

Abstract
Aldosterone increases blood pressure (BP) by stimulating sodium (Na) reabsorption within the distal nephron and collecting duct (CD). Aldosterone also stimulates endothelin-1 (ET-1) production that acts within the CD to inhibit Na reabsorption via a negative feedback mechanism. We tested the hypothesis that this renal aldosterone-endothelin feedback system regulates electrolyte balance and BP by comparing the effect of a high-salt (NaCl) diet and mineralocorticoid stimulation in control and CD-specific ET-1 knockout (CD ET-1 KO) mice. Metabolic balance and radiotelemetric BP were measured before and after treatment with desoxycorticosterone pivalate (DOCP) in mice fed a high-salt diet with saline to drink. CD ET-1 KO mice consumed more high-salt diet and saline and had greater urine output than controls. CD ET-1 KO mice exhibited increased BP and greater fluid retention and body weight than controls on a high-salt diet. DOCP with high-salt feeding further increased BP in CD ET-1 KO mice, and by the end of the study the CD ET-1 KO mice were substantially hypernatremic. Unlike controls, CD ET-1 KO mice failed to respond acutely or escape from DOCP treatment. We conclude that local ET-1 production in the CD is required for the appropriate renal response to Na loading and that lack of local ET-1 results in abnormal fluid and electrolyte handling when challenged with a high-salt diet and with DOCP treatment. Additionally, local ET-1 production is necessary, under these experimental conditions, for renal compensation to and escape from the chronic effects of mineralocorticoids.
Keywords: sodium reabsorption, desoxycorticosterone, blood pressure, aldosterone escape, renal, aldosterone, endothelin
the kidney is normally the primary organ that responds to changes in dietary sodium (Na) intake with commensurate changes in Na excretion to maintain normal Na balance. Derangements in the renal response to such changes can result in either Na depletion and hypotension, or excessive Na retention. Abnormal aldosterone activity for the degree of Na intake is an important element in the pathogenesis of many of these conditions. Aldosterone, produced by the adrenal cortex, is the most potent salt (NaCl)-retaining corticosteroid, and its action is opposed by multiple mechanisms, including endothelin-1 (ET-1) in the renal collecting duct (CD) (24). Aldosterone increases systemic blood pressure (BP) in part by increasing Na absorption via the apical epithelial Na channel (ENaC) and basolateral Na,K-ATPase. Aldosterone action is opposed by humoral, autocrine, and paracrine systems that include the natriuretic peptides atrial natriuretic peptide and brain natriuretic peptide, nitric oxide (NO), prostaglandins, and ET-1, which increase renal Na and water excretion to lower BP (20, 21, 23, 26, 39, 47).
Previously, we identified the ET-1 gene (Edn1) as an early aldosterone-responsive gene in a mouse inner medullary CD cell line (14). Further studies in our laboratory confirmed this action in vivo and demonstrated that aldosterone increases transcription of ET-1 via the mineralocorticoid receptor (MR) interacting directly with the Edn1 promoter at specific hormone response elements (38). Recently, we reported the first direct measurement of the quantitative effect of the acute application of ET-1 on Na reabsorption in the in vitro microperfused CD (30). In the presence of ET-1, ENaC-mediated Na reabsorption was significantly inhibited, which supports several other studies examining the effects of ET-1 in the CD (3, 4, 40, 48). Collectively, these studies suggest that the stimulation of ET-1 production by aldosterone acts locally within the CD as a negative feedback mechanism to inhibit Na and water reabsorption.
CD-specific ET-1 knockout (CD ET-1 KO) mice have salt-sensitive hypertension (1, 24). Studies suggest that relative overactivity of the renin-angiotensin-aldosterone system is responsible for the pathogenicity of this mouse model. Despite being hypertensive on a high-salt diet, the CD ET-1 KO mice did not exhibit greater suppression of plasma renin activity or aldosterone levels than control mice (1, 11). Ge et al. (11) further demonstrated that either angiotensin II or MR antagonism normalized BP in the CD ET-1 KO mice under normal salt conditions and partly corrected the hypertension during high-salt intake.
