

Abstract
Aggregation of copper-zinc superoxide dismutase (SOD1) is a defining feature of familial ALS caused by inherited mutations in the sod1 gene, and misfolded and aggregated forms of wild-type SOD1 are found in both sporadic and familial ALS cases. Mature SOD1 owes its exceptional stability to a number of post-translational modifications as follows: formation of the intramolecular disulfide bond, binding of copper and zinc, and dimerization. Loss of stability due to the failure to acquire one or more of these modifications is proposed to lead to aggregation in vivo. Previously, we showed that the presence of apo-, disulfide-reduced SOD1, the most immature form of SOD1, results in initiation of fibrillation of more mature forms that have an intact Cys-57–Cys-146 disulfide bond and are partially metallated. In this study, we examine the ability of each of the above post-translational modifications to modulate fibril initiation and seeded growth. Cobalt or zinc binding, despite conferring great structural stability, neither inhibits the initiation propensity of disulfide-reduced SOD1 nor consistently protects disulfide-oxidized SOD1 from being recruited into growing fibrils across wild-type and a number of ALS mutants. In contrast, reduction of the disulfide bond, known to be necessary for fibril initiation, also allows for faster recruitment during seeded amyloid growth. These results identify separate factors that differently influence seeded growth and initiation and indicate a lack of correlation between the overall thermodynamic stability of partially mature SOD1 states and their ability to initiate fibrillation or be recruited by a growing fibril.
Keywords: amyloid, amyotrophic lateral sclerosis (ALS) (Lou Gehrig disease), neurodegenerative disease, protein aggregation, protein misfolding, superoxide dismutase (SOD)
Introduction
Familial amyotrophic lateral sclerosis (FALS)3 caused by mutations in the SOD1 gene shows similarities to other members of the large class of neurodegenerative disorders that are characterized by gradual aggregation of specific proteins and subsequent cell death in characteristic regions of the central nervous system. Some prominent examples are amyloid-β deposits in Alzheimer disease, α-synuclein and hyperphosphorylated Tau protein deposits in Parkinson disease, prion protein deposits in the transmissible spongiform encephalopathies, and huntingtin deposits in Huntington disease (1). In the case of SOD1-linked FALS, protein deposits have been identified in affected tissues from patients and from ALS-SOD1 transgenic mice consisting largely of aggregated copper,zinc superoxide dismutase protein (Cu,Zn-SOD, SOD1 protein).
Our current understanding of this class of diseases is that the implicated proteins misfold, aggregate, and ultimately deposit as extracellular plaques or intracellular inclusions in the afflicted regions of the CNS. Although the exact structures contributing to these heterogeneous protein aggregates are unknown, such diseases are nevertheless usually referred to as “amyloid diseases” because fibrillar properties characteristic of cross-β-sheet amyloid are often detected in them and because each of the isolated proteins implicated in causing disease shows a strong tendency to form cross-β-sheet amyloid fibrils when aggregation is studied in vitro (2). Although aggregated proteins are frequently abundant in these amyloid diseases, many lines of evidence suggest that it is not the aggregates themselves that are responsible for cell death but rather it is some form of soluble oligomers of the protein that are cytotoxic and start a cascade of events that ultimately lead to cell death (3–5). A unifying theme is that aggregated protein species can travel from cell to cell thereby transmitting disease in a prion-like manner (2, 6). Current therapeutic strategies for amyloid diseases in general are focused on preventing or reversing protein aggregation as well as removing or detoxifying the diffusible species (2).
Although the exact structural characteristics of the aggregated protein species that transmit these amyloid diseases are unknown, one compelling hypothesis is that cross-β-amyloid fibrils or protofibrils are formed but eventually become fragmented and that these fragments are the agents that transmit the diseases by traveling to and penetrating new cells and seeding new protein aggregation at that location leading ultimately to cell death. There is considerable evidence that SOD1-associated FALS propagates in this manner and also that wild-type human SOD1 as well as ALS-mutant SOD1 can be recruited into the newly formed aggregates (7–12). Studies on mouse models of ALS overexpressing SOD1 strongly suggest that the aggregates consist largely of disulfide-reduced SOD1 (13) and that its conversion to the aggregated state occurs shortly after its synthesis in the cell (14). The ability of human SOD1 to form cross-β-amyloid fibrils is an intrinsic property of the wild-type protein, and overexpression of even the wild-type human SOD1 protein in mice causes ALS-like symptoms, similar to what is seen in the SOD1-ALS transgenic mice expressing ALS-mutant SOD1 at lower levels (15).
Both wild-type and ALS-mutant human SOD1 can be induced to aggregate in vitro using a variety of different treatments, including low concentrations of reducing agents such as DTT or glutathione (16–19), low pH (18, 20), structural perturbants such as trifluoroethanol and elevated temperature (21, 22), oxidative modification induced by hydrogen peroxide (23), and by long term air exposure leading to cross-linking of non-disulfide cysteines (24). Some of these conditions, notably low pH, elevated temperatures, trifluoroethanol, and incubation in reducing agents, generate aggregates that bear a remarkable resemblance to amyloid fibrils. Unfortunately conditions such as low pH or elevated temperatures are likely to lead to scrambling or loss of bound metals in partially metallated SOD1 and can lead to dimer dissociation (25–27). Such methods are therefore unsuitable for studies of partially metallated forms of SOD.
The SOD1 protein is normally a dimer, with each monomeric subunit folded into an eight-stranded Greek key β-barrel containing a copper and a zinc ion. Each subunit also contains a disulfide bond formed between two of the four cysteine residues present in each polypeptide (28). The mature dimeric form of wild-type SOD1, which contains a disulfide bond, a copper ion, and a zinc ion in each subunit, is one of the most stable proteins known, melting at an astonishingly high 94 °C in differential scanning calorimetry (DSC) experiments. Successive loss of metal ions lowers the melting temperature until the completely demetallated (apo) dimeric form melts at 52 °C. Reduction of the Cys-57–Cys-146 disulfide bond converts the dimeric apoprotein to the monomeric form and lowers the melting point further to 42 °C (29). The monomeric disulfide-reduced, metal-free form of SOD1 protein is the least stable form of SOD1 known to still maintain the β-barrel fold (30). The loss of stability upon removal of bound metal ions from SOD1 is accompanied by an increased propensity to aggregate, as has been demonstrated in multiple studies that showed that unmetallated apo-SOD1 is highly prone to aggregation and can form both soluble oligomers and insoluble amyloid fibrils under physiologically relevant aqueous conditions at pH 7 (24, 31, 32). In contrast, metallated SOD1 appears more resistant to aggregation under many of these conditions. These results are supported by studies of aggregated and soluble SOD1 from the spinal cords of mice expressing WT and FALS SOD1 mutants that showed that metallated SOD1 is largely protected from aggregation and exists in the soluble state (14).
We previously reported a procedure for in vitro aggregation of authentic ALS-mutant and wild-type SOD1 proteins into cross-β-sheet amyloid fibrils under mild conditions similar to those present in human cells (31, 33). In this model system, both wild-type and ALS-mutant SOD1 proteins were found to aggregate readily, adopting amyloid fibrillar structure as monitored using dyes such as thioflavin T that fluoresce when bound to amyloid fibrils (31). We report here our new studies of the ability of partially mature forms of SOD1 that form during the post-translational folding process in the cell to form amyloid fibrils either by spontaneous aggregation of the soluble protein or by aggregation induced by addition of preformed fibrillar seeds. Contrary to our expectations from a wealth of studies showing the large increase in stability provided by zinc binding (34–37), we find that the presence of zinc does not affect fibril initiation of SOD1 and only marginally affects fibril-seeded growth. In addition, we find that dimer dissociation is not necessary for either fibril initiation or seeded growth. In contrast, the disulfide bond has a large ability to modulate both these processes. We also examined how mutations affect the propensity of these partially mature states to participate in fibrillation. Finally, we tested the effects of introducing into human cells in culture preformed wild-type SOD1 fibril seeds prepared according to our protocols, and we found that these fibrils are indeed toxic to such cells. This study thus yields additional insights into the way wild-type and ALS-mutant SOD1 might participate in “prion-like” cell-to-cell propagation as proposed by Prusiner (6) and Münch et al. (12).
Experimental Procedures
SOD1 Expression and Purification
Wild-type and FALS SOD1 mutants were expressed in Saccharomyces cerevisiae and purified according to published procedures (31). C6A, C111S SOD1 (AS-SOD1) was expressed in Escherichia coli and purified using a similar procedure. Purified SOD1 was demetallated by dialysis in a Slide-a-Lyzer (Pierce, 10,000 Da molecular mass cutoff) against 10 mm EDTA, 50 mm NaCl, pH 3.8, as described (35) except that, in the last step, 20 mm potassium phosphate, pH 7.0, was used as the dialysis buffer. Apo-SOD1 was flash-frozen in liquid nitrogen and stored at −20 °C prior to use. Metal content of apo- and metallated SOD1 was determined by ICP-MS. Typically, apo-SOD1 contained ≤0.1 eq of copper and zinc per dimer.
Metal Titration
Metal titrations in SOD1S-S were carried out according to the procedures outlined by Goto et al. (38). Briefly, apo-SOD1 was dialyzed against 100 mm sodium acetate, pH 5.5, and concentrated to about 300 μm using a YM-3 microconcentrator (Millipore/Fisher). One equivalent of zinc or cobalt chloride was added from an aqueous 10 mm stock solution in 2–4 equal aliquots at intervals of an hour with slow stirring using a 3-mm micromagnet at 4 °C. When necessary, the 2nd eq of cobalt was added in a similar fashion and stirred overnight at 4 °C. Cobalt binding was monitored by UV-visible spectroscopy between 400 and 750 nm using a microcuvette. Once metal binding had saturated, the reaction was transferred to a YM-10 microconcentrator, concentrated to 200 μl, and overlaid with an equal volume of 20 mm potassium phosphate, pH 7.4. The solution was concentrated, and the wash step was repeated 2–3 additional times to remove any unbound metal. For cobalt binding to SOD12SH, the disulfide bond in apo-SOD1S-S in 10 mm potassium phosphate, pH 7, was reduced by incubation in 10 mm TCEP overnight at room temperature. Disulfide-reduced apo-SOD1 was concentrated to about 300 μm in a YM-3 microconcentrator. The buffer concentration was adjusted to 100 mm potassium phosphate, pH 7, and cobalt chloride was then added in 0.5 eq aliquots every hour with slow stirring on an ice-water slurry. After the last addition of metal, the reaction was stirred for an additional 3–5 h until cobalt binding was saturated as observed by UV-visible spectroscopy. The reaction was centrifuged at 16,000 × g for 10 min, and the supernatant containing Co(II)-SOD12SH was used in fibrillation reactions. In all cases, metal binding was quantified by using a molar absorptivity of 10,800 m−1 cm−1 at 280 nm for SOD1 and 370 m−1 cm−1 for each equivalent of cobalt bound to a subunit.
