

Abstract
Fatty liver is associated with endoplasmic reticulum stress and activation of the hepatic unfolded protein response (UPR). Reduced hepatic expression of the UPR regulator X-box binding protein 1 spliced (XBP1s) is associated with human nonalcoholic steatohepatitis (NASH), and feeding mice a high-fat diet with fructose/sucrose causes progressive, fibrosing steatohepatitis. This study examines the role of XBP1 in nonalcoholic fatty liver injury and fatty acid-induced cell injury. Hepatocyte-specific Xbp1-deficient (Xbp1−/−) mice were fed a high-fat/sugar (HFS) diet for up to 16 wk. HFS-fed Xbp1−/− mice exhibited higher serum alanine aminotransferase levels compared with Xbp1fl/fl controls. RNA sequencing and Gene Ontogeny pathway analysis of hepatic mRNA revealed that apoptotic process, inflammatory response, and extracellular matrix structural constituent pathways had enhanced activation in HFS-fed Xbp1−/− mice. Liver histology demonstrated enhanced injury and fibrosis but less steatosis in the HFS-fed Xbp1−/− mice. Hepatic Col1a1 and Tgfβ1 gene expression, as well as Chop and phosphorylated JNK (p-JNK), were increased in Xbp1−/− compared with Xbp1fl/fl mice after HFS feeding. In vitro, stable XBP1-knockdown Huh7 cells (Huh7-KD) and scramble control cells (Huh7-SCR) were generated and treated with palmitic acid (PA) for 24 h. PA-treated Huh7-KD cells had increased cytotoxicity measured by lactate dehydrogenase release, apoptotic nuclei, and caspase3/7 activity assays compared with Huh7-SCR cells. CHOP and p-JNK expression was also increased in Huh7-KD cells following PA treatment. In conclusion, loss of XBP1 enhances injury in both in vivo and in vitro models of fatty liver injury. We speculate that hepatic XBP1 plays an important protective role in pathogenesis of NASH.
Keywords: nonalcoholic steatohepatitis, steatosis, unfolded protein response, fatty liver, endoplasmic reticulum stress
nonalcoholic fatty liver disease (NAFLD) is currently the most common cause of abnormal liver chemistry tests in the United States and the Western world. NAFLD represents a spectrum of diseases associated with the accumulation of triglyceride in hepatocytes, ranging from isolated hepatic steatosis to nonalcoholic steatohepatitis (NASH) with inflammation, progressive liver injury, fibrosis, and cirrhosis (2, 6). With the rapid rise in the prevalence of obesity and the metabolic syndrome, NAFLD is estimated to affect more than 30 million people in the United States, with 5% developing cirrhosis. In fact, NASH is expected to be the leading indication for liver transplantation in the United States within the next 10 years (1, 17). The mechanisms underlying the progression from isolated hepatic steatosis to NASH are poorly understood although proinflammatory cytokines (9), oxidative stress, mitochondrial dysfunction (25), and genetic factors are believed to contribute to the pathogenesis.
Endoplasmic reticulum (ER) stress and the activation of the unfolded protein response (UPR) are associated with multiple liver diseases, including NAFLD (16, 21). ER stress is characterized by the accumulation of unfolded or misfolded proteins in the ER. Three UPR pathways are activated through the transmembrane ER sensors: inositol requiring enzyme 1α (IRE1α), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6). These pathways increase ER content, degrade misfolded proteins, reduce the synthesis of new proteins entering the ER, and ultimately restore ER homeostasis. IRE1α is the most evolutionarily conserved signaling component of the UPR, and its endoribonuclease activity is responsible for the unconventional splicing of XBP1 into the physiologically active transcription factor XBP1 spliced (XBP1s). XBP1s regulates several downstream genes that promote protein folding and ER-associated degradation (ERAD). The hepatic IRE1α/XBP1 pathway is also important for lipid metabolism since deficiency of hepatic Xbp1 in mice leads to decreased hepatic lipid synthesis and secretion. Although previous studies have focused on the role of XBP1 in hepatic lipid metabolism, the role of hepatic XBP1 in liver injury and lipotoxicity in fatty liver disorders remains poorly understood.
Several genetic and dietary mouse models have been used to study the pathogenesis of NAFLD and NASH (13, 19), although most of these animal models do not develop progressive steatohepatitis in the milieu of the metabolic syndrome. It was recently shown that wild-type mice fed a high-fat, sucrose/fructose diet develop steatohepatitis with fibrosis, as well as obesity, insulin resistance, and diabetes, similar to human patients with NASH. Mice fed this diet also develop ER stress and lipoapoptosis (7). In this study, we investigate the role of hepatocyte XBP1 in progressive fatty liver injury using this metabolic syndrome dietary model in mice with a liver-specific deficiency of Xbp1. Furthermore, we utilize liver-derived Huh7 cells deficient in XBP1 to determine the role of XBP1 in palmitic acid lipotoxicity.
MATERIALS AND METHODS
Materials
Hoechst 33258, palmitic acid, and fatty acid-free bovine serum albumin were purchased from Sigma (St. Louis, MO), puromycin was obtained from Millipore (Billerica, MA), and paraformaldehyde (16%) was supplied by Thermo Scientific (Waltham, MA). The following antibodies were purchased from the indicated vendors: XBP1 from Proteintech (Chicago, IL); GAPDH, phospho-JNK, JNK, phospho-ERK, ERK, phospho-p38, p38, cleaved caspase 3 from Cell Signaling Technology (Danvers, MA); β-actin from Sigma (St. Louis, MO); goat anti-mouse IgG-horseradish peroxidase (HRP) and goat anti-rabbit IgG-HRP antibodies from Santa Cruz Biotechnology (Santa Cruz, CA).
