primary hepatocytes. A, real-time PCR analysis of FOXP1, G6PC, PEPCK, and PGC-1α expression levels in the liver of db/db mice (top panel), and FOXP1 expression level in the liver of ob/ob mice (middle panel) and their control mice. Western blotting analysis of FOXP1 protein amounts in the liver of db/db, ob/ob, and C57BL/6J mice (bottom panel). (n = 6 each group). B, Western blotting analysis of FOXP1 protein amounts in primary mouse hepatocytes infected with Ad-GFP or Ad-FOXP1 in the presence/absence of Dex and Fsk. C, real-time PCR analysis of G6PC, PEPCK, and PGC-1α expression levels in primary mouse hepatocytes treated as in B. D, measurement of cellular glucose output in primary mouse hepatocytes infected with Ad-GFP or Ad-FOXP1 in the presence of Dex and Fsk. Results are presented as mean ± S.D. of triplicate experiments. *, p < 0.05; **, p < 0.01.
FOXP1 Suppresses the Gluconeogenic Program in Primary Hepatocytes
To test the functional significance of FOXP1 expression in the gluconeogenic program in vitro, we first generated a recombinant adenovirus expressing FOXP1 (Ad-FOXP1). Adenovirus-mediated expression of FOXP1 in primary mouse hepatocytes significantly inhibited the expression of PGC-1α, PEPCK, and G6PC in the absence or presence of Fsk and Dex (Fig. 1, B and C). Consistent with the finding that FOXP1 inhibits the gluconeogenic program in hepatocytes, overexpression of FOXP1 by Ad-FOXP1 infection markedly reduced glucose production in primary mouse hepatocytes (Fig. 1D).
FOXP1 Inhibits Hepatic Gluconeogenesis in Vivo
Given the dramatic effects of FOXP1 on the expression of PEPCK and G6PC in primary hepatocytes, we sought to examine the effect of FOXP1 gain-of function on glucose metabolism in vivo. Tail vein injection of Ad-FOXP1 into C57 BL/6J mice led to an modest increase in the FOXP1 expression levels in the liver (Fig. 2, A and B), but did not affect FOXP1 expression in the other tissues examined, including muscle and abdominal white adipose tissue (data not shown). Consistent with the results from primary hepatocytes, the expression levels of gluconeogenic genes, including PEPCK and G6PC, were decreased in the livers of Ad-FOXP1-injected mice (Fig. 2, A and B). As a result, Ad-FOXP1-infected mice had significantly lower fasting plasma glucose levels compared with control adenovirus Ad-GFP-treated mice (Fig. 2, C and D). Moreover, we also examined the blood glucose levels in mice following the intraperitoneal injection of sodium pyruvate. The PTT confirmed that hepatic gluconeogenesis was decreased in Ad-FOXP1-infected mice (Fig. 2E), indicating that the decrease in hepatic gluconeogenesis contributes to the lower glucose levels. Additionally, the GTTs indicated that hepatic overexpression of FOXP1 caused a significant enhancement in glucose excursion after glucose challenge (Fig. 2F). However, overexpression of FOXP1 did not significantly influence the expression of genes involved in lipogenesis (SREBP-1c, FAS, and ACC) or fatty acid oxidation (PPARα, MCAD, and CPT1a) (data not shown). Correspondingly, Ad-FOXP1 infection did not markedly affect hepatic triglyceride (TG) levels (Table 2). In addition, hepatic FOXP1 overexpression caused no major differences in body weight, serum cholesterol, or TG levels (Table 2).
FIGURE 2.

Overexpression of FOXP1 in the liver of C57BL/6J mice inhibits hepatic gluconeogenesis. Male C57BL/6J mice were injected with Ad-GFP of Ad-FOXP1 as described under “Experimental Procedures.” 5–7 days after infection, mice were sacrificed and plasma and liver tissues were collected for further analysis. A, real-time PCR analysis of mRNA levels of FOXP1, PEPCK, and G6PC in the liver of Ad-GFP or Ad-FOXP1-injected C57BL/6J mice (n = 7 each group). B, Western blotting analysis of protein levels of FOXP1, PEPCK, and α-Tubulin in the liver of Ad-GFP or Ad-FOXP1-injected C57BL/6J mice (n = 7 each group). C and D, blood glucose levels of 6-h fasted (C) and 16-h fasted (D) C57BL/6J mice on day 5 after injection with the indicated adenovirus (n = 7 each group). E, pyruvate tolerance test (n = 7 each group). F, glucose tolerance test (n = 7 each group). *, p < 0.05; **, p < 0.01, mice injected with Ad-GFP versus mice injected with Ad-FOXP1.
TABLE 2.