Fibril Initiation Assays
All solutions for fibrillation reactions were filtered through a 0.22-μm syringe filter prior to use. Apo-SOD1S-S was reduced by incubation with 10 mm TCEP overnight on the bench top, and the monomeric disulfide-reduced form was purified by SEC-HPLC. SOD12SH was concentrated to about 300–500 μm and either used directly in fibrillation assays or titrated with cobalt or zinc chloride, when necessary. Initiation reactions in Fig. 1 contained 5.0 μm apo- or metallated SOD12SH and 45 μm apo in 10 mm potassium phosphate, pH 7. Those in Fig. 3 contained 7.5 μm Co-SOD12SH and 42.5 μm apo- or cobalt-bound mutant SOD1S-S. Fibrillation assays were performed as described previously (31). Briefly, to monitor fibrillation, ThT was added to 40 μm from a 4 mm stock, and 600 μl of each reaction was divided into three aliquots of 200 μl each in separate wells of a 96-well plate to which a Teflon ball (1/8 inch in diameter) had been added. The plate was agitated at 300 rpm (3 mm rotation diameter) in a Fluoroskan plate reader (Thermo) at 37 °C. Fluorescence measurements were recorded every 15 min using at λex = 444 nm, λem = 485 nm, with an integration time of 200 μs. All reactions were performed in replicates of three or more.
FIGURE 1.

Fibril initiation kinetics of disulfide-reduced WT SOD1 is similar in the presence of Zn(II) or Co(II). A, fibrillation of 45 μm apo-WTS-S initiated by the presence of 5 μm apo-, 1Zn-, or 2Zn(II)-WT SOD12SH. Data are shown as normalized ThT fluorescence. B, visible spectra of 250 μm WT SOD12SH bound to 1, 1.8, or 4 eq of Co(II) in 100 mm potassium phosphate, pH 7, containing 2 mm TCEP. C, fibrillation of 45 μm apo-WTS-S initiated by the presence of 5 μm (10%) apo-WT2SH (○), 1.6 Co-WT2SH (●), or 2.9 Co-WT2SH (triangle).
FIGURE 3.
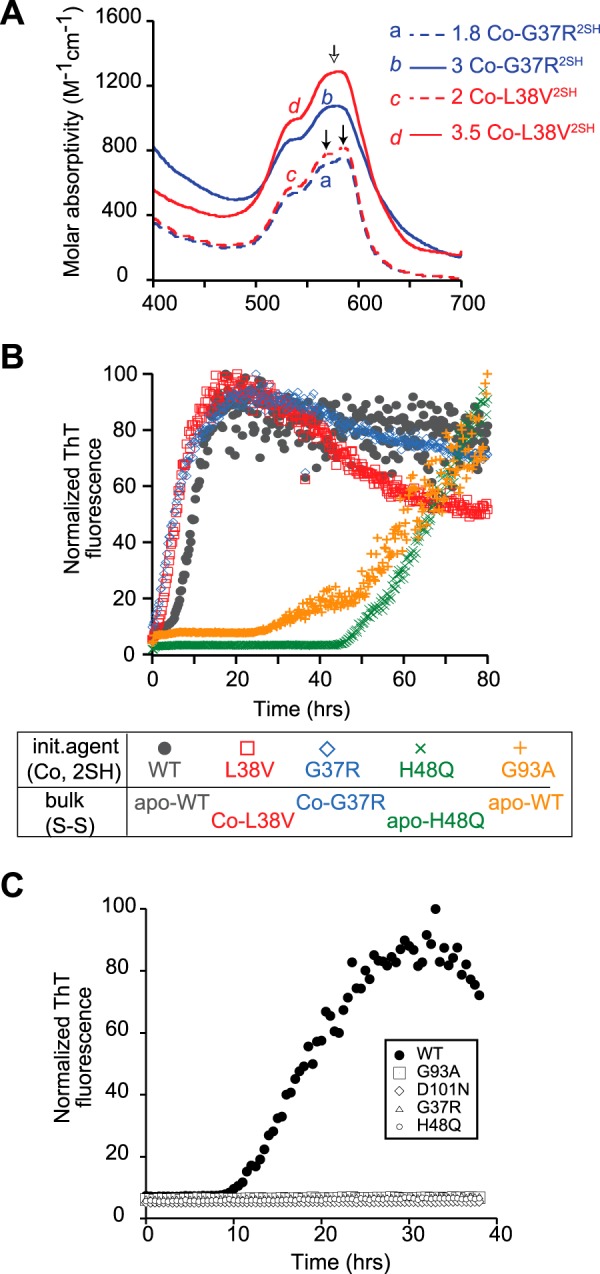
Co(II) binding and fibril initiation in ALS-associated SOD1 mutants in the disulfide-reduced form. A, visible spectra of 250 μm 2Co-L38V2SH (red dashed line) and 2Co-G37R2SH (blue dashed line). The arrows indicate the distinct peaks between 550 and 600 nm indicative of cobalt binding in the zinc site, whereas the copper site is unoccupied. Cobalt added in excess of 2 eq is bound in the copper site as shown in the top two chromatograms for 3.5 Co-G37R2SH (red solid line) and 3 Co-L38V2SH (solid blue line). The arrow shows the new peak arising from the broadening and blue shift of the features indicated in the previous two traces. B, fibril initiation kinetics by cobalt-bound, disulfide-reduced SOD1 mutants. All reactions contained 7.5 μm Co-SOD12SH and 42.5 μm apo or cobalt-bound mutant SOD1S-S. The reaction pairs were 2Co-WT2SH with apo-WTS-S (black circle), 2Co-L38V2SH with 2Co-L38VS-S (red square), 1.7Co-G37R2SH with 2Co-G37R2SH (blue diamond), Co-H48Q2SH with 1.7Co-H48QS-S (green x) and 2Co-G93A2SH with apo-WT2SH (orange +). C, fibril initiation kinetics as measured by ThT fluorescence for reactions consisting of 7.5 μm apo-WT2SH incubated with 42.5 μm apo-WTS-S (solid circle), apo-G37RS-S (open triangle), apo-H48QS-S (open circle), apo-G93AS-S (open square), and apo-D101N (open diamond).
Fibril Seeded Growth Assays
Seeds were prepared from fibrils generated from apo-SOD1S-S incubated in 5 mm DTT with agitation. Fibrils extracted from a 96-well microplate were centrifuged in a microcentrifuge tube at 16,800 × g for 15 min. The supernatant was pipetted out, and the fibrils were resuspended in the same volume (180 μl) of 2 m guanidinium hydrochloride, 20 mm potassium phosphate, pH 7, by pipetting up and down and vortexing at medium speeds and incubated at 37 °C for 90 min. They were then sonicated for 30 min in a bath sonicator (Branson Scientific, 100 watts) and used within 30 min. Seeding reactions were assembled using 10–20% by volume of fibril seeds prepared by the above protocol, 45 μm apo- or metallated SOD1 and 40 μm ThT in 10 mm potassium phosphate, pH 7. In reactions containing metallated SOD1, the buffer was 100 mm potassium phosphate, pH 7. When apo- or metallated SOD12SH was used, the concentration of TCEP in the reaction was limited to 50 μm or less to prevent secondary fibril initiation. Fibrillation was carried out as described above and in Ref. 31. Data were fitted to Equation 1,
where lag phase was calculated as tm − t.
SEC-HPLC and HPLC-ICP-MS
Disulfide-oxidized and -reduced apo-SOD1, which are dimeric and monomeric, respectively, were separated on a 7.5-mm × 30-cm TSK G2000 SW column (Toyosoda, Japan) equipped with a guard column on an Agilent 1200 HPLC using a mobile phase containing 50 mm sodium chloride, 50 mm potassium phosphate, pH 6.7, 5 mm DTT. To prepare samples for the initiation experiments, 0.5–1 mg of apo-SOD1S-S reacted overnight with 10 mm TCEP was loaded at a concentration of 3–5 mg/ml. SOD12SH collected from HPLC was concentrated using YM-3 microconcentrators to a final concentration of 300–500 μm for subsequent experiments. The metallation state of cobalt-bound SOD12SH was determined by in-line HPLC-ICP-MS according to previously published procedures (14). Briefly, a 7.5-mm × 30-cm TSK G2000 SW column was equilibrated in a mobile phase of 25 mm potassium phosphate, pH 5.7, 20 mm sodium chloride, 0.1 mm TCEP. The eluent stream was directed to the source of the ICP-MS using the high matrix interface (Agilent). ICP-MS analysis was run in the helium gas mode, with time-resolved analysis, to minimize signal suppression due to possible high salt content within the sample. Elution of the SOD1 polypeptide was monitored using absorbance at 280 nm, and the ICP-MS generated trace chromatograms for cobalt, copper, and zinc concentrations from the ICP-MS ion extract signal. The two chromatograms were offset by a delay of a minute due to the tube length that directed the HPLC eluent into the ICP-MS source.
Kinetics of Disulfide Reduction
50 μm apo- or metallated SOD1S-S was incubated in the presence of 5 mm TCEP in 10 mm potassium phosphate, pH 7, at 37 °C in a total volume of 150–200 μl. At the indicated intervals, 10-μl aliquots were reacted with 5 mm AMS in buffer containing 100 mm HEPES, pH 7.2, and 2.5% SDS at 37 °C for 1 h. The reactions were stopped with the addition of 2× SDS-PAGE loading dye (without reducing agents) and frozen at −20 °C until analysis by SDS-PAGE. Gels were stained with Coomassie Brilliant Blue, scanned, and analyzed using Adobe Photoshop CS4 software.
Acrylodan Labeling
Apo-SOD1 (50–100 μm stock) was reacted with a 5-fold molar excess of acrylodan overnight at 4 °C. Labeled SOD1 was washed by repeated steps (steps 3 and 4) of concentration and dilution with 10 mm potassium phosphate, pH 7, in a YM-3 microconcentrator. The degree of labeling was quantified by comparing the absorbance of the sample at 365 and 270 nm.
Cell Culture Viability Assay
SH-SY5Y cells (ATCC) were plated at a density of 20,000 cells per well in a 96-well plate and cultured in DMEM/F-12 (Invitrogen) containing 10% FBS and 1% penicillin/streptomycin. After 1 day, the medium was replaced with fresh medium containing 15 μm retinoic acid (Sigma) to induce a neuronal phenotype. After 5 days, cells were prepared for fibril transfection by replacing media with antibiotic-free DMEM/F-12 containing 1% FBS 1 h before fibril transfection.