During experimentally induced primary hyperaldosteronism, a process known as “aldosterone escape” prevents chronic Na retention (2, 23, 25, 27, 37, 41, 46, 47).1 Administration of exogenous aldosterone in the presence of adequate Na intake results in initial Na retention. However, this effect is transient, and after a period of positive Na balance, renal excretion adjusts to match Na intake, although blood pressure remains elevated. Various mechanisms have been proposed to mediate or contribute to aldosterone escape, but relatively few studies have quantified their contribution.
Here, we have conducted complete metabolic balance experiments that to our knowledge are the first full quantitative assessments of the role of a single gene product to aldosterone escape. If CD ET-1 KO mice have impaired feedback control of the renin-angiotensin-aldosterone axis, treatment with mineralocorticoid should result in abnormal Na conservation. In the present study, we addressed this question by treating CD ET-1 KO mice on a high-salt diet with the long-acting aldosterone analog desoxycorticosterone pivalate (DOCP) and measured electrolyte balance and BP response.
The current findings are consistent with a role for CD ET-1 as a component of a local feedback system that regulates mineralocorticoid-stimulated renal Na absorption. CD ET-1 KO mice exhibit increased fluid and Na retention and BP when challenged with a high-salt diet. On this diet, DOCP treatment resulted in acute Na retention in controls but not in CD ET-1 KO mice. This was followed by restoration of neutral Na balance in controls (escape), whereas CD ET-1 KO mice exhibited progressive Na retention. These data demonstrate that the absence of ET-1, specifically in the CD, impairs the renal response to a Na load and identifies ET-1 as a necessary component of mineralocorticoid escape. We conclude that disruption of a renal aldosterone-endothelin feedback mechanism prevents adequate renal compensation for the effects of chronic mineralocorticoids on electrolyte balance.
MATERIALS AND METHODS
Animals.
All animal use was in compliance with the American Physiological Society's Guiding Principles in the Care and Use of Animals, and animal use protocols were approved by the North Florida/South Georgia Veterans Administration Institutional Animal Care and Use Committee. Mice homozygous for floxed ET-1 were imported from University of Utah Health Sciences Center, Salt Lake City, UT, and were a kind gift from Dr. Donald Kohan. The floxed ET-1 mice were bred with AQP2-Cre transgenic females (kind gift from Dr. David Weiner, University of Florida) also homozygous for floxed ET-1. Mice were genotyped as follows; genomic DNA was prepared according to standard methods from tail samples and amplified by PCR in a single reaction using the following oligonucleotides: ET-1 forward (5′-AGC AAG CTG TGC TGC CCA AAG A-3′), ET-1 reverse (5′-GAC TGC CTA TTC CTG GCT ATG G-3′), AQP2 forward (5′-CCT CTG CAG GAA CTG GTG CTG G-3′), and CreTag reverse (5′-GCG AAC ATC TTC AGG TTC TGC GG-3′). CD ET-1 KO mice yielded bands at 349 (floxed ET-1) and 671 bp (Cre transgene).
Age-matched C57BL/6J control and CD ET-1 KO adult male mice were implanted with PA-C10 radio transmitters [Data Sciences International (DSI), St. Paul, MN], placed in metabolic cages (Techniplast, Exton, PA) for 19 days, fed a 2% NaCl (high-salt) gelled diet ad libitum [45% TD99191 (0.2% Na), 54% water, 1% agar, and 1.8% Na as NaCl; Teklad, Madison, WI], and given normal saline (0.9% NaCl) to drink without access to solute-free water. On day 8, mice were treated with DOCP (0.07 mg/g im). Each day, body weight, food intake, and saline intake were measured and urine and feces collected for [Na] and [K] analysis using a digital flame analyzer (model no. 2655-00; Cole-Parmer, Chicago, IL). BP and heart rate (HR) were continually recorded every 10 min for 2 min. A moving average was generated each hour using DSI Dataquest ART Analysis Software version 4.3. On day 19, blood was collected via the abdominal aorta under isoflurane anesthesia. Plasma was analyzed for [Na] and [K], as described above. In a separate experiment, CD ET-1 KO mice were adapted to either a normal (0.2%) Na diet or a high-salt gelled diet and given the choice of a water bottle or a saline bottle for 8 days. The volume of water and saline consumed daily was recorded and averaged for days 6–8.