Metabolic characteristics of 7-day-treated C57BL/6J mice with adenovirus Ad-GFP (control) or Ad-FOXP1
Data are mean ± S.D. (n = 7 per group).
| Ad-GFP | Ad-FOXP1 | |
|---|---|---|
| Parameters | ||
| Body weight (g) | 23.18 ± 0.72 | 21.81 ± 1.68 |
| Liver/body weight (×100) | 5.04 ± 0.43 | 5.29 ± 0.31 |
| Blood parameters | ||
| Triglyceride (mg/dl) | 29.42 ± 2.50 | 27.54 ± 0.88 |
| Cholesterol (mg/dl) | 92.26 ± 8.01 | 91.56 ± 6.84 |
| Liver parameter | ||
| Triglyceride (mg/g) | 16.48 ± 4.00 | 16.22 ± 4.24 |
FOXP1 Rescue in Diabetic Mouse Livers Alleviates Hyperglycemia
Given that insulin resistance associated with the metabolic syndrome leads to FOXO-dependent increases in hepatic gluconeogenesis (6, 13) and that FOXP1 has the ability to inhibit hepatic gluconeogenesis and lower plasma glucose in normal mice, we next explored the therapeutic potential of this transcription repressor for diabetic mice. We hypothesized that decreased FOXP1 expression may contribute to hyperglycemia in mouse models of diabetes and FOXP1 rescue would be sufficient to restore glucose homeostasis. To test this hypothesis, we performed FOXP1 genetic reconstitution experiments in db/db mouse model. Injection of db/db mice with Ad-FOXP1 also led to an increase in FOXP1 expression in liver, subsequently reducing expression of gluconeogenic genes (Fig. 3, A and B) and lowering fasting blood glucose levels compared with the control Ad-GFP-infected db/db mice (Fig. 3, C and D). To further characterize this response, we performed PTT and GTT experiments on these mice. As a result, the PTT demonstrated that de novo hepatic glucose production was also reduced in Ad-FOXP1-treated db/db mice (Fig. 3E). Adenovirus-mediated overexpression of FOXP1 in db/db mouse liver improved glucose intolerance, as indicated by GTT experiments (Fig. 3F). Overexpression of FOXP1 in db/db mice did not significantly affect body weight, ratio of liver weight to body weight, or circulating free fatty acid compared with Ad-GFP-infected db/db control mice (Table 3). However, FOXP1 overexpression significantly reduced hepatic TG and serum cholesterol levels (Table 3). Similar suppressive effects of FOXP1 on hepatic gluconeogenesis were observed in DIO mice (Fig. 4). Additionally, hepatic overexpression of FOXP1 in DIO mice also decreased hepatic TG and serum cholesterol levels (Table 4). Taken together, our data reveal that increasing the expression levels of hepatic FOXP1 in the liver is able to suppress hepatic gluconeogenesis in diabetic mice, thus reducing blood glucose levels and improving glucose intolerance.
FIGURE 3.

Overexpression of FOXP1 in the liver of db/db mice alleviates hyperglycemia. Male db/db mice were injected with Ad-GFP of Ad-FOXP1 as described under “Experimental Procedures.” A, real-time PCR analysis of mRNA levels of FOXP1, PEPCK, G6PC, and PGC-1α in the liver of Ad-GFP or Ad-FOXP1-injected db/db mice (n = 6 each group). B, Western blotting analysis of protein levels of FOXP1, PEPCK, and α-Tubulin in the liver of Ad-GFP or Ad-FOXP1-injected db/db mice (n = 6 each group). C and D, blood glucose levels of 6-h fasted (C) and 16-h fasted (D) db/db mice on day 5 after injection with the indicated adenovirus (n = 6 each group). E, pyruvate tolerance test (n = 6 each group). F, glucose tolerance test (n = 6 each group). *, p < 0.05; **, p < 0.01, mice injected with Ad-GFP versus mice injected with Ad-FOXP1.
TABLE 3.
Metabolic characteristics of 7-day treated db/db mice with adenovirus Ad-GFP (control) or Ad-FOXP1
Data are mean ± S.D. (n = 6 per group).
| Ad-GFP | Ad-FOXP1 | |
|---|---|---|
| Parameters | ||
| Body weight (g) | 45.30 ± 1.99 | 42.88 ± 2.28 |
| Liver/body weight (×100) | 5.39 ± 0.34 | 5.42 ± 0.19 |
| Blood parameters | ||
| Cholesterol (mg/dl) | 148.90 ± 5.16 | 106.32 ± 9.15a |
| Triglyceride (mg/dl) | 27.09 ± 8.85 | 39.58 ± 8.91a |
| Free fatty acid (mm) | 1.55 ± 0.11 | 1.44 ± 0.12 |
| Liver parameter | ||
| Triglyceride (mg/g) | 61.41 ± 8.14 | 37.25 ± 10.66a |
a p < 0.01.