Immediately after sonication, fibril seeds were diluted to 0.5 μg/μl in 20 mm HEPES, pH 7, incubated at room temperature for 15 min, and added to cells in each well of a 96-well plate. After incubation for 2 h at 37 °C, the media containing fibrils were aspirated out, and fresh DMEM/F-12 containing 10% FBS, 1% penicillin/streptomycin, and 15 μm retinoic acid was added to each well. Cells were incubated at 37 °C for 24 h before examining their viability using the Cell Titer Blue Assay (Promega) according to the manufacturer's instructions.
Results
In the early stages of spontaneous amyloid fibril formation, a soluble normally folded protein is converted into a kinetically unfavorable prefibrillar oligomer, termed the “nucleus,” in one or more kinetically distinct steps. The nucleus stabilizes itself by recruiting more soluble protein and rearranging it to a β-rich form to generate larger structures that fragment to form additional structures capable of recruiting soluble protein. This cyclic process is thought to be the chief reason for the exponential decrease in soluble protein concentration and a corresponding rise in cross-β-rich fibrillar structures (16, 17). The end products of this process are insoluble amyloid fibrils comprising one or more protofibrils, each containing multiple protein molecules in the characteristic cross-β-structure. In this paper, the processes leading to the formation of the nucleus, characterized by a flat thioflavin T fluorescence signal during the first stages of the fibrillation process, are referred to as initiation. The processes thereafter that include cycles of elongation of the nucleus by the recruitment of soluble protein followed by fragmentation of the elongated fibril, resulting in a characteristic exponential growth in ThT fluorescence, are referred to as seeded amyloid growth. The process of initiation during fibrillation of a soluble normally folded protein can be bypassed by adding small quantities of preformed fibrils to the soluble protein. The fibril ends, or seeds, provide fresh surfaces where the soluble protein can attach and undergo conformational changes to adopt a β-rich structure, thereby elongating the fibril. Longer fibrils cleave spontaneously, generating more recruiting surfaces, until the remaining soluble protein is converted to amyloid fibrils. Thus, adding small quantities of preformed fibrils to soluble protein is one way to study the elongation-fragmentation cycles characteristic of seeded amyloid growth.
Spontaneous Fibrillation
Previously, we showed that monomeric apo-, disulfide-reduced WT-SOD1, represented as apo-WT2SH (the unmodified polypeptide released from the ribosome before it acquires the earliest post-translational modification) could initiate fibrillation of Cys-57–Cys-146 disulfide-intact forms of the protein (WTS-S), either apo or zinc-bound, at sub-stoichiometric amounts. This activity required the presence of thiols on cysteine residues at both positions 57 and 146, as single mutants apo-C57S or -C146S were unable to initiate fibrillation of apo-WT-SOD1 nor was the S-alkylated form of apo, disulfide-reduced WT (31).
As part of our goal to understand how maturation of SOD1 can modulate initiation, we sought to determine whether other immature forms of SOD1 could likewise cause initiation. Zinc has been postulated to bind early to the nascent disulfide-reduced SOD1 polypeptide and has recently been shown to do so in the cytoplasm of E. coli and mammalian cells (39–43). Although apo-WT2SH adopts a β-barrel fold with highly disordered loop regions, upon the binding of zinc, it is converted to a more stable, dimeric form in which the previously disordered loops of each subunit are more structurally constrained (30, 44). Because of the change in oligomeric status, stability, and structure upon zinc binding, we wanted to examine the ability of zinc to modulate the fibril-initiating ability of apo-WT2SH. Zinc-bound derivatives of WT2SH, prepared by the addition of 1 or 2 eq of zinc chloride to apo-WT2SH, were added in small amounts to apo-WTS-S to determine whether metallation at the zinc site could modulate the kinetics of spontaneous fibrillation of disulfide-reduced SOD1. Surprisingly, we found both 1Zn-WT2SH and 2Zn-WT2SH to be as efficient at initiating fibrillation as apo-WT2SH (Fig. 1A).
One possibility for these unexpected results could be from adventitious zinc binding to WT2SH at several possible locations, besides its native site, such as the copper site or the free thiol groups in any of the four-cysteine residues (45, 46). The different binding modes cannot be distinguished easily because zinc is spectroscopically silent. Preparation of zinc derivatives of disulfide-reduced SOD1 is further complicated by the unstable nature of the disulfide-reduced form of SOD1 in the presence of dioxygen, during which it slowly converts to a mix of intra- and intermolecular disulfide-containing species (data not shown). Therefore, real time monitoring of the bound metal ion is necessary during metal titration so that the location and number of bound metal ions can be determined in an expeditious manner. These limitations can be overcome by using the classical approach of substituting Co2+ for Zn2+. Co(II) binds SOD1 identically to zinc using the same amino acid side chains in a similar geometry, when added at pH 5.5 (45, 47, 48), but it has the advantage of being spectroscopically active because of its partially filled d orbitals. Co(II) bound in the zinc site produces a characteristic UV-visible spectrum with three distinct peaks between 500 and 600 nm (38, 47), as we observed when Co(II) was added to apo-WTS-S at pH 5.5. A similar spectrum was observed when Co(II) was added to apo-WT2SH. When Co(II) was titrated at pH 7, it first appeared to bind the two zinc sites in the dimer (Fig. 1B, 1 and 2 eq). Further addition of Co(II) resulted in binding in the copper sites resulting in a slight broadening and blue shift of the major peak in the spectrum (Fig. 1B, 4 eq) as has been observed previously (45, 48).
In spontaneous fibrillation experiments, when 1Co-WT2SH was included with apo-WTS-S at low substoichiometric amounts (10% molar ratio), fibrillation was initiated efficiently with a lag time of about 10 h that is comparable with initiation by an equivalent amount of apo-WT2SH (Fig. 1C). The binding of zinc to disulfide-reduced SOD1 converts it from a monomer to a dimer and stabilizes the structure considerably, as evident from a melting point shift of 42–58 °C in DSC experiments (44). Prior reports have indicated that in dimeric SOD1 loaded with cobalt or zinc in the zinc site, a fraction of these metal ions migrates from the zinc site to the copper site at neutral pH (45). To address the possibility that the migration of cobalt to the copper site is responsible for 1Co-WT2SH being able to initiate fibrillation, we also tested the initiating abilities of 3Co-WT2SH, which must have at least one zinc site filled (Fig. 1C) and 4Co-WT2SH (Fig. 2A) with all zinc and copper sites loaded with cobalt in the dimer. Both were comparable with 1Co-WT2SH in their fibril initiation abilities, suggesting that metal occupancy in the zinc or copper sites of the disulfide-reduced SOD1 does not prevent it from acting as an initiating agent. The surprising ability of this species to initiate fibrillation suggested that the additional stability afforded by the binding of cobalt was not enough to prevent the onset of fibrillation.
FIGURE 2.
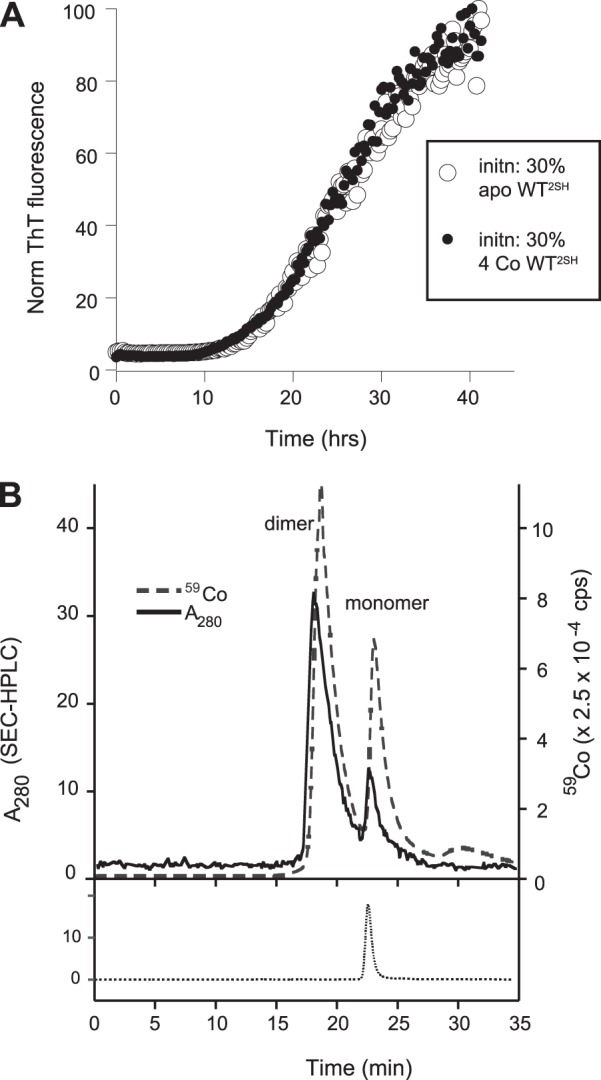
4Co(II)-WT2SH (i.e. 2Co/WT2SH monomer) is as competent as apo-WT2SH in initiating fibrillation and is largely dimeric under fibrillation conditions. A, ThT fluorescence showing fibrillation of 35 μm apo-WTS-S in the presence of 15 μm apo-WT2SH (○) or 4Co(II)-WT2SH (●). B, overlay of chromatograms from an HPLC-ICP-MS experiment showing HPLC absorbance at 280 nm corresponding to the SOD1 polypeptide (solid line) and ion extract count for cobalt by ICP-MS (dashed line) from an injection of 15 μm 1.8Co-WT2SH/monomer. The peak eluting at 19 min corresponds to the dimeric form, and the one eluting at 23 min corresponds to the monomeric form. At the bottom is shown the chromatogram from an injection of apo-WT2SH, which elutes as a monomer.
A number of studies have suggested that dimer dissociation of SOD1 to generate monomeric species is necessary for fibrillation, and it is the monomer that aggregates (49, 50). However, efficient initiation by Co(II)-WT2SH during spontaneous fibrillation experiments suggested that this metal-bound dimeric form might be as efficient as the monomeric apo-disulfide-reduced form. These experiments involved the mixing of low concentrations (2.5–10 μm) of Co(II)-WT2SH with significantly larger concentrations of apo-WTS-S (40–47 μm). Because the disulfide-reduced partially metallated form is likely to form a weaker dimer than a disulfide-intact partially metallated form, it is conceivable that dimeric Co(II)-WT2SH could dissociate in solution into a monomeric form that might act as the initiating agent. To address this possibility, we compared equivalent concentrations of apo-WT2SH and Co(II)-WT2SH for their fibrillation initiating abilities in apo-WTS-S (Fig. 2A). We found that 10 μm Co(II)-WT2SH was as efficient at initiating fibrillation of apo-WT2SH as 10 μm apo-WT2SH as demonstrated by their identical lag times. We analyzed Co(II)-WT2SH by in-line LC-ICP where the output of an SEC column was analyzed directly for its metal content by ICP-MS (14). An injection of 10 μm 1.8 Co(II)-WT2SH yielded a large peak at 19 min corresponding to the dimeric form, and a smaller peak at 23 min corresponding to the monomeric form of WT2SH, each of which was loaded with cobalt as suggested by the in-line ICP chromatogram (Fig. 2B). Thus, under fibrillation conditions, Co(II)-WT2SH appeared to exist predominantly in the dimeric state. Given the ability of this sample to initiate fibrillation of apo-WTS-S as efficiently as apo-WT2H, this experiment strongly suggested that disulfide-reduced SOD1, existing in a dimeric state due to being metal-loaded, is as efficient as monomeric apo-WT2H at initiating fibrillation. However, a scenario in which the small amount of the monomeric cobalt-bound, disulfide-reduced form initiates fibrillation and its subsequent capture in fibrils drives the dimer to monomer equilibrium forward resulting in sustained initiation by the monomeric form cannot be ruled out.