Animal Use and Treatment
C57BL/6-Xbp1fl/fl mice with loxP sites flanking exon 2 of the Xbp1 gene were kindly provided by Dr. Laurie H. Glimcher (Cornell University, Ithaca, NY) (18). These mice were bred with C57BL/6-Albumin-Cre mice (Jackson Laboratory, Bar Harbor, ME) that express Cre-recombinase in albumin-producing hepatocytes. All mice were housed on a 14-h light, 10-h dark cycle with free access to food and water. Male Xbp1fl/fl and Xbp1−/− mice (10 wk old) were randomly assigned to receive either standard chow diet with water or a high-fat diet (AIN-76 Western Diet, Test Diet) supplemented with drinking water containing 42 g/l of 55% fructose/45% glucose by weight (high fructose, corn syrup equivalent; HFS) for up to 16 wk. The mice were fasted for 4 h prior to euthanization, blood was obtained by cardiac puncture, and the liver was removed and rinsed with ice-cold saline, sectioned, and either immediately fixed in formalin or snap-frozen in liquid nitrogen. All protocols and procedures were approved by the Northwestern University Institutional Animal Care and Use Committee guidelines.
Biochemical Analysis
Serum alanine aminotransferase (ALT) was measured by using a spectrophotometric assay according to the manufacturer's protocol (Teco Diagnostics, Anaheim, CA). Fasting blood glucose was measured by the glucose oxidase method using a reflectance glucometer (One Touch Ultra 2, LifeScan, Milpitas, CA). Fasting insulin was measured by using a mouse insulin ELISA kit according to the manufacturer's protocol (ALPCO Diagnostics, Salem, NH). Hepatic homogenates were prepared as previously described (12), and homogenate triglyceride and cholesterol were determined by using an Infinity spectrophotometric assay according to the instructions of the manufacturer (Thermo Scientific, Waltham, MA). Liver hydroxyproline content was measured with a hydroxyproline colorimetric assay kit (Sigma, St. Louis, MO) according to manufacturer's instruction.
Histology
Specimens were fixed in 10% neutral buffered formalin, paraffin embedded, and sectioned. Staining with hematoxylin and eosin (H&E), Masson's trichrome, and Oil Red O was performed (Northwestern University Mouse Histology and Phenotyping Laboratory). All histology was assessed by an investigator blinded to treatment group.
Cell Culture
Human hepatoma Huh7 cells were kindly provided by Dr. Saul Karpen (Emory University, Atlanta, GA) and cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, l-glutamine, and penicillin-streptomycin at 37°C with 5% CO2. To establish stable knockdown cell lines, Huh7 cells were transfected with either scramble shRNA or XBP1 shRNA (OriGene, Rockville, MD) by use of Lipofectamine LTX (Invitrogen Life Technologies) according to the manufacturer's instruction. At 48 h after transfection, cells were put into selection media containing 2 μg/ml puromycin. Single clones were achieved by limiting dilution, expanded in selection media, and assessed for XBP1 mRNA levels. Palmitic acid was dissolved in isopropanol in a stock solution of 40 mM. In all palmitic acid experiments, serum-free DMEM containing 1% fatty acid-free bovine serum albumin was used.
RNA Extraction and qPCR
Total RNA was extracted from frozen liver or cell culture by using TRIzol reagent according to according to the manufacturer's protocol (Invitrogen Life Technologies, Carlsbad, CA), and 1 μg of total RNA was reverse transcribed to cDNA with the qScript cDNA synthesis kit (Quanta Bioscience, Gaithersburg, MD). Quantitative real-time PCR (qPCR) was then performed by using QuantiTect SYBR Green PCR Master Mix (Qiagen, Valencia, CA) with the Applied Biosystems Prism 7300 Sequence Detection System (Applied Biosystems, Foster City, CA). Real-time data were collected for 40 cycles of 95°C, 10 s; 60°C, 1 min. Relative expression of the gene of interest was estimated by the ΔΔCt method using β2-microglobulin or 18s as a reference gene. Samples were analyzed in duplicate, and experiments were repeated a minimum of three times. All primers were synthesized by Integrated DNA Technology (Coralville, CA).
Library Construction, Sequencing, and Transcriptome Analysis
After RNA isolation, library construction and sequencing were performed at the Beijing Genomics Institute (BGI; Beijing, China). Briefly, total RNA samples were treated with DNase I and mRNA enrichment by use of oligo(dT) magnetic beads. The mRNA was mixed with fragmentation buffer, the mRNA was fragmented into short fragments of ∼200 bp. Then the first strand of cDNA was synthesized by using random hexamer-primer. Buffer, deoxynucleotide triphosphates RNase H, and DNA polymerase I were added to synthesize the second strand. The double-stranded cDNA was purified with magnetic beads. End reparation and 3′-end single nucleotide A (adenine) addition were then performed. Finally, sequencing adaptors were ligated to the fragments. The fragments were enriched by PCR amplification. During the quality control step, Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System were used to qualify and quantify the sample libraries. Library construction (100 bp, paired-end) and sequencing were carried out at BGI and library products were sequenced on the Illumina HiSeq2000. The quality of DNA reads, in fastq format, was evaluated by using FastQC. Adapters were removed and reads of poor quality were filtered. The data were processed following the procedure described in Trapnell et al. (33). Briefly, the reads were aligned to the Mus musculus genome (mm10) by use of TopHat (v2.0.8b). Subsequently, the aligned reads, in conjunction with a gene annotation file for mm10 obtained from the University of California, Santa Cruz Genome Browser website, were used to determine the expression of known genes by using Cufflinks (v2.1.1). The individual transcript files generated by Cufflinks for each sample were merged into a single gene annotation file, which was then used to perform a differential expression analysis with the Cufflinks routine, cuffdiff. Differential expression was determined by cuffdiff using the procedure described before (33) with a false discovery rate cutoff value of 0.05. The results of the differential expression analysis were processed with cummeRbund. The differentially expressed genes were separated into those that were upregulated and those that were downregulated. Pathway analysis was performed on both gene lists by using GeneCoDis to identify Gene Ontogeny (GO) pathways that are enriched with genes that are upregulated and downregulated (5, 23, 30). The GEO accession number for the data set is GSE64824.
Western Blotting
Protein homogenates from frozen liver or Huh7 cells were isolated with T-Per protein extraction reagent (Thermo Scientific, Waltham, MA) containing protease inhibitor cocktails (Millipore, Billerica, MA) and Halt phosphatase inhibitor (Thermo Scientific). After protein quantification with Coomassie Plus protein assay reagent (Thermo Scientific), equal amounts of protein samples were subjected to immunoblotting for target proteins, and immunoreactive bands were visualized by using Amersham ECL Western Blotting Detection Reagents according to the manufacturer's protocol (GE Healthcare, Piscataway, NJ).