Fecal digestion.
Fecal samples were dried overnight at 80°C and digested as follows; representative samples (∼0.1 g) and a control blank were dissolved in 2 ml of 1:1 nitric acid-deionized water heated to 95°C for 5 min. After cooling, 1 ml of concentrated nitric acid was added, and the samples were refluxed for 10–15 min at 95°C. After cooling, 1 ml of deionized water and 1 ml of 30% hydrogen peroxide were added. The samples were heated until they were clear and effervescence had subsided. After cooling, 1 ml of 1:1 hydrochloric acid-deionized water and 2 ml of deionized water were added, and the samples were refluxed for 10 min at 95°C. The samples were quantitatively transferred to a 15-ml conical tube and diluted to 10 ml of total volume in deionized water.
Statistical analysis.
The results are expressed as means ± SE. One-way or two-way repeated-measures ANOVA and post hoc Tukey t-test were used to determine the effect of treatment (days 0–8, high-salt; days 9–19, high-salt/DOCP unless described otherwise) and genotype using Sigma Plot for Windows version 12. Paired and unpaired Student's t-tests were performed using Microsoft Excel Version 2010 for comparison of groups and when interactions were present. Differences between groups were considered statistically significant at P < 0.05.
RESULTS
High-salt diet intake.
The development of deoxycorticosterone acetate (DOCA) salt-induced hypertension in mice is influenced by the genetic background (16). The mouse strain used in the present study (C57BL/6) is more resistant to DOCA-induced increases in BP than other strains. Several studies reported robust DOCA-induced increases in BP in C57BL/6J mice when supplemented with NaCl in the drinking water (31, 43). Availability of dietary Na is an important consideration during mineralocorticoid treatment. To achieve a maximum hypertensive response, we chose a high-salt model that included 2% Na in a gelled diet and 0.9% NaCl in the drinking water (saline).
The CD ET-1 KO mice had a two-fold greater saline intake than the control mice (Fig. 1A) before and after treatment with DOCP when normalized for body weight. In a separate group of CD ET-1 KO mice, a preference study was performed (Fig. 1B). Given the option of water or saline, CD ET-1 KO mice drank small and equal volumes of water and saline each day on a normal Na diet (0.2%). However, the mice drank almost exclusively from the water bottle when adapted to the high-salt diet. During the first eight days of the high-salt diet, saline intake was significantly increased in CD ET-1 KO mice (repeated-measures ANOVA, F = 4.209, P < 0.01) but not in controls. Both genotypes increased their saline intake after DOCP treatment (control F = 76.126, CD ET-1 KO F = 11.056; P < 0.001). The CD ET-1 KO ate more of the high-salt gelled diet (2% Na) than control mice before and after DOCP treatment (Fig. 1C).
Fig. 1.

Saline and food intake, urine volume, and osmolality. Saline intake (A), preference (water vs. saline; B), and food intake (C) are shown as means ± SE normalized for body weight. Urinary output (D) and osmolality (E) are shown as means ± SE. Control, n = 9–10; collecting duct (CD)-specific endothelin-1 (ET-1) knockout (CD ET-1 KO) mice, n = 6. HS, average of days 6–8 of high-salt treatment; HS + DOCP, average of days 17–19 of DOCP (desoxycorticosterone pivalate) treatment. In A, C–E, *P < 0.05 vs. control within same treatment, unpaired Student's t-test; †P < 0.05 vs. HS treatment within same genotype, paired Student's t-test. In B, *P < 0.05 vs. water, same treatment, unpaired Student's t-test.