FIGURE 4.

Overexpression of FOXP1 in the liver of DIO mice inhibits hepatic gluconeogenesis and improves glucose intolerance. Male DIO mice were injected with Ad-GFP of Ad-FOXP1 as described under “Experimental Procedures.” A, real-time PCR analysis of mRNA levels of FOXP1, PEPCK, G6PC, and PGC-1α in the liver of Ad-GFP or Ad-FOXP1-injected DIO mice (n = 6 each group). B, Western blotting analysis of protein levels of FOXP1, PEPCK, and α-Tubulin in the liver of Ad-GFP or Ad-FOXP1 injected DIO mice (n = 6 each group). C, fasting (6 h) blood glucose levels of DIO mice on day 5 after injection with the indicated adenovirus (n = 6 each group). D, pyruvate tolerance test (n = 6 each group). E, glucose tolerance test (n = 6 each group). *, p < 0.05; **, p < 0.01, mice injected with Ad-GFP versus mice injected with Ad-FOXP1.
TABLE 4.
Metabolic characteristics of 7-day treated DIO mice with adenovirus Ad-GFP (control) or Ad-FOXP1
Data are mean ± S.D. (n = 6 per group).
| Ad-GFP | Ad-FOXP1 | |
|---|---|---|
| Parameters | ||
| Body weight (g) | 48.65 ± 5.59 | 43.60 ± 5.56 |
| Liver/body weight (100×) | 13.86 ± 1.12 | 14.10 ± 1.09 |
| Blood parameters | ||
| Cholesterol (mg/dl) | 144.06 ± 5.15 | 130.23 ± 8.30a |
| Triglyceride (mg/dl) | 63.12 ± 15.64 | 37.32 ± 8.31b |
| Free fatty acid (mm) | 1.03 ± 0.11 | 0.93 ± 0.44 |
| Liver parameter | ||
| Triglyceride (mg/g) | 86.84 ± 15.24 | 63.60 ± 8.56a |
a p < 0.05.
b p < 0.01.
FOXP1 Physically Interacts with FOXO1 in Vivo
Certain Forkhead factors have been shown to regulate gene expression as Forkhead heterodimers. For example, FOXG1 abolishes the FOXO-mediated induction of cell proliferation inhibitor P21 expression through the direct binding to FOXOs in vivo, acting as an antagonist of FOXO1 (24). This fact promoted us to explore the possibility that FOXP1 and FOXO1 may also form a complex in vivo. To this end, we transiently expressed FLAG-tagged FOXP1 and HA-tagged FOXO1 in HEK293 cells. Subsequently the transfected cells were lysed and subjected to immunoprecipitation. As a result, we observed that FOXP1 immunoprecipitation can also pull down FOXO1 (Fig. 5A), indicating that these two proteins are able to interact in vivo. In addition, we also tested whether they physically associate in intact liver. As expected, FOXO1 immunoprecipitation can also pull down FOXP1 in db/db mouse livers (Fig. 5B).
FIGURE 5.
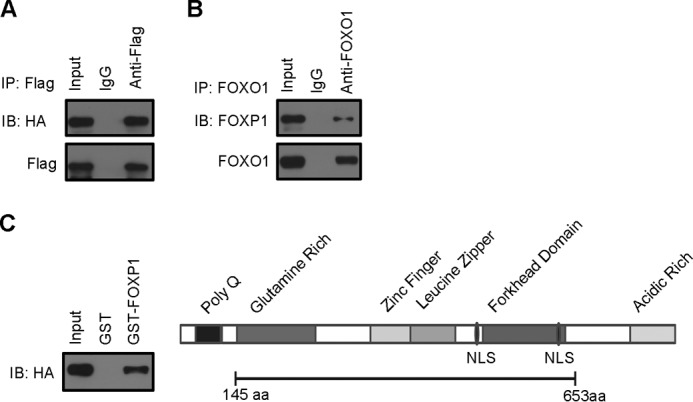
FOXP1 physically interacts with FOXO1 in vivo and in vitro. A, immunoblotting (IB) of HA and FLAG in FLAG immunoprecipitates (IP) from HEK293A cells expressing FLAG-tagged FOXP1 and HA-tagged FOXO1. B, immunoblotting of FOXO1 and FOXP1 in FOXO1 immunoprecipitates from liver tissue of db/db mice infected with Ad-FOXP1. C, GST pulldown experiments with anti-HA antibody as described under “Experimental Procedures” (left panel). Schematic representation of the FOXP1 protein is shown with the various functional domains indicated (right panel). GST-FOXP1 (amino acids 145–653) contains a zinc finger (amino acids 338–361), a leucine zipper (amino acids 378–403), a forkhead domain (amino acids 495–578).