Cytotoxicity and Apoptosis Analysis
LDH assay.
Cytotoxicity was measured by the release of lactate dehydrogenase (LDH) into the media by using CytoTox nonradioactive cytotoxicity assay kit according to the manufacturer's protocol (Promega, Madison, WI). A 10× lysis buffer was added to medium for measuring maximal LDH release. Data are presented as percent cytotoxicity, which is calculated as the ratio of the experimental LDH release to the maximal LDH release.
Morphological detection of apoptosis and caspase 3/7 activity assay.
Cells were grown on glass coverslips and were fixed in 4% paraformaldehyde for 5 min and rinsed in cold PBS twice. Then the cells were dried and stained with Hoechst 33258 (5 μg/ml) for 10 min, followed by rinsing with distilled water. After the coverslips were air dried, they were mounted with Aqua poly/Mount (Polysciences, Warrington, PA) and stored at −20°C. Nuclear morphology was evaluated with a fluorescence microscope. Cells with chromatin condensation and nuclear fragmentation were considered apoptotic; 400 random cells were counted for each condition. Apoptosis is presented as the percent of apoptotic cells in all counted cells. Apoptosis was also assayed by measuring caspase 3/7 activity by using Caspase-Glo 3/7 assay kit according to the manufacturer's protocol (Promega, Madison, WI).
Statistics
Each experiment was repeated a minimum of three times. Data are shown as means ± SD. Comparison between two groups was performed by Student's t-test. When more than two groups were compared, ANOVA was performed followed by Tukey's post hoc analysis using GraphPad Prism 6 software (GraphPad Software, Lo Jolla, CA). Statistical significance was defined as P values of less than or equal to 0.05.
RESULTS
Mice Lacking Hepatocyte Xbp1 Have Higher Serum ALT Level after HFS Feeding
To study the role of Xbp1 in the pathogenesis of NASH, we developed hepatocyte-specific Xbp1 knockout mice (Fig. 1A). Both control (Xbp1fl/fl) and knockout (Xbp1−/−) mice were fed either regular chow or a HFS diet for 4 or 16 wk. As shown in Fig. 1B, after 4 wk of feeding, serum ALT levels were significantly higher in HFS-fed Xbp1−/− mice (79.1 ± 13.2 U/l) compared with chow-fed Xbp1−/− mice (33.5 ± 7.1 U/l, P < 0.01). In contrast, there was no significant increase in serum ALT after 4 wk of HFS feeding in the Xbp1fl/fl mice. After 16 wk of feeding, ALT level increased in both Xbp1−/− (76.8 ± 7.5 U/l, P < 0.01) and Xbp1fl/fl mice (43.7 ± 17.2 U/l, P < 0.05) compared with chow-fed mice; however, HFS-fed Xbp1−/− mice had higher ALT than Xbp1fl/fl mice (P < 0.05). Baseline ALT levels of Xbp1−/− mice were slightly higher than Xbp1fl/fl mice (33.5 ± 7.1 vs. 20.0 ± 2.0 U/l, P < 0.01), although histology was normal in both genotypes fed chow. Chow-fed Xbp1fl/fl and Xbp1−/− mice had similar weight gain at 16 wk. Xbp1−/− mice fed a HFS diet for 4 wk gained less weight compared with Xbp1fl/fl mice (7.5 ± 2.3 vs. 3.2 ± 1.7 g, P < 0.05). The weight gain at 16 wk was similar in both groups fed HFS. Figure 1, C–F, describes the metabolic changes that occurred in Xbp1fl/fl and Xbp1−/− mice fed either chow or a HFS diet. The baseline levels of serum glucose, quantitative insulin sensitivity check index (QUICKI), hepatic triglyceride, and cholesterol were all similar between Xbp1fl/fl and Xbp1−/− mice. Xbp1−/− mice fed a HFS diet for 4 wk had lower serum glucose levels, higher QUICKI, lower hepatic triglycerides, and lower hepatic cholesterol levels compared with Xbp1fl/fl mice. Therefore, higher serum ALT levels after 4 wk HFS feeding occurred in the Xbp1−/− mice, despite lower hepatic lipid content. After 16 wk of HFS feeding, the metabolic changes became similar between the two genotypes.
Fig. 1.

Serum alanine aminotransferase (ALT) and metabolic changes in Xbp1fl/fl and Xbp1−/− mice fed a high-fat/sugar (HFS) diet. A: total liver mRNA from Xbp1fl/fl and Xbp1−/− mice was analyzed by quantitative PCR (qPCR) for Xbp1 exon 2 expression (n = 4). *P < 0.001 compared with Xbp1fl/fl mice. B–F: Xbp1fl/fl and Xbp1−/− mice were fed either chow or a HFS diet for 4 or 16 wk; n ≥ 4 in each group. Serum ALT activity (B), fasting serum glucose (C), the quantitative insulin sensitivity check index (QUICKI) (D), hepatic triglyceride (TG) (E), and hepatic cholesterol (F) levels were measured or calculated. *P < 0.05 compared with chow-fed Xbp1fl/fl mice. #P < 0.05 compared with chow-fed Xbp1−/− mice. ¶P < 0.05 compared with HFS-fed Xbp1fl/fl mice.
RNA-Seq Analysis Reveals Activation of Inflammation and Apoptosis Pathways in the Knockout Mice after HFS Feeding
Since differences of ALT elevation occurred by 4 wk of HFS feeding, we examined the gene profiling of the entire gene transcriptome after 4 wk of HFS diet. RNA sequencing (RNA-Seq) was performed on hepatic mRNA to investigate global hepatic gene expression after 4 wk on HFS diet in the four cohorts: chow-fed Xbp1fl/fl and Xbp1−/− mice; and HFS-fed Xbp1fl/fl and Xbp1−/− mice. Figure 2A demonstrates the GO pathways of apoptotic process, inflammatory response, and extracellular matrix structural constituent, which are differentially expressed in the four groups. There are 24, 15, and 7 differentially expressed genes identified in these pathways, respectively, when comparing Xbp1−/− to Xbp1fl/fl mice fed a HFS diet for 4 wk. Genotype-specific differential expression of these gene pathways was absent in chow-fed mice. Similarly, there are 23, 19, and 10 differentially expressed genes in the apoptotic process, inflammatory response, and extracellular matrix structural pathways, respectively, in the Xbp1−/− mice fed HFS compared with chow diet. This effect of diet was less pronounced when HFS and chow-fed Xbp1fl/fl mice are compared. A heat map of the gene expression levels of the genes from these GO pathways is depicted in Fig. 2B. These gene expression effects were most pronounced when the HFS diet is fed to the Xbp1−/− mouse genotype. Of note, changes in hepatic fatty acid synthesis gene expression detected by using RNA-Seq in the Xbp1−/− mice were similar to those previously reported (data not shown) (18).