Urinary volume and osmolality.
Commensurate with greater saline intake, CD ET-1 KO mice had nearly a twofold greater daily urinary volume than controls both before and after DOCP treatment (Fig. 1D). DOCP treatment increased the urinary volume in both genotypes. CD ET-1 KO mice had significantly lower urine osmolality than control mice before and after DOCP treatment (Fig. 1E), and DOCP treatment decreased urinary osmolality in both genotypes. Over the course of the first 8 days of the high-salt diet, the daily urinary volume was increased significantly (repeated-measures ANOVA: F = 4.646, P < 0.001), and the urine osmolality decreased significantly (P < 0.05, day 1 vs. days 4 and 5) within the CD ET-1 KO mice but not within the controls. Urine pH was not significantly different between genotypes either before or after DOCP treatment, except for on day 1 (data not shown).
Fluid balance and body weight.
Fluid balance and body weight were measured to assess whether treatment with high salt and DOCP caused more fluid retention (positive fluid balance) and body weight gain in the CD ET-1 KO mice compared with controls. Fluid balance was calculated as the difference between fluid intake and the urinary output normalized for body weight (Fig. 2A). After 3 days of acclamation to the high-salt diet and metabolic cages, the CD ET-1 KO mice came into steady state with constant fluid intake and urine output. Accordingly, analysis was performed on days 4–8 of the high-salt diet. When fed the high-salt diet, CD ET-1 KO mice exhibited greater positive fluid balance (Fig. 2A) and body weight gain than controls (Fig. 2B). After DOCP treatment, control mice exhibited an increase in fluid balance and body weight, whereas CD ET-1 KO mice maintained a chronic state of positive fluid balance and increased body weight.
Fig. 2.

Fluid balance (A) normalized for body weight and %change in body weight from day 3 (B) are shown as means ± SE. Control, n = 9–10; CD ET-1 KO, n = 6. HS, average of days 4–8 of high-salt treatment; HS + DOCP, average of days 17–19 of DOCP treatment. *P < 0.05 vs. control within same treatment, unpaired Student's t-test; †P < 0.05 vs. HS treatment within same genotype, paired Student's t-test.
Radiotelemetry.
We evaluated systolic BP and HR in response to the high-salt diet before and after DOCP treatment. Daytime (0600–1700) and nighttime (1800-0500) systolic BP measurements are shown in Fig. 3A, top and bottom, respectively. When placed on the high-salt diet, systolic BP was increased in the CD ET-1 KO mice compared with pretreatment recordings on a normal-salt diet during both the day and night. In contrast, the high-salt diet did not increase BP in the control mice. DOCP treatment increased systolic BP in the CD ET-1 KO mice during both day and night. DOCP treatment increased BP in the control mice during the resting phase (daytime) but failed to reach statistical significance during the active phase (nighttime).
Fig. 3.

Systolic blood pressure (BP) and heart rate. Systolic BP (A) and heart rate (B) are shown as means ± SE during the daytime (top) and nighttime (bottom). Control, n = 7; CD ET-1 KO, n = 4. NS, normal salt; HS, average of days 5–7 of high-salt treatment; HS + DOCP, average of days 15–18 of DOCP treatment. *P < 0.05 vs. NS within the same genotype; **P = 0.05 vs. NS within the same genotype; †P < 0.05 vs. HS within the same genotype.
Daytime HR significantly increased in both genotypes when placed on the high-salt diet (Fig. 3B). After DOCP treatment, HR decreased significantly in the control mice during both day and night.
Sodium balance.