To further confirm the direct physical interaction between FOXP1 and FOXO1, we used an in vitro interaction assay with recombinant glutathione S-transferase-FOXP1 fusion protein (GST-FOXP1) containing amino acids 145–663 of FOXP1 (Fig. 5C), consisting of a glutamine-rich domain, a zinc finger, a leucine zipper, and a forkhead domain (25). Our GST pulldown assay indicates that recombinant FOXP1 proteins can directly interact with FOXO1 and an internal fragment of FOXP1 (amino acid 145–663) mediates its interaction with FOXO1 (Fig. 5C). These results suggest that FOXP1, like FOXG1, may act as antagonist of FOXO1 through direct binding to FOXO1 to form heterodimers, blocking FOXO1 activity on regulating expression of gluconeogenic genes.
FOXP1 Competes with FOXO1 for Binding to Promoter of Gluconeogenic Gene
As FOXP1 and FOXO1 share a highly conserved DNA-binding domain, they recognize and bind to the same DNA-binding motif of target genes (26). FOXO1 has been shown to activate expression of PEPCK and G6PC through direct binding to IRE mapped in the promoters of these target genes (27, 28). However, in contrast to FOXO1, FOXP1 acts as a transcriptional repressor (14, 29). These data led us to hypothesize that FOXP1 may exert its inhibitory effects on gluconeogenic genes through competing with FOXO1 for direct binding to IRE of these genes, in addition to interaction with FOXO1 mentioned above. To test this hypothesis, we first performed a promoter-luciferase reporter gene assay in HepG2 cells. The promoter fragments of G6PC were cloned and fused to a luciferase reporter gene (referred to as G6PC-922-Luc). Transfection of the FOXP1 expression plasmid alone into HepG2 cells significantly inhibits the activity of G6PC-922-Luc. In contrast, transfection of the FOXO1 expression plasmid alone caused an ∼4-fold activation of G6PC-922-Luc. Moreover, cotransfection of FOXP1 plasmid abolished the stimulatory effects of FOXO1 on activity of G6PC-922-Luc (Fig. 6A), suggesting that FOXP1 and FOXO1 exert the opposite effects on the transcription of this gluconeogenic gene. To map the cis-acting region that mediates the FOXP1 inhibition of G6PC promoter-reporter gene activity, we performed cotransfection experiments with reporter constructs harboring serial deletions at the 5′-end of the G6PC promoter. Our results indicate that IRE (−186 bp to −158 bp) in the G6PC promoter is essential for FOXP1-mediated inhibition of its transcription (Fig. 6B). We also studied the effects of FOXP1 on activity of the PEPCK promoter-reporter gene and obtained similar results (Fig. 6A).
FIGURE 6.

FOXP1 antagonizes activity of FOXO1 on regulation of gluconeogenic genes expression through direct binding to IRE. A and B, luciferase reporter gene assay. A, the G6PC 922-bp and PEPCK promoter-driven luciferase constructs were cotransfected into HepG2 cells, together with pcDNA4 (control) or FOXP1 expression plasmid in the presence/absence of FOXO1 expression plasmid. B, a series of truncated G6PC promoters fused to the luciferase reporter gene were cotransfected into HepG2 cells, together with pcDNA4 (control) or FOXP1 expression plasmids. C, EMSA analysis of HEK293A cells transfected with FOXO1 or FOXP1 expression constructs. The protein extract was incubated with biotin-labeled DNA fragments encompassing IRE of G6PC promoter or cold probe. D, ChIP analysis on primary mouse hepatocytes infected with adenovirus overexpressing FOXO1 and assessed for FOXO1 occupancy on G6PC or PEPCK promoters (top panel). ChIP analysis on primary mouse hepatocytes were infected with adenovirus overexpressing FLAG-tagged FOXP1 and assessed for FOXP1 occupancy on G6PC or PEPCK promoters (bottom panel). E, ChIP analysis on primary mouse hepatocytes infected with Ad-FOXO1 in the presence/absence of Ad-FOXP1, and assessed for FOXO1 occupancy on G6PC promoter. F, Western blotting analysis of FOXP1, FOXO1 protein amounts (left panel), and real-time PCR analysis of the G6PC mRNA level (right panel) in primary mouse hepatocytes infected with Ad-GFP or Ad-FOXP1 in the presence/absence of Ad-FOXO1 following a 2-h treatment with Dex and Fsk. G, ChIP analysis on liver tissues of C57BL/6J and db/db mice, and assessed for endogenous FOXP1 occupancy on G6PC (top panel) or PEPCK promoter (bottom panel). Results are presented as mean ± S.D. of 3 replication experiments. *, p < 0.05; **, p < 0.01.