Fig. 2.

RNA sequencing (RNA-Seq) of liver mRNA indicates that inflammation, apoptosis, and extracellular matrix pathways are activated in the Xbp1−/− mice fed a HFS diet. Xbp1−/− and Xbp1fl/fl mice were fed chow or a HFS diet for 4 wk, and RNA-Seq and Gene Ontogeny (GO) analysis were performed on hepatic mRNA to identify differentially expressed pathways; n = 2, of 2 pooled samples. A: numbers of differentially expressed genes in 3 GO pathways were shown. B: log expression of differentially expressed genes in 3 GO pathways was depicted in a heat map format.
Xbp1−/− Mice Develop Greater Hepatic Fibrosis Compared with Xbp1fl/fl Mice in Response to HFS Feeding
RNA-Seq revealed differences in expression of several extracellular matrix-related genes in the cohorts of Xbp1−/− and Xbp1fl/fl mice fed a HFS diet for 4 wk. Therefore, we performed qPCR to further examine the effects on the expression of the fibrosis-related genes Col1a1, Tgfβ1, and α-Sma (Table 1). Hepatic Col1a1 gene expression was significantly elevated after both 4 and 16 wk of HFS feeding in Xbp1−/−, but only increased in Xbp1fl/fl mice after 16 wk of feeding. Additionally, Col1a1 expression was threefold higher in Xbp1−/− mice after 4 wk of HFS feeding and nearly twice as high after 16 wk, compared with HFS-fed Xbp1fl/fl mice. The profibrotic Tgfβ1 gene expression was also increased in both Xbp1−/− and Xbp1fl/fl mice after 16-wk HFS feeding, with significantly higher expression in the Xbp1−/− mice compared with Xbp1fl/fl mice. After 16-wk HFS feeding, α-Sma gene expression was significantly increased in Xbp1−/− mice, but not in the Xbp1fl/fl mice, compared with chow. Hepatic FGF21 has been shown to increase in NAFLD and NASH (10); therefore we examined its gene expression in the mouse livers. Hepatic Fgf21 mRNA expression was significantly increased in Xbp1−/− mice, but not in the Xbp1fl/fl mice, after 4-wk feeding compared with chow (Table 1). Masson's trichrome staining demonstrated no fibrosis in HFS-fed Xbp1fl/fl mice and only minimal focal pericellular fibrosis in HFS-fed Xbp1−/− mice after HFS feeding for 4 wk (Fig. 3). After 16 wk of HFS feeding, there was a marked increase in hepatic fibrosis in the Xbp1−/− mice, with only minimal fibrosis in Xbp1fl/fl mice. The hydroxyproline content was higher in Xbp1−/− mice compared with Xbp1fl/fl mice after 4-wk HFS feeding (Fig. 3B). H&E staining demonstrated no hepatocyte ballooning in Xbp1fl/fl mice and few ballooned cells in Xbp1−/− mice after HFS feeding for 4 wk. Both groups had mild ballooning cells after 16 wk of HFS feeding. After 4 wk on the HFS diet, Xbp1−/− mice showed mild inflammation (< 2 foci/×200), whereas no inflammation was present in Xbp1fl/fl mice. After 16 wk, both groups showed mild inflammation. Consistently, mRNA expression of the macrophage markers CD68 and F4/80 (Emr1) was significantly increased in Xbp1−/− mice, but not in Xbp1fl/fl mice after HFS-feeding (Table 1). H&E staining also showed less hepatic steatosis in the Xbp1−/− livers (5–33%) compared with the Xbp1fl/fl livers at both time points (>33–66%) (n ≥ 4). Oil Red O staining demonstrated predominantly macrovesicular fat in the livers of both genotypes fed HFS, with less steatosis in the Xbp1−/− mice livers (Fig. 3C).
Table 1.
Gene expression in Xbp1fl/fl and Xbp1−/− mice fed a HFS diet
|
Xbp1fl/fl |
Xbp1−/− |
|||||
|---|---|---|---|---|---|---|
| Genes | Chow | HFS 4 wk | HFS 16 wk | Chow | HFS 4 wk | HFS 16 wk |
| Fibrosis | ||||||
| Col1a1 | 1.01 ± 0.11 | 2.97 ± 0.98 | 3.20 ± 1.80* | 2.33 ± 0.86 | 9.11 ± 4.80†‡ | 8.48 ± 4.12†§ |
| Tgfβ1 | 0.95 ± 0.20 | 1.21 ± 0.24 | 2.28 ± 0.55* | 1.69 ± 0.51 | 1.95 ± 0.44‡ | 4.41 ± 2.09†§ |
| α-Sma | 1.02 ± 0.24 | 1.35 ± 0.41 | 2.66 ± 1.46 | 1.11 ± 0.33 | 1.46 ± 0.51 | 2.96 ± 0.57† |
| Inflammatory markers | ||||||
| Cd68 | 0.90 ± 0.09 | 1.05 ± 0.16 | 1.11 ± 0.14 | 0.71 ± 0.01 | 1.78 ± 0.11†‡ | 1.20 ± 0.14† |
| Emr1 | 1.33 ± 0.37 | 1.59 ± 0.40 | 1.85 ± 0.36 | 1.12 ± 0.11 | 2.18 ± 0.12†‡ | 1.60 ± 0.20† |
| UPR pathway | ||||||
| Chop | 1.01 ± 0.16 | 1.81 ± 0.46 | 1.71 ± 0.57 | 1.42 ± 0.33 | 6.95 ± 2.77†‡ | 2.39 ± 0.69 |
| Bip | 1.00 ± 0.08 | 0.81 ± 0.21 | 1.07 ± 0.36 | 0.43 ± 0.06* | 0.52 ± 0.15‡ | 0.77 ± 0.18 |
| Ire1α | 1.21 ± 0.15 | 0.99 ± 0.13 | 0.94 ± 0.11* | 1.45 ± 0.21 | 0.89 ± 0.09† | 1.31 ± 0.36 |
| Xbp1s | 1.01 ± 0.13 | 1.24 ± 0.72 | 0.60 ± 0.12 | n.d. | n.d. | n.d. |
| Atf6 | 0.94 ± 0.05 | 0.90 ± 0.09 | 1.23 ± 0.4 | 0.72 ± 0.03* | 0.57 ± 0.02†‡ | 1.02 ± 0.21† |
| Atf4 | 1.02 ± 0.21 | 1.18 ± 0.32 | 1.85 ± 0.80 | 1.31 ± 0.54 | 1.85 ± 0.53 | 2.72 ± 0.52† |
| Other | ||||||
| Fgf21 | 1.45 ± 1.01 | 1.61 ± 0.78 | 3.18 ± 1.86 | 1.68 ± 1.16 | 5.96 ± 2.70†‡ | 4.08 ± 1.43 |
Values are means ± SD. HFS, high-fat/sugar diet.