We also tested a number of FALS-associated SOD1 mutants for their ability to initiate fibrillation. They were selected based on our knowledge of their biophysical behavior, position of the mutation (A4V, G37R, G93A, L38V, and D101N being wild-type like and H48Q being a metal-binding region mutant), prevalence in human patients (A4V), and availability of transgenic mouse models (G93A and G37R). Each of the mutants tested was capable of binding Co(II) in the zinc site. Additionally, in each of them, Co(II) added in excess of 2 eq/dimer showed a spectroscopic signature of binding in the copper site (Fig. 3A). Although there have been reports of the mutants A4V, G93A, and L38V being mismetallated to varying extents, we did not observe any evidence of Co(II) binding in the copper site before it saturated the zinc site (38). Each of the mutants tested was capable of initiating fibrillation in the Co(II)-bound form (Fig. 3B). The lag phases of these reactions were similar to the fibrillation reactions where the initiating agent was the apo-form of the same mutant in its disulfide-reduced form, suggesting that, despite stabilizing the overall structure, binding of Co(II) has no inhibitory effect on the ability of disulfide-reduced SOD1 mutants to initiate fibrillation.
Spontaneous fibrillation of Cys-57–Cys-146 disulfide-intact WT SOD1 requires the presence of a substoichiometric amount of the disulfide-reduced form of WT SOD1. In our hands, a small amount of disulfide-reduced mutant SOD1 could initiate fibrillation when mixed with a larger amount of disulfide-intact WT SOD1 that did not form fibrils on its own. However, we found the converse not to be true. When small amounts (5–20%) of disulfide-reduced WT SOD1 were mixed with larger amounts of disulfide-intact SOD1 mutant, we observed no increase in the ThT fluorescence in these reactions (Fig. 3C). When significantly higher amounts of WT SOD12SH (40–50%) were added to mutant SOD1S-S, fibrillation did occur, and the fibrils contained both WT and mutant forms of SOD1. However, the initiating ability of WT SOD12SH in these reactions is unclear because SOD12SH can spontaneously fibrillate on its own. Therefore, WT2SH fibrils may have formed first, which then acted as seeds to recruit mutant SOD1. The fate of WT SOD12SH in these reactions and the precise mechanism of initiation by SOD12SH warrant further investigation. Overall, these experiments suggest an important role for disulfide-reduced forms of SOD1 mutants in promoting the beginning of fibrillation when co-expressed with wild-type SOD1, as is the case in most FALS patients.
Seeded Amyloid Growth
Seeds are commonly generated by sonication-driven shearing of mature fibrils to generate active ends (28). They are then incubated with soluble protein, and ThT fluorescence is monitored as a measure of the rate of fibril growth. To examine the ability of various forms of SOD1 to support seeded amyloid growth, we used fibrils generated by incubating apo-WTS-S in the presence of 5 mm DTT. These fibrils lacked intermolecular disulfide bonds as shown by denaturation in the presence of thiol-alkylating agent N-ethylmaleimide and subsequent HPLC-MS (31). When seeds prepared from these fibrils were added at 10–15 mol % to soluble apo-WTS-S in the absence of reducing agents, fibril growth occurred with a lag phase of about 20 h as shown by ThT fluorescence (Fig. 4A; Table 1). Because apo-WTS-S incubated on its own in the absence of reducing agents did not fibrillate even after 7 days of incubation (data not shown), this result suggested that fibril seeds thus prepared were active and efficient, although the lag phase was long compared with that of spontaneous fibrillation of apo-WTS-S in the presence of 5 mm DTT (4–5 h).
FIGURE 4.
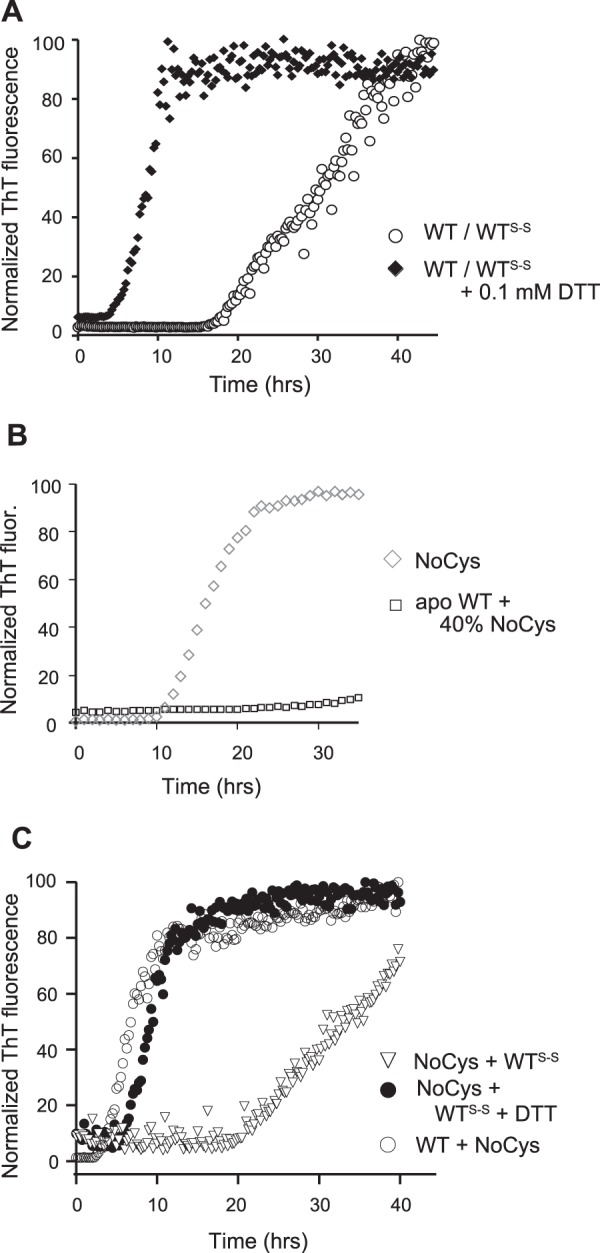
Role of cysteine residues in fibril-seeded growth of SOD1. A, ThT fluorescence during seeded fibrillation of 45 μm apo-WTS-S (○) or apo-WTS-S and 0.1 mm DTT (♦) in the presence of 10% (by volume) WT fibril seeds. B, NoCys SOD1 fibrillates spontaneously but cannot initiate fibrillation of apo-WTS-S. Fibrillation was measured as ThT fluorescence over time. Reactions consisted of all NoCys-SOD1 (diamonds) or 40% NoCys-SOD1 and 60% apo-WT-SOD1S-S (squares). C, seeded growth kinetics of recruitment of apo-WTS-S with NoCys-SOD1 fibril seeds (▿), apo-WTS-S with NoCys SOD1 fibril seeds, and 0.1 mm DTT (●) and NoCys-SOD1 in the presence of WT-SOD1 fibril seeds (○).
TABLE 1.
Summary of experiments exploring the role of the disulfide bond in seeded growth experiments
+ denotes fibrillation with a lag time of around 20 h; +++ denotes fibrillation with a shorter lag time of around 5 h.
| Bulk protein | Initiating species | Seed | DTT | Fibril formation | Ref. |
|---|---|---|---|---|---|
| Initiation | |||||
| WTS-S | No | 31, 32 | |||
| WT2SH | Yes | This work | |||
| NoCys | Yes | This work | |||
| WTS-S | WT2SH | Yes | 32 | ||
| WTS-S | NoCys | No | This work | ||
| Seeded growth | Lag phase | ||||
| WTS-S | WTS-S | + | This work | ||
| WTS-S | WTS-S | 0.1 mm | +++ | This work | |
| WTS-S | NoCys | + | This work | ||
| WTS-S | NoCys | 0.1 mm | +++ | This work | |
| NoCys | WTS-S | +++ | This work | ||
Having established that SOD1 fibril seeds could recruit soluble Cys-57–Cys-146 disulfide-intact SOD1, albeit with a longer lag phase, we asked whether reducing any intramolecular disulfide bonds in the fibrils would accelerate the reaction. We found that the addition of a small amount of DTT (0.1 mm) accelerated the kinetics of this reaction significantly, as shown by a shorter lag phase (Fig. 4A). Because the DTT added was below the minimum concentration required for spontaneous fibrillation of soluble WTS-S under reducing conditions (0.25 mm) (31), the increase in ThT fluorescence should not have arisen from spontaneous fibrillation of WTS-S initiated by WT2SH that was formed in situ. Therefore, the added DTT was acting on either intramolecular disulfide bonds in SOD1 in solution or in the seeds, and reduction of these bonds either in the seeds or in the solution was accelerating the kinetics of this reaction. Although a tempting way to address this question is to add WT2SH to WTS-S seeds and monitor ThT fluorescence, a small lag phase would be meaningless because disulfide-reduced apo-SOD1 fibrillates spontaneously and may do so before being recruited by the fibril seeds.
To compare the seeded growth efficiency of WT2SH with respect to the disulfide-intact form, we used the NoCys mutant where all four cysteine residues were mutated to serines or alanines. Apo NoCys-SOD1 has been shown to behave in a similar fashion structurally and biophysically to WT2SH and can spontaneously fibrillate in the absence of reducing agents (32, 44), but it does not initiate fibrillation of WTS-S (Fig. 4B; Table 1). When seeds prepared from NoCys fibrils were added to WTS-S, the lag phase was about 20 h, similar to WTS-S added to WTS-S seeds, but the presence of 0.1 mm DTT lowered the lag phase to about 4 h (Fig. 4C). In this case, the added DTT can have an effect only on the added soluble WTS-S by reducing the disulfide bond because the seeds composed of NoCys-SOD1 do not have any cysteines. Therefore, this experiment suggested that disulfide-reduced SOD1 is recruited faster than disulfide-intact SOD1 by fibril seeds and that the faster recruitment does not require the presence of free thiols in the reduced protein.