To determine whether FOXP1 directly binds to IRE identified in the G6PC gene promoter in vitro, we performed electrophoretic mobility shift assays. As expected, FOXO1 proteins, as a positive control, reacted with the labeled oligonucleotide probes, resulting in shifted bands (Fig. 6C). Like FOXO1, FOXP1 proteins also bound to the labeled probe (Fig. 6C), indicating that FOXP1 can directly bind to IRE in the promoter of gluconeogenic gene.
To examine whether FOXP1 proteins may be recruited to the promoter region of these gluconeogenic genes, we performed chromatin immunoprecipitation (CHIP) assays. To this end, mouse primary hepatocytes were isolated and infected with adenoviruses expressing FLAG-tagged FOXP1 and FOXO1 (as a positive control). As expected, the PEPCK and G6PC promoter fragments containing the IRE could be amplified from the precipitates obtained when using FOXO1-specific antibody, but not when using normal mouse immunoglobulin G (IgG; negative control) in cells infected with Ad-FOXO1 (Fig. 6D, top panel). Likewise, FOXP1 proteins also bound the same promoter regions of PEPCK and G6PC genes (Fig. 6D, bottom panel). Notably, recruitments of FOXO1 to promoters of gluconeogenic genes were markedly decreased by co-expression of FOXP1 in primary hepatocytes (Fig. 6E). Consistent with this effect, overexpression of FOXP1 completely blocked FOXO1 induction of the G6PC expression in primary hepatocytes (Fig. 6F). These data support the notion that FOXP1 can compete with FOXO1 for binding to promoters of gluconeogenic genes and inhibit their expression as a transcriptional repressor.
Finally, we explored whether endogenous FOXP1 proteins bind to the promoter region of gluconeogenic genes in mouse livers. Our ChIP results confirmed that and showed less endogenous FOXP1 protein amount associated with these promoters in liver of db/db mice compared with wild type mice (Fig. 6G).
In addition, we found that hepatic FOXP1 protein levels are dynamically regulated in normal mice by the nutritional status and its expression levels are higher under feeding conditions compared with fasting status (data not shown). Based on these data, we propose a model of the mechanism of FOXP1 action on hepatic gluconeogenesis (Fig. 7). In this model, FOXP1 regulates expression of gluconeogenic genes by a two-pronged mechanism. On one hand, FOXP1 can directly interact with FOXO1 in vivo, blocking a limited pool of FOXO1 binding to promoter of gluconeogenic genes to activate their transcription. On the other hand, FOXP1, as a transcription repressor, can compete with FOXO1 for binding to IRE of gluconeogenic genes, thereby inhibiting expression of these genes and hepatic gluconeogenesis. Under certain pathophysiological conditions (insulin resistance and diabetes), FOXP1 protein levels decrease, allowing FOXO1 binding to gluconeogenic genes, contributing to hepatic glucose production. However, under the feeding condition, FOXP1 expression is increased, blocking FOXO1 activity on hepatic gluconeogenesis and maintaining glucose homeostasis.
FIGURE 7.

Proposed model of FOXP1-mediated repression of hepatic gluconeogenesis. Under pathophysiological conditions (left panel) FOXP1 expression levels were decreased, allowing FOXO1 binding to IRE of gluconeogenic genes, subsequently activating their expression. However, under the feeding condition (right panel), increased FOXP1 proteins directly bind to IRE in the promoters of gluconeogenesis genes occupied by FOXO1 or physically interacts with FOXO1, preventing FOXO1 binding, thereby inhibiting expression of hepatic gluconeogenic genes.
Discussion
FOXO1 has a well defined role in the regulation of hepatic gluconeogenesis and this transcription factor exerts its function by binding to the consensus sequence of IRE, T(G/A)TTT(T/G)(G/T), in the promoters of PEPCK and G6PC genes (8).