P < 0.05 compared to Xbp1fl/fl chow;
P < 0.05 compared to Xbp1−/− chow;
P < 0.05 compared to Xbp1fl/fl HFS 4 wk;
P < 0.05 compared to Xbp1fl/fl HFS 16 wk; n.d., not determined.
Fig. 3.

Xbp1−/− mice fed a HFS diet have increased fibrosis. Xbp1−/− and Xbp1fl/fl mice were fed either with chow or a HFS diet for 4 or 16 wk. A: hematoxylin and eosin (H&E; top) and Masson's trichrome staining (bottom) of liver sections (magnification: ×200) from each group were shown. Numbers on the left indicate liver sections from 3 different mice. B: liver hydroxyproline levels were measured. *P = 0.05 compared with HFS-fed Xbp1fl/fl mice. C: representative Oil Red O staining were shown from Xbp1−/− and Xbp1fl/fl mice fed with either chow or a HFS diet for 16 wk. Magnification: top, ×100; bottom, ×200.
Xbp1−/− Mice Fed a HFS Diet Have Increased CHOP Expression and JNK Phosphorylation
Enhanced hepatic expression of the transcription factor C/EBP homologous protein (CHOP) and phosphorylated c-Jun NH2-terminal kinase (p-JNK) have been associated with human and animal models of progressive NASH (24, 27). We therefore evaluated hepatic Chop and p-JNK expression in Xbp1−/− and Xbp1fl/fl mice fed chow or HFS diet. Chop mRNA levels were similar in chow-fed Xbp1−/− and Xbp1fl/fl mice. However, HFS feeding for 4 wk increased hepatic Chop gene expression approximately fourfold in Xbp1−/− mice (Table 1). Similarly, p-JNK expression was significantly higher in the HFS-fed Xbp1−/− mice compared with HFS-fed Xbp1fl/fl mice after 4 wk (Fig. 4). After 16 wk of feeding, both Chop and p-JNK expression were increased and similar between Xbp1−/− and Xbp1fl/fl mice. Table 1 also shows the expression of other UPR genes after HFS feeding. To further determine whether other MAPK were affected, we examined the hepatic expression of phosphorylated ERK and p38. As shown in Fig. 4, phosphorylated p38 level did not change significantly after HFS feeding in either Xbp1fl/fl mice or Xbp1−/− mice. Phosphorylated ERK expression was decreased after 16-wk HFS feeding in both Xbp1fl/fl mice and Xbp1−/− mice.
Fig. 4.

Xbp1−/− mice fed a HFS diet have an increased hepatic level of phosphorylated JNK. Xbp1fl/fl and Xbp1−/− mice were fed either chow or a HFS diet for 4 or 16 wk. Liver homogenates from each group were pooled (n ≥ 4 in each group) and analyzed by Western blot using antibodies against phosphorylated and total JNK, ERK, and p38. GAPDH and β-actin were used as a loading control. Vertical lines indicate reassembly of noncontiguous gel lanes.
PA Induces XBP1s Expression in Huh7 Cells
Fatty acid lipotoxicity is important in the pathogenesis of NASH and treating liver cells with palmitic acid induces UPR pathway activation and apoptosis (20). To further determine the mechanisms by which XBP1 deficiency augments liver injury in steatohepatitis, we subsequently treated Huh7 cells with stable XBP1 knockdown (Huh7-KD) and scramble shRNA (Huh7-SCR) with 400 μM PA for 24 h. Figure 5 demonstrates that treating the Huh7-SCR cells with PA induced both XBP1s gene and protein expression, whereas there was no change in the Huh7-KD cells.
Fig. 5.
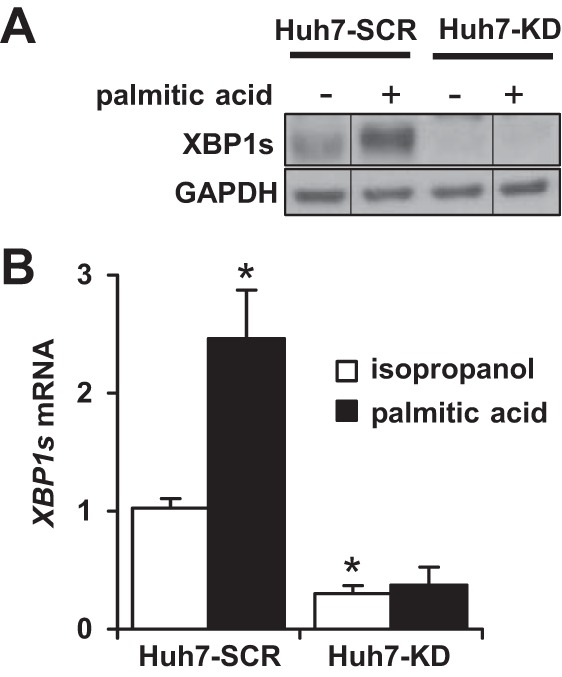
Palmitic acid increases XBP1s expression in Huh7 cells. Huh7 cells were stably transfected with either XBP1 shRNA (Huh7-KD) or scramble shRNA (Huh7-SCR). Both groups were treated with either isopropanol (vehicle) or palmitic acid (400 μM) for 24 h. A: whole cell lysates were immunoblotted with antibody against XBP1s. GAPDH was used as a loading control. B: qPCR was performed to assess the mRNA abundance of XBP1s. The results are presented as relative expression over vehicle-treated Huh7-SCR cells. *P < 0.001 compared with vehicle-treated Huh7-SCR cells (n = 5). Vertical lines indicate reassembly of noncontiguous gel lanes.