Next, we looked at the effect of zinc on SOD1-seeded growth. As before, cobalt was used as a zinc substitute and was added at pH 5.5 to ensure selective occupancy of the zinc site. We examined both Co-SOD1S-S and apo-SOD1S-S for their ability to be recruited by fibril seeds. Co-WTS-S was recruited somewhat more slowly than apo-WTS-S as shown by a longer lag phase (Fig. 5A), and this was true irrespective of whether one or both of the zinc sites were occupied by cobalt in the dimer. The same effect was also observed for 1Zn and 2Zn-WTS-S (per dimer), although the location of the bound zinc could not be directly ascertained in these experiments. However this was not a consistent effect in mutants. For some mutants such as A4V, L38V, H48Q, and D101N, contrary to our expectations, Co-SOD1S-S was actually recruited faster than the apo-form. In mutants G93A and G37R, cobalt binding slowed down the kinetics of recruitment as shown by a slightly longer lag phase. Despite the enormous stability and tighter structure gained upon zinc binding, in no case did zinc completely inhibit fibril growth (Fig. 5B).
FIGURE 5.
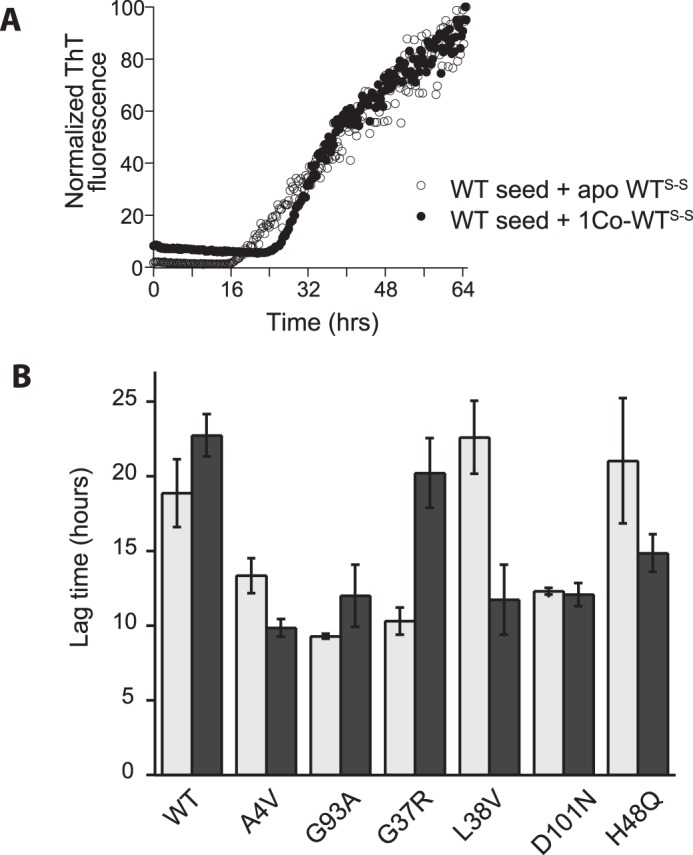
Role of cobalt in modulating fibril-seeded growth kinetics of SOD1. A, ThT fluorescence during seeded fibrillation of 45 μm apo-WTS-S (○) or Co-WTS-S (●) in the presence of 10% (by volume) WT fibril seeds. B, lag time of seeded fibrillation reactions of various SOD1 mutants in their apo, disulfide-intact (light gray), or cobalt-bound, disulfide-intact (dark gray) states. The equivalents of cobalt bound per dimer were 1.3 eq (WT), 0.8 eq (A4V), 0.9 eq (G93A), 1.5 eq (G37R), 1.4 eq (L38V, 0.9 eq (D101N), and 1.6 eq (H48Q).
Effects of Mutations on Disulfide Bond Reduction
Assuming that fibrillation in vivo is started by disulfide-reduced forms of mutant SOD1, as was shown to occur in vitro in the previous section, it becomes important to understand the factors that might lead to an accumulation of this form of SOD1. Tiwari and Hayward (51) have already shown that disulfide bond reduction occurs at lower DTT concentrations in mutant SOD1 than in wild-type SOD1, leading to the conclusion that the disulfide bond in mutants is more easily reduced than that in WT SOD1. To understand this process better, we investigated the kinetics of disulfide reduction in apo-SOD1 under mildly reducing conditions akin to those in the cytoplasm of cells. Both wild-type SOD1 and FALS mutants were examined by incubation in the presence of 5 mm TCEP at pH 7 followed by thiol alkylation using AMS. SDS-PAGE of the reactions showed that SOD1S-S migrated as a set of two bands corresponding to the unmodified polypeptide and SOD1 modified by a single AMS, possibly at the non-disulfide Cys-111, which is solvent-accessible at pH 7. Occasionally a higher band corresponding to alkylation at both the non-disulfide cysteines (Cys-6 and Cys-111) was observed. SOD12SH migrated higher than these bands due to additional modifications at Cys-57 and Cys-146 (Fig. 6A). We found that almost all the mutants examined were reduced much faster than WTS-S suggesting that the disulfide bond of ALS mutants was kinetically more accessible to reducing agents than that of WT SOD1 in the absence of bound metals (Fig. 6A).
FIGURE 6.
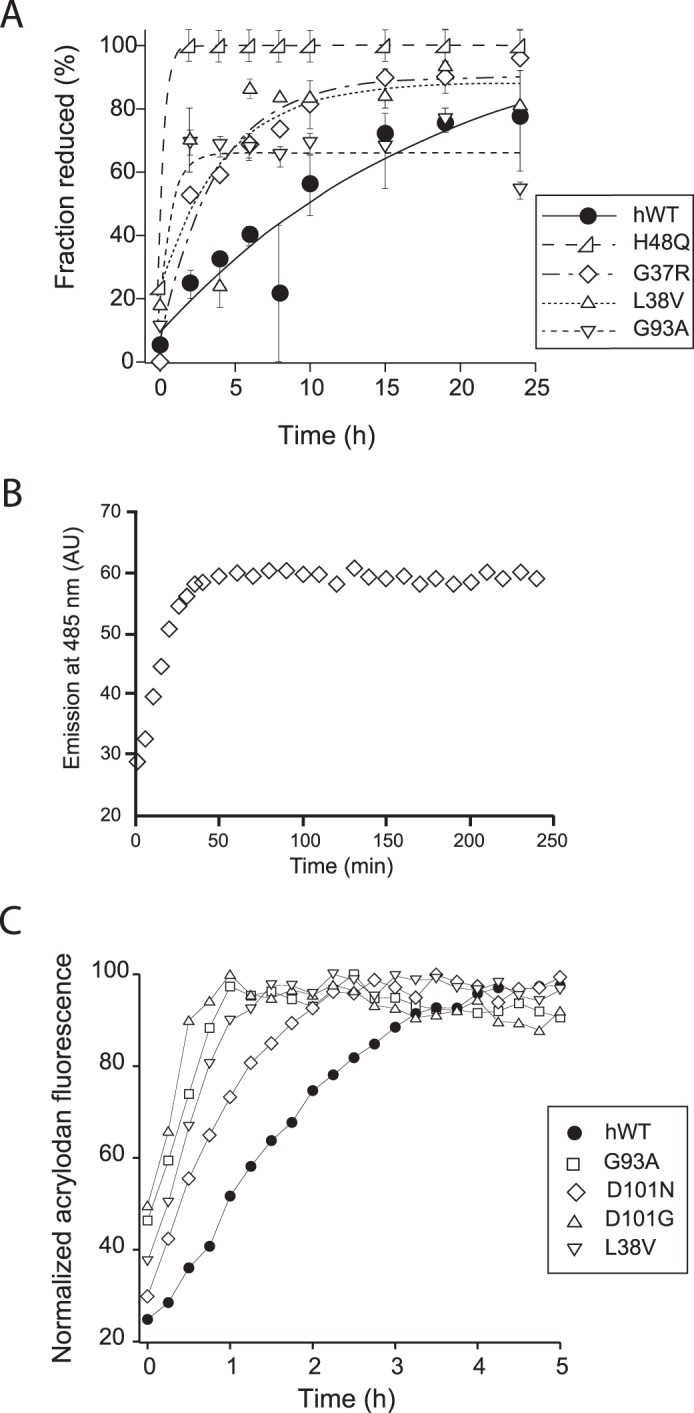
Kinetics of disulfide bond reduction in WT and mutant SOD1. A, apo-SOD1 was incubated with 5 mm TCEP at 37 °C, and at indicated intervals, aliquots were reacted with AMS and quantified by SDS-PAGE and densitometry analysis. The mutants examined were apo-WTS-S (●), apo-G37RS-S (♢), apo-L38VS-S (▵), apo-H48QS-S (▷), and apo-G93AS-S (▿). B, acrylodan fluorescence can be used to track the disulfide bond reduction of SOD1 in real time. 20 μm apoWTS-S conjugated with acrylodan was incubated with 100 mm DTT at 37 °C. without agitation. Reduction of the disulfide bond was verified by SEC-HPLC. C, acrylodan fluorescence as a function of time in reactions containing WT or mutant SOD1 conjugated to acrylodan and incubated in conditions promoting fibrillation (5 mm DTT, with agitation at 37 °C). The mutants examined are apo-WTS-S (●), apo-L38VS-S (▿), apo-H48QS-S and apo-G93AS-S (□), apo-D101N (♢), and apo-D101G (▵) (14).
To determine whether this phenomenon could be observed under fibrillation conditions, we developed a fluorescence-based assay for monitoring reduction in situ. Acrylodan is a thiol-specific fluorophore whose fluorescence is highly sensitive to the polarity of its local environment, making it particularly suitable for probing protein conformation changes (52). SOD1 labeled with acrylodan at Cys-111 showed an increased fluorescence at 485 nm (λex = 395 nm) when incubated in the presence of DTT until the fluorescence reached a maximum corresponding to complete reduction of the disulfide bond (Fig. 6B). When acrylodan-labeled WT and mutant SOD1 were incubated under fibrillation conditions (pH 7, 37 °C with agitation) in the presence of 5 mm DTT, all mutants examined showed a faster increase in acrylodan fluorescence than apo-WTS-S before reaching saturation (Fig. 6C). Thus, the disulfide bond of demetallated SOD1 mutants was kinetically more susceptible to reduction under fibrillation conditions, compared with wild-type SOD1. Interestingly, the disulfide bond of 1- or 2Zn-SOD1S-S, WT or mutant, was highly resistant to reduction even in the presence of much higher concentrations of TCEP (up to 50 mm) and/or upon longer incubation. These experiments suggested that in the metal-free state, disulfide-intact mutant SOD1 is more prone to sample a conformation in which the disulfide bond is easily reduced, and the binding of zinc reduces the ability of SOD1 to sample this state in both mutant and WT SOD1.