Direct assessment of Na handling was performed, measuring complete daily Na balance as the difference between total Na intake and excretion. We evaluated urinary and fecal excretion individually. In doing so, we were able to recognize distinct differences in Na handling in the CD ET-1 KO and control mice. Urinary Na excretion (Fig. 4A) was significantly greater in CD ET-1 KO mice compared with controls before and after DOCP treatment. CD ET-1 KO mice exhibited an increase in urinary Na excretion before and after DOCP treatment, whereas the control mice did not increase urinary Na excretion until after DOCP treatment. Control mice, but not CD ET-1 KO, exhibited a significant decrease in urinary Na excretion on day 11, 3 days after DOCP injection. Fecal Na excretion (Fig. 4B) was ∼20-fold less than urinary Na excretion. Fecal Na excretion was significantly higher in the CD ET-1 KO mice than controls on the first 2 days of the high-salt diet and 3 days after DOCP injection on day 11.
Fig. 4.
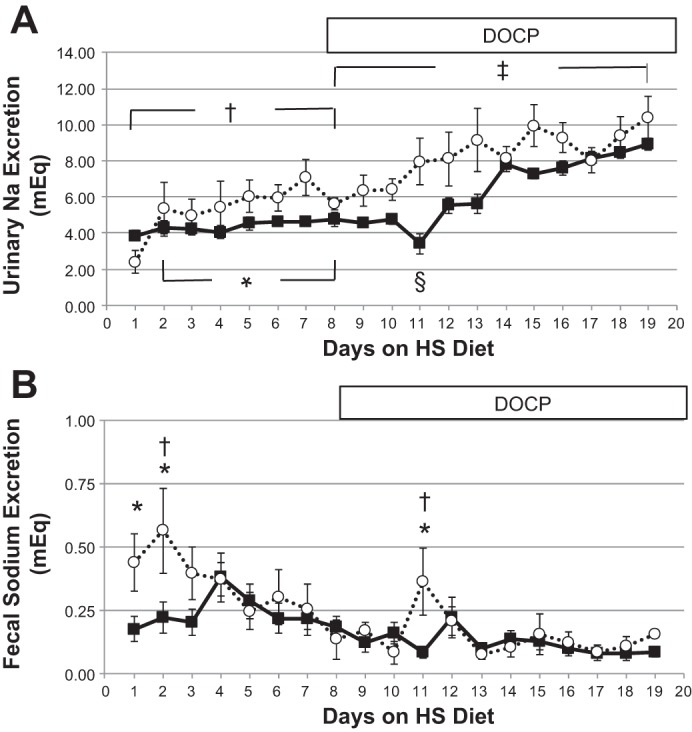
Sodium excretion. Urinary Na excretion (A) and fecal Na excretion (B) are shown as means ± SE. Solid lines are controls, and dashed lines are CD ET-1 KO. Control, n = 9–10; CD ET-1 KO, n = 6. In A, *P < 0.05, significant effect of genotype, repeated-measures ANOVA; †P < 0.05, significant effect of treatment within CD ET-1 KO, repeated-measures ANOVA; ‡P < 0.05, significant effect of genotype and treatment, repeated-measures ANOVA; §P < 0.05, significant effect of treatment within control, repeated-measures ANOVA, post hoc day 11 vs. days 8 and 12–19. In B, *P < 0.05, significant effect of genotype, repeated-measures ANOVA post hoc, days 1, 2, and 11; †P < 0.05, significant effect of treatment within CD ET-1 KO, repeated-measures ANOVA, post hoc, day 2 vs. days 8, 10, and 13–19 and day 11 vs. days 10, 13, 14, and 17.
Daily Na balance was calculated as the difference between the total Na intake and output (Fig. 5, A and B). Neither daily nor cumulative Na balance (Fig. 5, C and D) was different between controls and the CD ET-1 KO mice before DOCP treatment. However, DOCP treatment profoundly increased daily and cumulative Na balance in the CD ET-1 KO mice, resulting in a significant difference between the genotypes. DOCP treatment did not increase the daily Na balance in the control mice (which exhibited only a transient positive balance 3 days after DOCP injection) but did increase their cumulative Na balance.
Fig. 5.