Loss of XBP1 Augments Palmitic Acid-Induced Apoptosis and Cytotoxicity in Huh7 Cells
To evaluate the role of XBP1 in PA-induced cell death, we initially measured cytotoxicity using LDH release into the media. Huh7-KD cells had a 5% higher baseline cytotoxicity compare to Huh7-SCR cells. After 24 h of treatment with 400 μM PA, cytotoxicity increased to 21% in Huh7-SCR cells, whereas PA-treated Huh7-KO cells exhibited over 50% cell death (P < 0.001) (Fig. 6A). Since LDH release does not distinguish between apoptosis and necrosis, we then investigated PA-induced apoptosis by assessing nucleus condensation/fragmentation and caspase 3/7 activity. H33258 staining reveals that PA-induced cell apoptosis occurred in both Huh7-SCR and Huh7-KD cells, but PA-treated Huh7-KD cells displayed significantly more apoptosis compared with Huh7-SCR cells (65 ± 3 vs. 38 ± 4%, P < 0.01) (Fig. 6B). PA-treated Huh7-KD cells also had higher caspase 3/7 activity than PA-treated Huh7-SCR cells (P = 0.05) (Fig. 6C). Western blots further confirmed that cleaved-caspase 3 protein expression was greater in PA-treated Huh7-KD cells compared with Huh7-SCR cells (Fig. 6D). We next examined CHOP and p-JNK expression in PA-treated Huh7-KD and Huh7-SCR cells. Figure 6E shows that although CHOP gene expression was increased by PA treatment in both groups of cells, it was significantly greater in PA-treated Huh7-KD cells compared with PA-treated Huh7-SCR cells (P = 0.01). PA-induced p-JNK expression was also enhanced in the Huh7-KD cells treated with PA (Fig. 6F). These results indicate that lack of XBP1 augments cell death, apoptosis, and cell injury induced by palmitic acid. Furthermore, the palmitic acid-induced increases in CHOP and p-JNK expression in Huh-7 cells lacking XBP1 were similar to those detected in HFS-treated Xbp1−/− mice.
Fig. 6.
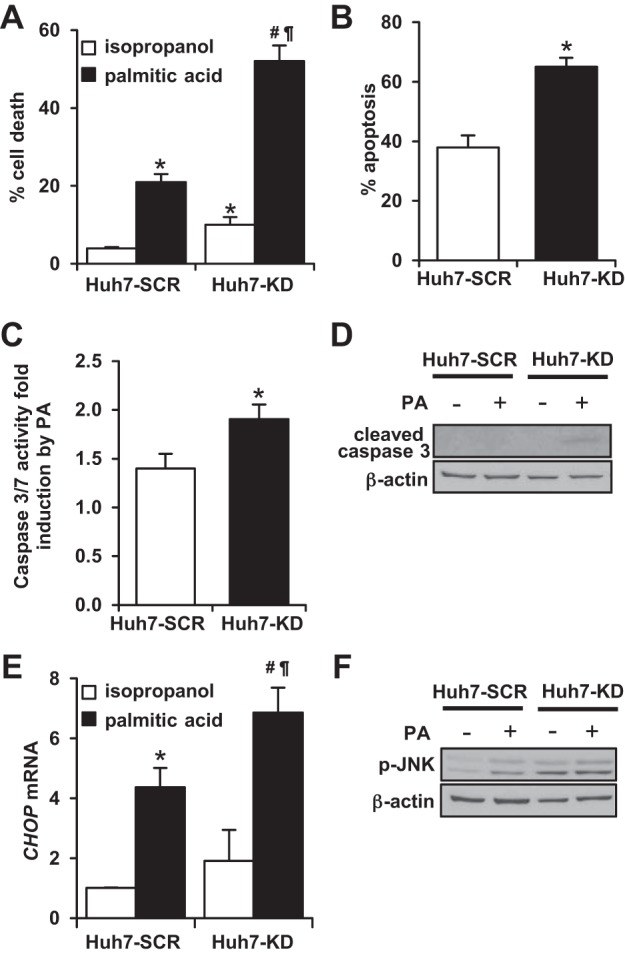
XBP1 knockdown Huh7 cells have enhanced cytotoxicity and apoptosis by palmitic acid treatment. Huh7-SCR (scramble) and Huh7-KD (XBP1 knockdown) cells were treated with either isopropanol (vehicle) or palmitic acid (PA, 400 μM) for 24 h. A: cytotoxicity was examined by measuring cellular release of LDH. *P < 0.001 compared with vehicle-treated Huh7-SCR cells (n = 4). #P < 0.001 compared with vehicle-treated Huh7-KD cells. ¶P < 0.001 compared with PA-treated Huh7-KD cells. B: apoptosis was monitored by H33258 nucleus staining (n = 3 experiments). *P < 0.01 compared with PA-treated Huh7-SCR cells. C: caspase 3/7 activity was measured using luciferase activity and expressed as PA-induced fold induction over vehicle-treated cells (n = 3). *P = 0.01 compared with Huh7-SCR cells. D: cleaved caspase 3 expression was examined by Western blot using whole cell lysates. β-Actin was used as a loading control. E: qPCR was performed to evaluate CHOP mRNA levels (n = 3). *P < 0.001 compared with vehicle-treated Huh7-SCR cells. #P < 0.005 compared with vehicle-treated Huh7-KD cells. ¶P = 0.01 compared with PA-treated Huh7-KD cells. F: Huh7-SCR and Huh7-KD cells were treated with either isopropanol or PA (200 μM) for 24 h. Phosphorylated JNK expression was examined by Western blotting. β-Actin was used as a loading control.