Toxicity in Vivo
To test the relevance of our experimental SOD1 fibrillation model to human cells, we examined whether the presence of fibril seeds altered cell viability. SH-SY5Y cells that had been treated with retinoic acid to induce a neuronally differentiated state were exposed to increasing concentrations of fibril seeds prepared as for in vitro fibrillation (Fig. 7). Although the viability of cells exposed to a very low amount of fibril seeds (1 μg) was comparable with the buffer control, cells exposed to higher amounts of fibril seeds showed a dose-dependent loss of viability, consistent with a toxic property of fibril seeds. Wild-type human SOD1 fibrils prepared with DTT using our laboratory protocol were also demonstrated to be toxic to THP1 cells (8).
FIGURE 7.
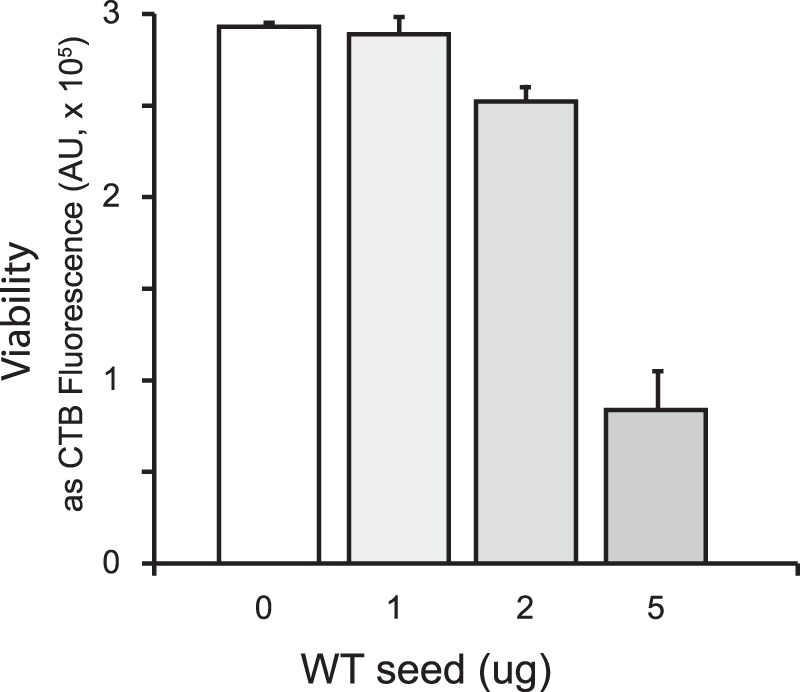
Toxicity of fibril seeds. SY5Y cells plated in 96-well plates and induced to differentiate into a neuronal phenotype were treated with the indicated concentration of SOD1 fibril seeds prepared as described above. After 2 h, the medium was replaced, removing excess seeds, and incubation was continued for 24 h, after which viability was determined. Viability is shown as Cell Titer Blue Assay fluorescence, in arbitrary units (AU).
Discussion
The key findings of our study are as follows: 1) any form of disulfide-reduced SOD1, irrespective of cobalt or zinc status, is competent to initiate spontaneous fibrillation; 2) the Cys-57–Cys-146 disulfide bond has the highest ability to modulate SOD1 fibrillation because the presence of this bond inhibits fibril initiation completely and slows down seeded amyloid growth considerably; 3) occupancy of the zinc site has a variable effect on the rate of seeded amyloid growth across different SOD1 mutants, and 4) mutants have a higher propensity to exist in an initiation-competent state in a reducing environment. These findings suggest that most possible intermediates on the maturation pathway can be recruited by growing fibrils. The prominent exception is the fully mature holo-form of SOD1 containing a copper atom, a zinc atom, and a disulfide bond per subunit of the dimer, which can neither initiate fibrillation nor sustain it.
An unexpected finding in our study was the complete inability of cobalt and zinc to protect SOD1 from all steps of fibrillation. In the disulfide-reduced state, cobalt-bound SOD1 is as efficient as apo-SOD1 in initiating fibrillation. The effects of zinc and cobalt binding to disulfide-intact SOD1 have been well documented. Compared with apo-WTS-S, the binding of the first zinc to the dimer results in a more thermodynamically stable protein that melts at a higher temperature in DSC experiments and has a more tightly packed structure resulting in slower hydrogen-deuterium exchange (37). The binding of the second zinc raises the melting transition to even higher temperatures (37). The effects of cobalt or zinc binding to the disulfide-reduced form are less understood. Although apo-SOD12SH is monomeric, zinc binding converts SOD12SH to a dimer that has a higher thermodynamic stability compared with the apo-form as shown by a higher melting temperature in DSC experiments (44). Recent unpublished work from our laboratory4 suggests that it also has a tighter structure in several β-strands and loops as shown by lower hydrogen-deuterium exchange. However, these structural changes upon metal ion binding do not prevent disulfide-reduced SOD1 from acting as an efficient fibril-initiating agent, nor do they hinder the ability of disulfide-reduced SOD1 to be incorporated into growing fibrils with a much smaller lag time compared with the disulfide-intact form. The comparable efficiency of initiation by dimeric cobalt-bound SOD12SH and apo-monomeric SOD12SH also suggests that dimer dissociation is not necessary for initiating spontaneous fibrillation. This conclusion differs from those of several earlier studies that concluded that a large component of the destabilization of SOD1 structure leading to its aggregation arose from dimer dissociation (23, 27, 53, 54).
Two groups have considered disulfide scrambling as part of the initiation process (55), and indeed, the presence of the disulfide-forming cysteines Cys-57 and Cys-146 in their reduced states is necessary for initiation (56). However, this is not true for seeded amyloid growth in our experiments. NoCys-SOD1, used as a “cysteineless” model for disulfide-reduced WT-SOD1 because of their similar thermal stabilities as shown in DSC experiments, was recruited as efficiently as the disulfide-reduced form of WT SOD1. Its comparable rate of recruitment to that of disulfide-reduced SOD1 during seeded amyloid growth suggests that it is not the cysteine residues but the conformational flexibility of the disulfide-reduced state that allows this form of SOD1 to be recruited at a faster rate.
Apo-WT-SOD12SH can form a dimer in two ways as follows: by disulfide formation leading to apo-WTS-S that melts at 52 °C in DSC experiments or by the acquisition of zinc leading to the (E2, Zn-E) WT2SH (where E is an empty metal-binding site), which melts at 58 °C (35, 44). Despite the higher melting temperature of the disulfide-reduced dimeric form arising from a stronger dimer, a more tightly packed β-barrel, or both, we find that it is recruited far more efficiently by the fibril seed as demonstrated by a shorter lag time. These results reinforce the lack of correlation between overall stability of SOD1 and its aggregation propensity.
One mechanism by which a more stable form of the protein can be more fibrillation-prone compared with a less stable one is local unfolding. In this scenario, one or more β-strand from one SOD1 monomer or dimer unfolds enough to establish new contacts with a β-strand of another SOD1 molecule while keeping the rest of the β-barrel intact. It is possible for such a phenomenon to be independent of the overall stability of the SOD1 dimer. Indeed, local unfolding of segments of SOD1 has been observed for wild-type SOD1 as well as a number of mutants in both the disulfide-intact and -reduced states, and the extent of unfolding was poorly correlated with the overall stability of the protein as evident from the melting temperature in DSC experiments (57, 58). The inability of the fully metallated, Cu2Zn2-SOD1 to participate in either fibril initiation or seeded growth suggests that local unfolding events that allowed the formation of intermolecular contacts in the less mature forms of SOD1 are blocked in the fully mature enzyme.
In transgenic mouse models of ALS overexpressing SOD1 mutants, the SOD1-rich aggregates are composed almost entirely of disulfide-reduced SOD1 (13). This is an important although not unexpected finding because the abnormally high level of SOD1 expression probably leads to a saturation of the SOD1 folding apparatus resulting in the buildup of disulfide-reduced SOD1 over time. In ALS patients, however, the status of the disulfide in SOD1 is not known. Because they express endogenous amounts of SOD1, the chaperones responsible for SOD1 folding should not be overwhelmed. Therefore, the relative amounts of disulfide-reduced versus -intact forms of SOD1 should be comparable. Under such circumstances, the recruitment of Cys-57–Cys-146 disulfide-intact forms of SOD1 by the seed or growing fibril becomes a more likely scenario and may play an important role in the buildup of SOD1 aggregates in these patients.
Inside the cell, the newly synthesized, completely immature SOD1 polypeptide undergoes a series of post-translational modifications that convert it to the exceptionally stable, fully active form, which is highly resistant to aggregation. Although the exact sequence of events remains unknown, recent papers suggest that the first step is binding of a single zinc ion (42, 43). The formation of the copper-bound disulfide-intact form can now occur via one of two pathways. In the predominant pathway that has been observed in many eukaryotes, including yeast and human, the protein CCS (copper chaperone for SOD1) transfers a copper ion and inserts a disulfide bond in a concerted mechanism whereupon the disulfide-intact, copper- and zinc-bound SOD1 forms a dimer (59). CCS also appears to enhance the formation of the intrasubunit disulfide bond without a concomitant copper insertion leading to the formation of disulfide-intact, zinc-bound SOD1 (42, 43). Additionally, a chaperone-independent pathway has been observed in mammalian cells and in some multicellular organisms like Caenorhabditis elegans, whereby SOD1 acquires a disulfide bond independent of and, probably preceding, the binding of copper to form the fully active dimeric form (60, 61). Our results also show that a number of intermediates in these two pathways that have an intact disulfide bond with varying zinc equivalents can sustain seeded fibril growth. These maturation steps occur in at least two locations, the cytoplasm, where synthesis takes place, and the mitochondrial intermembrane space, into which SOD1 is imported in an unfolded state, so more than one location exists where fibril initiation and elongation could occur.
Based on our results identifying specific intermediates on the SOD1 folding pathway that can initiate or sustain fibrillation, we propose a model for the SOD1 fibrillation pathway in vivo when WT and mutant SOD1 are coexpressed, as is the case for most FALS patients (Fig. 8). Spontaneous fibrillation is initiated in one or more steps by the formation of an amyloid nucleus from a very early intermediate in the maturation process, most likely the zinc-bound disulfide-reduced mutant SOD1 (Fig. 8A). For metal-binding mutants that cannot bind zinc, the apo-form of disulfide-reduced SOD1 forms the amyloid nucleus or seed. The nucleus subsequently becomes stabilized by recruitment of additional soluble WT or mutant SOD1 and elongates to form a mature fibril in multiple steps. Seeded growth proceeds this way as well, and the rate at which this occurs depends on the disulfide status of SOD1 that interacts with the nucleus (Fig. 8B). For interactions with disulfide-reduced SOD1 that is apo or partially metallated with zinc, the fibril nucleus is rapidly converted to a mature fibril. However, if the nucleus interacts with disulfide-intact, apo, or partially metallated forms, the mature fibril forms on a much slower time scale. Fully mature SOD1 containing a copper ion, a zinc ion, and a disulfide bond in each subunit of the dimer does not participate in any aspect of fibrillation.