Members of the FOX family share a highly conserved DNA-binding domain. In this study we found that FOXP1 can bind to IRE in the promoters of PEPCK and G6PC in vitro and in vivo. In addition, we show that FOXP1 competes with FOXO1 for binding IRE to modulate expression of these gluconeogenic genes. In contrast to FOXOs, which activates target genes, FOXP1 acts as a transcriptional repressor and its repressive function is mediated by the recruitment of specific chromatic modifying proteins, including histone deacetylases (14). We speculated that FOXP1 may also recruit these proteins to the promoters of gluconeogenic genes, decreasing histone acetylation levels and inhibiting expression of these genes. Further studies are required as confirmation. Of note, whereas we showed that overexpression of FOXP1 in primary hepatocytes inhibited expression of gluconeogenic genes and lowered the glucose concentration in the media (Fig. 1, C and D), we cannot rule out the possibility that FOXP1 may also affect glycogenolysis of hepatocytes, thereby contributing to the difference in glucose levels in the media. In addition to directly binding to and activating its target gene, FOXO1 can also regulate transcriptional responses via a DNA binding-independent action. It has been demonstrated that FOXO1 can associate with various unrelated transcription factors, regulating activation or repression of diverse target genes (30). Puigserver et al. (7) previously showed that FOXO1 physically interacts with PGC-1α, synergistically stimulating expression of gluconeogenic genes, and insulin regulates PGC-1α function through inactivating FOXO1 activity. In this study our co-IP assay and GST pulldown experiment suggest that FOXP1 and FOXO1 can directly interact and form a complex in vivo. The internal fragment of FOXP1 used in the GST pulldown assay contains a glutamine-rich domain, a zinc finger, a leucine zipper, and a forkhead domain. Thus, FOXP1 may physically interact with FOXO1, preventing the limited pool of FOXO1 binding to IRE of gluconeogenic genes. However, it remains unclear which domain of FOXO1 is responsible for interaction with FOXP1. Further studies are needed to answer this question.
Although our FOXP1-overexpressing mouse model somewhat phenocopies FOXO1-deficient mice, we did not observe marked glycemic phenotype in C57BL/6J mice with hepatic FOXP1 knockdown (data not shown). One possible interpretation of these data is that the organism can cope with FOXP1 deficiency due to a compensatory effect of some factors in either the liver or other tissues. Notably, FOXP1 inhibits the expression of gluconeogenic genes by a two-pronged mechanism wherein FOXP1 can directly interact with FOXO1, blocking FOXO1 binding to the promoter of gluconeogenic genes; meanwhile FOXP1, as a member of Forkhead family of transcription factors, can compete with FOXO1 to bind IRE of gluconeogenic genes, thereby inhibiting expression of these genes and hepatic gluconeogenesis. In fact, similar outcomes have been observed upon deletion of other transcription factors or cofactors that participate in hepatic gluconeogenesis. For example, PGC-1α is proposed to be a master regulator of hepatic gluconeogenesis, activating an entire program of key gluconeogenic enzymes, including PEPCK and G6PC, through coactivating hepatocyte nuclear factor 4 (HNF4) and FOXO1 (1, 7). However, ablation of PGC-1α did not markedly affect gluconeogenesis in vivo and this phenotype has been ascribed to the compensatory increase of C/EBPβ (31, 32).
FOXO1 also plays a role in regulating hepatic lipid homeostasis. Transgenic mice with FOXO1 overexpression in liver display increased hepatic TG content, resulting from enhanced SREBP-1 expression and hepatic lipogenesis (33). Similarly, adenoviral delivery of constitutively nuclear FOXO1 to mouse liver results in steatosis because of increased TG synthesis and decreased fatty acid oxidation (16). In this study we show that overexpression of FOXP1 decreased the hepatic TG contents in db/db and DIO mice. However, we did not observe inhibitory effects of FOXP1 on TG levels in normal C57BL/6J mouse livers. Moreover, FOXP1 overexpression in db/db mouse livers reduced blood cholesterol level, but increased blood TG levels, which are not consistent with that in DIO and normal mice (Tables 2–4), reflecting the genetic difference of mouse models. Further studies are needed to clarify the molecular mechanism underlying FOXP1-mediated changes in hepatic or serum TG and cholesterol levels in different mouse models.
Under certain pathophysiological conditions, such as insulin resistance and diabetes, hepatic FOXP1 expression is decreased, implying that FOXP1 may be involved in these processes. Decreased FOXP1 expression may be a contributing factor for increased hepatic gluconeogenesis in the insulin resistance state. The anti-diabetic drug metformin is proposed to act through the suppression of hepatic glucose production, although its action mechanism remains unclear. Thus, the suppression of gluconeogenesis is feasible for treatment of diabetes. In summary, our study provides a novel therapeutic target for treatment of type 2 diabetes, and small molecules that induce increases in FOXP1 levels may have the potential for treatment of this disease.
Author Contributions
Y. Z., N. G., and Y. C. designed and performed the experiments and wrote the manuscript. X. W., Q. C., and T. J. participated in the execution of experiments. Y. C. and A. C. participated in the construction of plasmids and data analysis. X. D. and H. Y. contributed to the animal experiment. S. Z. and F. F. participated in the revision of the manuscript.
This work was supported by the Major State Basic Research Development Program of China 973 program Grant 2012CB517504, National Natural Science Foundation of China Grants 81471049 and 81170763, and Natural Science Foundation of Beijing Grant 5142018. No potential conflicts of interest relevant to this article were reported.