DISCUSSION
The ER lumen is the major site for protein synthesis and folding. Obesity, insulin resistance, and hepatic lipid accumulation can lead to excess unfolded and misfolded proteins in the ER lumen that can induce ER stress. In response to ER stress, signaling pathways of the UPR are activated in an adaptive response to manage the increased demand. Dysregulation of the hepatic UPR response and XBP1s expression has been previously associated with human NASH (16, 24). The evolutionarily most conserved UPR pathway is the IRE1α/XBP1 signaling pathway, which is a cytoprotective response that increases ER chaperone production and ERAD in the liver and other tissues (8).
Hepatic XBP1 (termed Xbp1 in the mouse) regulates hepatic lipid and glucose metabolism, and previous in vivo murine models of hepatic Xbp1 deficiency have demonstrated a regulatory function of Ire1α/Xbp1 in lipid and lipoprotein metabolism (18, 28, 36). However, the role of XBP1 in hepatic injury during NASH remains unclear. Studies in human NAFLD indicate that reduced hepatic XBP1s is associated with the progressive form of NASH, but not with isolated hepatic steatosis (24). However, this correlation has not been shown to be causative or mechanistically involved in the pathogenesis of NASH. Therefore, this study investigates the role of XBP1 in the pathogenesis of progressive NASH and hepatic lipotoxicity.
Although most high-fat diets cause mice to develop isolated hepatic steatosis without inflammation, high-fat diets with sucrose and fructose (corn syrup equivalent) cause progressive, fibrosing steatohepatitis (7, 32, 34). We demonstrate that, when fed the HFS diet, mice lacking hepatocyte Xbp1 develop more hepatocellular damage and fibrosis than control mice. This indicates that Xbp1 has a protective role in preventing or lessening the severity of murine steatohepatitis, and diminished hepatic expression of Xbp1 predisposes the mice to a more severe injury phenotype. Using unbiased RNA-Seq and GO pathway analysis, we further demonstrated a concomitant enhanced activation of inflammation and apoptosis pathways. The regulation of these signaling pathways is important in both human and experimental models of steatohepatitis and liver injury. RNA-Seq data also demonstrated increased expression of extracellular matrix components in the HFS diet-fed Xbp1−/− mice compared with Xbp1fl/fl mice; consistent with the histological evidence of increased fibrosis after 16 wk of HFS diet. Furthermore, hepatic hydroxyproline content was greater in Xbp1−/− mice after 4 wk of HFS feeding, as was the expression of both hepatic Col1a1 and its canonical regulator Tgfβ1. Hepatic FGF21 has been shown to potentially play a protective role in steatohepatitis (11, 38). The increased hepatic Fgf21 expression in Xbp1−/− mice livers may potentially act as a compensatory mechanism to lessen the liver injury and fibrotic response. It is important to note that the phenotypic expression of more severe progressive, fibrosing steatohepatitis occurs when both environmental (diet) and genetic (loss of hepatocyte Xbp1) factors simultaneously occur. Chow-fed Xbp1−/− mice have normal histology and little evidence of hepatic inflammation or injury, yet HFS-fed Xbp1−/− mice have more injury than Xbp1fl/fl mice fed the same diet. This environmental-genetic interaction is characteristic of human NASH, where genetically predisposed individuals develop the disease when stressed by obesity, insulin resistance, and the metabolic syndrome (15, 26, 29, 35, 37, 39). Of note, the enhanced injury in the Xbp1−/− mice after 4 wk of HFS diet occurs despite the fact that the livers from these mice had significantly less steatosis.
Hepatic pathology developed significantly sooner after HFS feeding in the Xbp1−/− mice compared with Xbp1fl/fl mice. Differences in the kinetics of hepatic responses to liver injury have been reported with other liver knockout mice (3, 4, 14). Nonetheless, the development of the increased hepatic fibrosis in Xbp1−/− mice after 16 wk of HFS feeding indicates that they develop more progressive liver disease. Longer HFS feeding durations will be required to determine whether the liver injury progresses to cirrhosis.
The enhanced hepatic injury and fibrosis that develop in mice lacking hepatocyte Xbp1 demonstrate a causative role of hepatocyte Xbp1 deficiency in worsening NASH. This injury was associated with increased expression of p-JNK, although p-p38 and p-ERK were not increased. To further support the role of XBP1 in the pathogenesis of NASH, we also showed that liver cell cultures lacking XBP1 are more sensitive to palmitic acid lipoapoptosis. Liver lipotoxicity is mediated by signaling networks triggered by free fatty acids, which cause cellular steatosis and apoptosis. JNK signaling and UPR pathways mediate the lipoapoptosis induced by fatty acids. JNK is a downstream target of IRE1α/XBP1 that is associated with inflammation and apoptosis, and CHOP is a proapoptotic signaling molecule that sensitizes cells to apoptosis (22, 31). Our results indicate that both CHOP expression and p-JNK are increased in HFS-fed Xbp1−/− mice and in palmitic acid-treated Huh7 cells that lack XBP1. These in vivo and in vitro data are consistent with the expression levels detected in human liver biopsies from patients with NASH (16, 24). However, the causative roles of CHOP and JNK remain unclear and need further investigation.
NASH is currently a leading cause of liver disease that can lead to cirrhosis, hepatocellular carcinoma, and need for liver transplantation. Because of the epidemic of obesity, the prevalence of NASH and its sequelae will increase in the future. Although NASH is associated with the metabolic syndrome, the pathogenesis and mechanisms responsible for disease progression remain poorly understood. Human NASH has been associated with diminished hepatic XBP1s expression, and our data support a causative role of hepatic Xbp1 in the pathogenesis of murine steatohepatitis. Furthermore, we also demonstrate a role of XBP1s signaling in liver cell culture lipotoxicity. Our in vivo and in vitro findings of enhanced p-JNK and CHOP expression in hepatocytes lacking Xbp1 are also consistent with reports in patients with NASH. These data demonstrate that, in the presence of the environmental dietary stressor of a HFS diet and the metabolic syndrome, hepatocyte XBP1s signaling is important for the development and severity of experimental NASH. Since hepatocyte XBP1 appears to have a protective role in the pathogenesis of NASH, it is a potential therapeutic target for pharmacological or genetic treatments for this common form of liver disease with few effective therapies.