FIGURE 8.
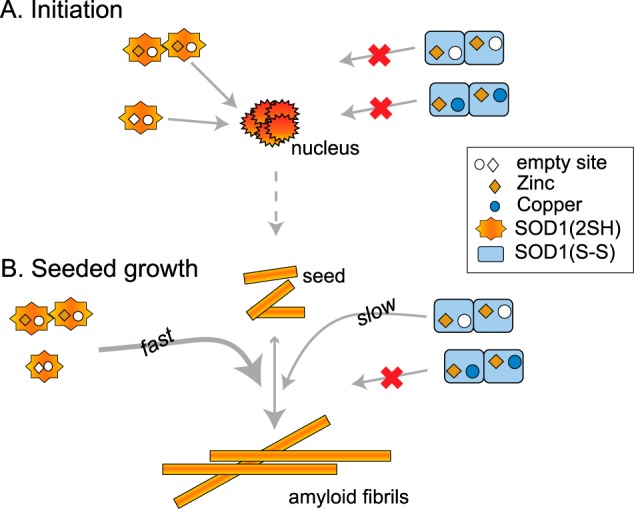
Proposed mechanism of SOD1 aggregation driven by intermediates on the maturation pathway. See text for description.
It is reasonable to expect that if toxic species are formed during the course of fibrillation, either as direct intermediates or as by-products of the process, then conditions that promote fibrillation are likely to enhance the formation of such species. Thus, small molecules that interfere with fibrillation pathways or intermediates are under investigation (62). Although WT SOD1 alone can participate in all steps in this pathway, the expression of a mutant SOD1 would strongly promote aggregation because of the following: (a) its higher propensity to exist in the disulfide-reduced apo-state and (b) its ability to initiate fibrillation of WT even when present in minute quantities. The second feature is especially important for disease as it guarantees that fibril-seeded growth can be sustained even when the coexpressed mutant has a short half-life. Indeed, coexpression of WT exacerbates the disease profile of mice expressing mutants, and the contrast in disease profile is dramatic for short-lived mutants like A4V and L126Z (63–65). It is important to remember that the identity of the toxic species is unknown; hence, the relative contributions of spontaneous fibrillation versus seeded amyloid formation to this process are unclear. Recent observations that not all ALS mutations are fully penetrant (66) and that overexpression of wild-type human SOD1 in mice leads to ALS symptoms (15) highlight the stochastic nature of initiation of disease and the potential for wild-type human SOD1 to play a role in sporadic cases of ALS.
There is now considerable evidence, including the toxicity studies we report above, showing that cells internalize externally added fibrils and that some form of SOD1 aggregates can move from cell to cell and even from animal to animal (7–9, 11, 12) and induce aggregation of partially mature states of SOD1. This phenomenon, the spreading of the aggregation via the transfer of seeds, offers a mechanism for the spreading of the neurodegeneration phenotype from a small initial lesion, which is a hallmark of the disease. Initiation is clearly critical to disease onset, but it is the spreading of the pathology that generates symptoms in patients. Our mechanistic understanding of the requirements for aggregation coupled with the knowledge that seeds are transferred from cell to cell should contribute to development of treatments for this disease. Based on current knowledge of the folding pathway in the cell and the tight coupling between the oligomerization state of SOD1 and its metal occupancy or disulfide status, a number of specific, partially mature states of SOD1 are considered highly likely to exist in the cell (35, 42, 44). Thus, our efforts at containing the progress of the disease are best concentrated on halting the cell-to-cell transfer of aggregates or on inhibiting the seeded growth process. Because fully mature SOD1 does not aggregate, one way to accomplish the latter goal could be to stimulate rapid achievement of full metallation and to maintain the enzyme in its fully metallated state. A small molecule approach would be to find agents that stabilize SOD1 in a similar manner to prevent its recruitment or, alternatively, that block the ability of seeds to recruit fresh material.
Author Contributions
M. C. designed and carried out the majority of the studies. E. N. carried out and analyzed the experiment in Fig. 6 and provided other technical assistance. C. D. S. designed vectors and a protein purification system to produce mutant proteins. E. B. G. prepared the figures and helped interpret data. J. S. V. and J. P. W. directed the overall experimental design and made substantial contributions to data interpretation. M. C., E. B. G., J. S. V., and J. P. W. wrote and critically revised the paper, and all authors reviewed the results and agreed on the conclusions.
Acknowledgments
We thank Armando Durazo and Kevin Barnese for purifying specific SOD1 mutants and Herman Lelie for help with the HPLC-ICP-MS experiments. We thank David Borchelt for insightful discussions.
This work was supported by National Institutes of Health Program Project Grant P01 NS049134 from NINDS. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
A. Durazo, unpublished data.
- FALS
- familial amyotrophic lateral sclerosis
- ALS
- amyotrophic lateral sclerosis
- TCEP
- tris(2-carboxyethyl)phosphine
- ICP-MS
- inductively coupled plasma-mass spectrometry
- DSC
- differential scanning calorimetry
- SEC
- size-exclusion chromatography
- ThT
- thioflavin T
- AMS
- 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid.
References
- 1.Chiti F., and Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 2.Eisele Y. S., Monteiro C., Fearns C., Encalada S. E., Wiseman R. L., Powers E. T., and Kelly J. W. (2015) Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug. Discov. 14, 759–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caughey B., and Lansbury P. T. (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298 [DOI] [PubMed] [Google Scholar]
- 4.Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soto C. (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60 [DOI] [PubMed] [Google Scholar]
- 6.Prusiner S. B. (2012) A unifying role for prions in neurodegenerative diseases. Science 336, 1511–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ayers J. I., Fromholt S., Koch M., DeBosier A., McMahon B., Xu G., and Borchelt D. R. (2014) Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol. 128, 791–803 [DOI] [PubMed] [Google Scholar]
- 8.Bhatia N. K., Srivastava A., Katyal N., Jain N., Khan M. A., Kundu B., and Deep S. (2015) Curcumin binds to the pre-fibrillar aggregates of Cu/Zn superoxide dismutase (SOD1) and alters its amyloidogenic pathway resulting in reduced cytotoxicity. Biochim. Biophys. Acta 1854, 426–436 [DOI] [PubMed] [Google Scholar]
- 9.Chia R., Tattum M. H., Jones S., Collinge J., Fisher E. M., and Jackson G. S. (2010) Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS ONE 5, e10627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furukawa Y., Kaneko K., Watanabe S., Yamanaka K., and Nukina N. (2013) Intracellular seeded aggregation of mutant Cu,Zn-superoxide dismutase associated with amyotrophic lateral sclerosis. FEBS Lett. 587, 2500–2505 [DOI] [PubMed] [Google Scholar]
- 11.Grad L. I., Guest W. C., Yanai A., Pokrishevsky E., O'Neill M. A., Gibbs E., Semenchenko V., Yousefi M., Wishart D. S., Plotkin S. S., and Cashman N. R. (2011) Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. U.S.A. 108, 16398–16403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Münch C., O'Brien J., and Bertolotti A. (2011) Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. U.S.A. 108, 3548–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karch C. M., Prudencio M., Winkler D. D., Hart P. J., and Borchelt D. R. (2009) Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc. Natl. Acad. Sci. U.S.A. 106, 7774–7779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lelie H. L., Liba A., Bourassa M. W., Chattopadhyay M., Chan P. K., Gralla E. B., Miller L. M., Borchelt D. R., Valentine J. S., and Whitelegge J. P. (2011) Copper and zinc metallation status of copper-zinc superoxide dismutase from amyotrophic lateral sclerosis transgenic mice. J. Biol. Chem. 286, 2795–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graffmo K. S., Forsberg K., Bergh J., Birve A., Zetterström P., Andersen P. M., Marklund S. L., and Brännström T. (2013) Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 51–60 [DOI] [PubMed] [Google Scholar]
- 16.Harper J. D., and Lansbury P. T. Jr. (1997) Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66, 385–407 [DOI] [PubMed] [Google Scholar]
- 17.Knowles T. P., Waudby C. A., Devlin G. L., Cohen S. I., Aguzzi A., Vendruscolo M., Terentjev E. M., Welland M. E., and Dobson C. M. (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537 [DOI] [PubMed] [Google Scholar]
- 18.Oztug Durer Z. A., Cohlberg J. A., Dinh P., Padua S., Ehrenclou K., Downes S., Tan J. K., Nakano Y., Bowman C. J., Hoskins J. L., Kwon C., Mason A. Z., Rodriguez J. A., Doucette P. A., Shaw B. F., and Valentine J. S. (2009) Loss of metal ions, disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase. PLoS ONE 4, e5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan P. K., Chattopadhyay M., Sharma S., Souda P., Gralla E. B., Borchelt D. R., Whitelegge J. P., and Valentine J. S. (2013) Structural similarity of wild-type and ALS-mutant superoxide dismutase-1 fibrils using limited proteolysis and atomic force microscopy. Proc. Natl. Acad. Sci. U.S.A. 110, 10934–10939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DiDonato M., Craig L., Huff M. E., Thayer M. M., Cardoso R. M., Kassmann C. J., Lo T. P., Bruns C. K., Powers E. T., Kelly J. W., Getzoff E. D., and Tainer J. A. (2003) ALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilization. J. Mol. Biol. 332, 601–615 [DOI] [PubMed] [Google Scholar]
- 21.Stathopulos P. B., Rumfeldt J. A., Scholz G. A., Irani R. A., Frey H. E., Hallewell R. A., Lepock J. R., and Meiering E. M. (2003) Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc. Natl. Acad. Sci. U.S.A. 100, 7021–7026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vassall K. A., Stubbs H. R., Primmer H. A., Tong M. S., Sullivan S. M., Sobering R., Srinivasan S., Briere L.-A., Dunn S. D., Colón W., and Meiering E. M. (2011) Decreased stability and increased formation of soluble aggregates by immature superoxide dismutase do not account for disease severity in ALS. Proc. Natl. Acad. Sci. U.S.A. 108, 2210–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rakhit R., Crow J. P., Lepock J. R., Kondejewski L. H., Cashman N. R., and Chakrabartty A. (2004) Monomeric Cu,Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J. Biol. Chem. 279, 15499–15504 [DOI] [PubMed] [Google Scholar]
- 24.Banci L., Bertini I., Durazo A., Girotto S., Gralla E. B., Martinelli M., Valentine J. S., Vieru M., and Whitelegge J. P. (2007) Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: A possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. U.S.A. 104, 11263–11267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pantoliano M. W., Valentine J. S., Mammone R. J., and Scholler D. M. (1982) The pH dependence of metal ion binding to the native zinc site of bovine erythrocuprein (superoxide dismutase). J. Am. Chem. Soc. 104, 1717–1723 [Google Scholar]