- PEPCK
- phosphoenolpyruvate kinase
- PGC-1α
- peroxisome proliferative activated receptor γ co-activator 1α
- FOXP1
- forkhead box P1
- DIO
- diet-induced obese
- G6PC
- glucose-6-phosphatase, catalytic
- Ad-GFP
- adenovirus-containing green fluorescent protein
- Ad-FOXP1
- adenovirus expressing FOXP1
- GTT
- glucose tolerance test
- PTT
- pyruvate tolerance test
- Fsk
- forskolin
- Dex
- dexamethasone
- TG
- triglyceride
- SREBP-1c
- sterol regulatory element-binding protein-1c
- FAS
- fatty acid synthase
- ACC
- acetyl-CoA carboxylase
- PPARα
- peroxisome proliferator-activated receptor-a
- CPT1a
- carnitine palmitoyltransferase 1a
- MCAD
- medium chain acyl-CoA dehydrogenase
- IRE
- insulin response element.
References
- 1.Yoon J. C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C. R., Granner D. K., Newgard C. B., and Spiegelman B. M. (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138 [DOI] [PubMed] [Google Scholar]
- 2.Saltiel A. R., and Kahn C. R. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 [DOI] [PubMed] [Google Scholar]
- 3.Pilkis S. J., and Granner D. K. (1992) Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu. Rev. Physiol. 54, 885–909 [DOI] [PubMed] [Google Scholar]
- 4.Herzig S., Long F., Jhala U. S., Hedrick S., Quinn R., Bauer A., Rudolph D., Schutz G., Yoon C., Puigserver P., Spiegelman B., and Montminy M. (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179–183 [DOI] [PubMed] [Google Scholar]
- 5.Koo S. H., Flechner L., Qi L., Zhang X., Screaton R. A., Jeffries S., Hedrick S., Xu W., Boussouar F., Brindle P., Takemori H., and Montminy M. (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437, 1109–1111 [DOI] [PubMed] [Google Scholar]
- 6.Nakae J., Biggs W. H. 3rd, Kitamura T., Cavenee W. K., Wright C. V., Arden K. C., and Accili D. (2002) Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat. Genet. 32, 245–253 [DOI] [PubMed] [Google Scholar]
- 7.Puigserver P., Rhee J., Donovan J., Walkey C. J., Yoon J. C., Oriente F., Kitamura Y., Altomonte J., Dong H., Accili D., and Spiegelman B. M. (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423, 550–555 [DOI] [PubMed] [Google Scholar]
- 8.Barthel A., Schmoll D., and Unterman T. G. (2005) FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 16, 183–189 [DOI] [PubMed] [Google Scholar]
- 9.Biggs W. H. 3rd, Meisenhelder J., Hunter T., Cavenee W. K., and Arden K. C. (1999) Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. U.S.A. 96, 7421–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Accili D., and Arden K. C. (2004) FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 117, 421–426 [DOI] [PubMed] [Google Scholar]
- 11.Hall R. K., Yamasaki T., Kucera T., Waltner-Law M., O'Brien R., and Granner D. K. (2000) Regulation of phosphoenolpyruvate carboxykinase and insulin-like growth factor-binding protein-1 gene expression by insulin. The role of winged helix/forkhead proteins. J. Biol. Chem. 275, 30169–30175 [DOI] [PubMed] [Google Scholar]
- 12.Schmoll D., Walker K. S., Alessi D. R., Grempler R., Burchell A., Guo S., Walther R., and Unterman T. G. (2000) Regulation of glucose-6-phosphatase gene expression by protein kinase Balpha and the forkhead transcription factor FKHR. Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity. J. Biol. Chem. 275, 36324–36333 [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto M., Pocai A., Rossetti L., Depinho R. A., and Accili D. (2007) Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 6, 208–216 [DOI] [PubMed] [Google Scholar]
- 14.Lalmansingh A. S., Karmakar S., Jin Y., and Nagaich A. K. (2012) Multiple modes of chromatin remodeling by Forkhead box proteins. Biochim. Biophys. Acta 1819, 707–715 [DOI] [PubMed] [Google Scholar]
- 15.Banham A. H., Beasley N., Campo E., Fernandez P. L., Fidler C., Gatter K., Jones M., Mason D. Y., Prime J. E., Trougouboff P., Wood K., and Cordell J. L. (2001) The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res. 61, 8820–8829 [PubMed] [Google Scholar]
- 16.Wang B., Weidenfeld J., Lu M. M., Maika S., Kuziel W. A., Morrisey E. E., and Tucker P. W. (2004) Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development 131, 4477–4487 [DOI] [PubMed] [Google Scholar]