GRANTS
This work is supported by NIH R01DK093807 and the Max Goldenberg Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
X.L. and R.M.G. conception and design of research; X.L., A.S.H., B.E.L., and K.A.A. performed experiments; X.L., M.J.S., and R.M.G. analyzed data; X.L., M.J.S., and R.M.G. interpreted results of experiments; X.L. and M.J.S. prepared figures; X.L. and M.J.S. drafted manuscript; X.L. and R.M.G. edited and revised manuscript; X.L., A.S.H., B.E.L., M.J.S., K.A.A., and R.M.G. approved final version of manuscript.
REFERENCES
- 1.Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 129: 113–121, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 346: 1221–1231, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Behrens A, Sibilia M, David JP, Mohle-Steinlein U, Tronche F, Schutz G, Wagner EF. Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c-jun in the liver. EMBO J 21: 1782–1790, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borude P, Edwards G, Walesky C, Li F, Ma X, Kong B, Guo GL, Apte U. Hepatocyte-specific deletion of farnesoid X receptor delays but does not inhibit liver regeneration after partial hepatectomy in mice. Hepatology 56: 2344–2352, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmona-Saez P, Chagoyen M, Tirado F, Carazo JM, Pascual-Montano A. GENECODIS: a web-based tool for finding significant concurrent annotations in gene lists. Genome Biol 8: R3, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 55: 2005–2023, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H, Gores G. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol 301: G825–G834, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med 18: 59–68, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Diehl AM, Li ZP, Lin HZ, Yang SQ. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut 54: 303–306, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, Badman MK, Martinez-Chantar ML, Maratos-Flier E. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 139: 456–463, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher FM, Chui PC, Nasser IA, Popov Y, Cunniff JC, Lundasen T, Kharitonenkov A, Schuppan D, Flier JS, Maratos-Flier E. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology 147: 1073–1083.e6, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henkel AS, Dewey AM, Anderson KA, Olivares S, Green RM. Reducing endoplasmic reticulum stress does not improve steatohepatitis in mice fed a methionine- and choline-deficient diet. Am J Physiol Gastrointest Liver Physiol 303: G54–G59, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanuri G, Bergheim I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int J Mol Sci 14: 11963–11980, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karavia EA, Papachristou DJ, Kotsikogianni I, Giopanou I, Kypreos KE. Deficiency in apolipoprotein E has a protective effect on diet-induced nonalcoholic fatty liver disease in mice. FEBS J 278: 3119–3129, 2011. [DOI] [PubMed] [Google Scholar]
- 15.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, Vogt TF, Hobbs HH, Cohen JC. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 46: 352–356, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lake AD, Novak P, Hardwick RN, Flores-Keown B, Zhao F, Klimecki WT, Cherrington NJ. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol Sci 137: 26–35, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazo M, Hernaez R, Eberhardt MS, Bonekamp S, Kamel I, Guallar E, Koteish A, Brancati FL, Clark JM. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol 178: 38–45, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320: 1492–1496, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longato L. Non-alcoholic fatty liver disease (NAFLD): a tale of fat and sugar? Fibrogen Tissue Rep 6: 14, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem 281: 12093–12101, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol 54: 795–809, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21: 1249–1259, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nogales-Cadenas R, Carmona-Saez P, Vazquez M, Vicente C, Yang X, Tirado F, Carazo JM, Pascual-Montano A. GeneCodis: interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res 37: W317–W322, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134: 568–576, 2008. [DOI] [PubMed] [Google Scholar]
- 25.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med 52: 59–69, 2012. [DOI] [PubMed] [Google Scholar]
- 26.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 40: 1461–1465, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology 49: 87–96, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.So JS, Hur KY, Tarrio M, Ruda V, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Lichtman AH, Iwawaki T, Glimcher LH, Lee AH. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab 16: 487–499, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Speliotes EK, Butler JL, Palmer CD, Voight BF, Consortium G, Consortium MI, Nash CRN, Hirschhorn JN. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 52: 904–912, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A. GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res 40: W478–W483, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T, Iwaisako K, Seo S, Nakajima A, Ikai I, Uemoto S. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol 294: G498–G505, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol 295: G987–G995, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wada T, Kenmochi H, Miyashita Y, Sasaki M, Ojima M, Sasahara M, Koya D, Tsuneki H, Sasaoka T. Spironolactone improves glucose and lipid metabolism by ameliorating hepatic steatosis and inflammation and suppressing enhanced gluconeogenesis induced by high-fat and high-fructose diet. Endocrinology 151: 2040–2049, 2010. [DOI] [PubMed] [Google Scholar]
- 35.Wagenknecht LE, Scherzinger AL, Stamm ER, Hanley AJ, Norris JM, Chen YD, Bryer-Ash M, Haffner SM, Rotter JI. Correlates and heritability of nonalcoholic fatty liver disease in a minority cohort. Obesity 17: 1240–1246, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, Chen Z, Lam V, Han J, Hassler J, Finck BN, Davidson NO, Kaufman RJ. IRE1alpha-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab 16: 473–486, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willner IR, Waters B, Patil SR, Reuben A, Morelli J, Riely CA. Ninety patients with nonalcoholic steatohepatitis: insulin resistance, familial tendency, and severity of disease. Am J Gastroenterol 96: 2957–2961, 2001. [DOI] [PubMed] [Google Scholar]
- 38.Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, Vonderfecht S, Hecht R, Li YS, Lindberg RA, Chen JL, Jung DY, Zhang Z, Ko HJ, Kim JK, Veniant MM. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 58: 250–259, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zain SM, Mohamed R, Mahadeva S, Cheah PL, Rampal S, Basu RC, Mohamed Z. A multi-ethnic study of a PNPLA3 gene variant and its association with disease severity in nonalcoholic fatty liver disease. Human Genet 131: 1145–1152, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]