- 26.Lyons T. J., Liu H., Goto J. J., Nersissian A., Roe J. A., Graden J. A., Café C., Ellerby L. M., Bredesen D. E., Gralla E. B., and Valentine J. S. (1996) Mutations in copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proc. Natl. Acad. Sci. U.S.A. 93, 12240–12244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khare S. D., Caplow M., and Dokholyan N. V. (2004) The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 101, 15094–15099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valentine J. S., Doucette P. A., and Zittin Potter S. (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593 [DOI] [PubMed] [Google Scholar]
- 29.Potter S. Z., and Valentine J. S. (2003) The perplexing role of copper-zinc superoxide dismutase in amyotrophic lateral sclerosis (Lou Gehrig's disease). J. Biol. Inorg. Chem. 8, 373–380 [DOI] [PubMed] [Google Scholar]
- 30.Arnesano F., Banci L., Bertini I., Martinelli M., Furukawa Y., and O'Halloran T. V. (2004) The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J. Biol. Chem. 279, 47998–48003 [DOI] [PubMed] [Google Scholar]
- 31.Chattopadhyay M., Durazo A., Sohn S. H., Strong C. D., Gralla E. B., Whitelegge J. P., and Valentine J. S. (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. U.S.A. 105, 18663–18668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furukawa Y., Kaneko K., Yamanaka K., O'Halloran T. V., and Nukina N. (2008) Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in familial form of ALS. J. Biol. Chem. 283, 24167–24176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheng Y., Chattopadhyay M., Whitelegge J., and Valentine J. S. (2012) SOD1 aggregation and ALS: role of metallation states and disulfide status. Curr. Top. Med. Chem. 12, 2560–2572 [DOI] [PubMed] [Google Scholar]
- 34.Ding F., and Dokholyan N. V. (2008) Dynamical roles of metal ions and the disulfide bond in Cu,Zn superoxide dismutase folding and aggregation. Proc. Natl. Acad. Sci. U.S.A. 105, 19696–19701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doucette P. A., Whitson L. J., Cao X., Schirf V., Demeler B., Valentine J. S., Hansen J. C., and Hart P. J. (2004) Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant: insights into the molecular basis for dimer stability. J. Biol. Chem. 279, 54558–54566 [DOI] [PubMed] [Google Scholar]
- 36.Kayatekin C., Zitzewitz J. A., and Matthews C. R. (2008) Zinc binding modulates the entire folding free energy surface of human Cu,Zn superoxide dismutase. J. Mol. Biol. 384, 540–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potter S. Z., Zhu H., Shaw B. F., Rodriguez J. A., Doucette P. A., Sohn S. H., Durazo A., Faull K. F., Gralla E. B., Nersissian A. M., and Valentine J. S. (2007) Binding of a single zinc ion to one subunit of copper-zinc superoxide dismutase apoprotein substantially influences the structure and stability of the entire homodimeric protein. J. Am. Chem. Soc. 129, 4575–4583 [DOI] [PubMed] [Google Scholar]
- 38.Goto J. J., Zhu H., Sanchez R. J., Nersissian A., Gralla E. B., Valentine J. S., and Cabelli D. E. (2000) Loss of in vitro metal ion binding specificity in mutant copper-zinc superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 275, 1007–1014 [DOI] [PubMed] [Google Scholar]
- 39.Banci L., Barbieri L., Bertini I., Cantini F., and Luchinat E. (2011) In-cell NMR in E. coli to monitor maturation steps of hSOD1. PLoS ONE 6, e23561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruns C. K., and Kopito R. R. (2007) Impaired post-translational folding of familial ALS-linked Cu,Zn superoxide dismutase mutants. EMBO J. 26, 855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leinartaite L., Saraboji K., Nordlund A., Logan D. T., and Oliveberg M. (2010) Folding catalysis by transient coordination of Zn2+ to the Cu ligands of the ALS-associated enzyme Cu/Zn superoxide dismutase 1. J. Am. Chem. Soc. 132, 13495–13504 [DOI] [PubMed] [Google Scholar]
- 42.Banci L., Barbieri L., Bertini I., Luchinat E., Secci E., Zhao Y., and Aricescu A. R. (2013) Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat. Chem. Biol. 9, 297–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luchinat E., Barbieri L., Rubino J. T., Kozyreva T., Cantini F., and Banci L. (2014) In-cell NMR reveals potential precursor of toxic species from SOD1 fALS mutants. Nat. Commun. 5, 5502. [DOI] [PubMed] [Google Scholar]
- 44.Furukawa Y., and O'Halloran T. V. (2005) ALS mutations have the greatest destabilizing effect on the apo, reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 280, 17266–17274 [DOI] [PubMed] [Google Scholar]
- 45.Ming L. J., and Valentine J. S. (1990) NMR studies of cobalt (II)-substituted derivatives of bovine copper-zinc superoxide dismutase. Effects of pH, phosphate, and metal migration. J. Am. Chem. Soc. 112, 4256–4264 [Google Scholar]
- 46.Tiwari A., Liba A., Sohn S. H., Seetharaman S. V., Bilsel O., Matthews C. R., Hart P. J., Valentine J. S., and Hayward L. J. (2009) Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J. Biol. Chem. 284, 27746–27758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lyons T. J., Nersissian A., Goto J. J., Zhu H., Gralla E. B., and Valentine J. S. (1998) Metal ion reconstitution studies of yeast copper-zinc superoxide dismutase: the “phantom” subunit and the possible role of Lys7p. J. Biol. Inorg. Chem. 3, 650–662 [Google Scholar]
- 48.Ming L. J., Banci L., Luchinat C., Bertini I., and Valentine J. S. (1988) NMR study of cobalt (II)-substituted yeast and human copper-zinc superoxide dismutase. Inorg. Chem. 27, 728–733 [Google Scholar]
- 49.Mulligan V. K., Kerman A., Ho S., and Chakrabartty A. (2008) Denaturational stress induces formation of zinc-deficient monomers of Cu,Zn superoxide dismutase: implications for pathogenesis in amyotrophic lateral sclerosis. J. Mol. Biol. 383, 424–436 [DOI] [PubMed] [Google Scholar]
- 50.Svensson A.-K., Bilsel O., Kayatekin C., Adefusika J. A., Zitzewitz J. A., and Matthews C. R. (2010) Metal-free ALS variants of dimeric human Cu,Zn-superoxide dismutase have enhanced populations of monomeric species. PLoS ONE 5, e10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tiwari A., and Hayward L. J. (2003) Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J. Biol. Chem. 278, 5984–5992 [DOI] [PubMed] [Google Scholar]
- 52.Prendergast F. G., Meyer M., Carlson G. L., Iida S., and Potter J. D. (1983) Synthesis, spectral properties, and use of 6-acryloyl-2-dimethylaminonaphthalene (Acrylodan). A thiol-selective, polarity-sensitive fluorescent probe. J. Biol. Chem. 258, 7541–7544 [PubMed] [Google Scholar]
- 53.Lindberg M. J., Byström R., Boknäs N., Andersen P. M., and Oliveberg M. (2005) Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc. Natl. Acad. Sci. U.S.A. 102, 9754–9759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rakhit R., Robertson J., Vande Velde C., Horne P., Ruth D. M., Griffin J., Cleveland D. W., Cashman N. R., and Chakrabartty A. (2007) An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat. Med. 13, 754–759 [DOI] [PubMed] [Google Scholar]
- 55.Toichi K., Yamanaka K., and Furukawa Y. (2013) Disulfide scrambling describes the oligomer formation of superoxide dismutase (SOD1) proteins in the familial form of amyotrophic lateral sclerosis. J. Biol. Chem. 288, 4970–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leinartaitë L., and Johansson A. S. (2013) Disulfide scrambling in superoxide dismutase 1 reduces its cytotoxic effect in cultured cells and promotes protein aggregation. PLoS ONE 8, e78060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Durazo A., Shaw B. F., Chattopadhyay M., Faull K. F., Nersissian A. M., Valentine J. S., and Whitelegge J. P. (2009) Metal-free superoxide dismutase-1 and three different amyotrophic lateral sclerosis variants share a similar partially unfolded β-barrel at physiological temperature. J. Biol. Chem. 284, 34382–34389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shaw B. F., Durazo A., Nersissian A. M., Whitelegge J. P., Faull K. F., and Valentine J. S. (2006) Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J. Biol. Chem. 281, 18167–18176 [DOI] [PubMed] [Google Scholar]
- 59.Furukawa Y., Torres A. S., and O'Halloran T. V. (2004) Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 23, 2872–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carroll M. C., Girouard J. B., Ulloa J. L., Subramaniam J. R., Wong P. C., Valentine J. S., and Culotta V. C. (2004) Mechanisms for activating Cu- and Zn-containing superoxide dismutase in the absence of the CCS Cu chaperone. Proc. Natl. Acad. Sci. U.S.A. 101, 5964–5969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leitch J. M., Jensen L. T., Bouldin S. D., Outten C. E., Hart P. J., and Culotta V. C. (2009) Activation of Cu,Zn-superoxide dismutase in the absence of oxygen and the copper chaperone CCS. J. Biol. Chem. 284, 21863–21871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wright G. S., Antonyuk S. V., Kershaw N. M., Strange R. W., and Samar Hasnain S. (2013) Ligand binding and aggregation of pathogenic SOD1. Nat. Commun. 4, 1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng H. X., Shi Y., Furukawa Y., Zhai H., Fu R., Liu E., Gorrie G. H., Khan M. S., Hung W. Y., Bigio E. H., Lukas T., Dal Canto M. C., O'Halloran T. V., and Siddique T. (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7142–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jaarsma D., Teuling E., Haasdijk E. D., De Zeeuw C. I., and Hoogenraad C. C. (2008) Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci. 28, 2075–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu G., Ayers J. I., Roberts B. L., Brown H., Fromholt S., Green C., and Borchelt D. R. (2015) Direct and indirect mechanisms for wild-type SOD1 to enhance the toxicity of mutant SOD1 in bigenic transgenic mice. Hum. Mol. Genet. 24, 1019–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Synofzik M., Ronchi D., Keskin I., Basak A. N., Wilhelm C., Gobbi C., Birve A., Biskup S., Zecca C., Fernández-Santiago R., Kaugesaar T., Schöls L., Marklund S. L., and Andersen P. M. (2012) Mutant superoxide dismutase-1 indistinguishable from wild-type causes ALS. Hum. Mol. Genet. 21, 3568–3574 [DOI] [PubMed] [Google Scholar]