- 17.Hu H., Wang B., Borde M., Nardone J., Maika S., Allred L., Tucker P. W., and Rao A. (2006) Foxp1 is an essential transcriptional regulator of B cell development. Nat. Immunol. 7, 819–826 [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y., Li S., Yuan L., Tian Y., Weidenfeld J., Yang J., Liu F., Chokas A. L., and Morrisey E. E. (2010) Foxp1 coordinates cardiomyocyte proliferation through both cell-autonomous and nonautonomous mechanisms. Genes Dev. 24, 1746–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shu W., Lu M. M., Zhang Y., Tucker P. W., Zhou D., and Morrisey E. E. (2007) Foxp2 and Foxp1 cooperatively regulate lung and esophagus development. Development 134, 1991–2000 [DOI] [PubMed] [Google Scholar]
- 20.Luo J., Deng Z. L., Luo X., Tang N., Song W. X., Chen J., Sharff K. A., Luu H. H., Haydon R. C., Kinzler K. W., Vogelstein B., and He T. C. (2007) A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protoc. 2, 1236–1247 [DOI] [PubMed] [Google Scholar]
- 21.Orlicky D. J., and Schaack J. (2001) Adenovirus transduction of 3T3-L1 cells. J. Lipid Res. 42, 460–466 [PubMed] [Google Scholar]
- 22.Zhou Y., Lee J., Reno C. M., Sun C., Park S. W., Chung J., Lee J., Fisher S. J., White M. F., Biddinger S. B., and Ozcan U. (2011) Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat. Med. 17, 356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang R., Kong X., Cui A., Liu X., Xiang R., Yang Y., Guan Y., Fang F., and Chang Y. (2010) Sterol-regulatory-element-binding protein 1c mediates the effect of insulin on the expression of Cidea in mouse hepatocytes. Biochem. J. 430, 245–254 [DOI] [PubMed] [Google Scholar]
- 24.Seoane J., Le H. V., Shen L., Anderson S. A., and Massagué J. (2004) Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117, 211–223 [DOI] [PubMed] [Google Scholar]
- 25.Wang B., Lin D., Li C., and Tucker P. (2003) Multiple domains define the expression and regulatory properties of Foxp1 forkhead transcriptional repressors. J. Biol. Chem. 278, 24259–24268 [DOI] [PubMed] [Google Scholar]
- 26.Gabut M., Samavarchi-Tehrani P., Wang X., Slobodeniuc V., O'Hanlon D., Sung H. K., Alvarez M., Talukder S., Pan Q., Mazzoni E. O., Nedelec S., Wichterle H., Woltjen K., Hughes T. R., Zandstra P. W., Nagy A., Wrana J. L., and Blencowe B. J. (2011) An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 147, 132–146 [DOI] [PubMed] [Google Scholar]
- 27.O'Brien R. M., Noisin E. L., Suwanichkul A., Yamasaki T., Lucas P. C., Wang J. C., Powell D. R., and Granner D. K. (1995) Hepatic nuclear factor 3- and hormone-regulated expression of the phosphoenolpyruvate carboxykinase and insulin-like growth factor-binding protein 1 genes. Mol. Cell. Biol. 15, 1747–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onuma H., Vander Kooi B. T., Boustead J. N., Oeser J. K., and O'Brien R. M. (2006) Correlation between FOXO1a (FKHR) and FOXO3a (FKHRL1) binding and the inhibition of basal glucose-6-phosphatase catalytic subunit gene transcription by insulin. Mol. Endocrinol. 20, 2831–2847 [DOI] [PubMed] [Google Scholar]
- 29.Li S., Weidenfeld J., and Morrisey E. E. (2004) Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol. Cell Biol. 24, 809–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Vos K. E., and Coffer P. J. (2008) FOXO-binding partners: it takes two to tango. Oncogene 27, 2289–2299 [DOI] [PubMed] [Google Scholar]
- 31.Lin J., Wu P. H., Tarr P. T., Lindenberg K. S., St-Pierre J., Zhang C. Y., Mootha V. K., Jäger S., Vianna C. R., Reznick R. M., Cui L., Manieri M., Donovan M. X., Wu Z., Cooper M. P., Fan M. C., Rohas L. M., Zavacki A. M., Cinti S., Shulman G. I., Lowell B. B., Krainc D., and Spiegelman B. M. (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 119, 121–135 [DOI] [PubMed] [Google Scholar]
- 32.Leone T. C., Lehman J. J., Finck B. N., Schaeffer P. J., Wende A. R., Boudina S., Courtois M., Wozniak D. F., Sambandam N., Bernal-Mizrachi C., Chen Z., Holloszy J. O., Medeiros D. M., Schmidt R. E., Saffitz J. E., Abel E. D., Semenkovich C. F., and Kelly D. P. (2005) PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLos Biol. 3, e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qu S., Altomonte J., Perdomo G., He J., Fan Y., Kamagate A., Meseck M., and Dong H. H. (2006) Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology 147, 5641–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]