Sodium balance, plasma sodium, and osmolality. A: daily Na balance. C: cumulative Na balance. HS, average of days 6–8 of HS treatment; HS + DOCP, average of days 17–19 of DOCP treatment. B and D: daily and cumulative Na balance, respectively, are shown for days 1–19. Solid lines are controls, and dotted lines are CD ET-1 KO. Control, n = 9; CD ET-1 KO, n = 6. E and F: Plasma Na and osmolality, respectively, are shown. Control, n = 4–11; CD ET-1 KO, n = 3–7. All data are shown as means + SE. In A and C, *P < 0.05 vs. control within same treatment, unpaired Student's t-test; †P < 0.05 vs. HS treatment within same genotype, paired Student's t-test. In B, *P < 0.05 within control, day 1 vs. days 2, 3, 6, and 7, Tukey's test; †P < 0.05 within control, day 11 vs. days 9–12 and 14–19, Tukey's test; ‡P < 0.05, significant effect of genotype, repeated-measures ANOVA. In D, *P < 0.05, significant effect of treatment within both genotypes, repeated-measures ANOVA; †P < 0.05 significant effect of treatment within both genotypes, repeated-measures ANOVA; ‡P < 0.05, significant effect of genotype, repeated-measures ANOVA, post hoc days 17–19. In E and F, *P < 0.05 vs. control within same treatment, unpaired Student's t-test; †P < 0.05 vs. NS treatment within the same genotype, unpaired Student's t-test.
The control mice exhibited positive Na balance (Na retention) within 24 h of the beginning of the high-salt diet and rapidly returned to balance by day 2 (Fig. 5B). Control mice exhibited a sudden positive Na balance 3–5 days after the DOCP injection (mostly on day 11) and rapidly returned to balance. The CD ET-1 KO mice responded differently from control mice to DOCP treatment. Of importance is that they did not exhibit the abrupt positive Na balance observed in the control mice after DOCP treatment. In response to DOCP, CD ET-1 KO mice entered a prolonged positive balance that was significantly greater than controls.
Plasma Na and osmolality were measured on the final day of the experiment and compared with mice fed a normal Na diet and are shown in Fig. 5, E and F. Plasma Na and osmolality in the CD ET-1 KO mice were not different from controls when fed a normal Na diet. After a chronic high-salt diet and DOCP treatment, the CD ET-1 KO mice had a significant increase in plasma osmolality and plasma Na.
Potassium balance.
Aldosterone excess is commonly associated with hypokalemia, although this hypokalemia does not reflect potassium deficiency but rather transcellular potassium redistribution (15). We measured daily urinary and fecal K excretion and calculated daily and cumulative K balance throughout the duration of the high-salt/DOCP study. Urinary K excretion (Fig. 6A) was not significantly different between control and CD ET-1 KO mice on the high-salt diet. Despite constant food intake, both genotypes exhibited diminishing urinary K excretion and increasing K balance (Fig. 6, B and C) after DOCP treatment. Furthermore, the CD ET-1 KO mice exhibited less urinary K excretion and greater K retention than the control mice after DOCP treatment. Plasma K, shown in Fig. 6D, was not different between control and CD ET-1 KO mice on a normal-salt (0.2% Na) diet. With DOCP treatment and a high-salt diet, plasma K decreased significantly in both genotypes. Urinary and fecal K excretions over the entire duration of the study are shown in Fig. 7.
Fig. 6.

Potassium excretion and balance and plasma potassium. A: urinary K excretion. B and C: daily and cumulative K balance, respectively. Control, n = 9; CD ET-1 KO, n = 6. HS, average of days 6–8 of HS treatment; HS + DOCP, average of days 17–19 of DOCP treatment. D: Plasma K. Control, n = 8 NS and 4 HS; CD ET-1 KO, n = 8 NS and 6 HS. All data are shown as means ± SE. In A–C, *P < 0.05 vs. HS diet within same genotype, paired Student's t-test; †P < 0.05 vs. control, same treatment, unpaired Student's t-test. In D, *P < 0.05 vs. NS diet within same genotype, unpaired Student's t-test.
Fig. 7.
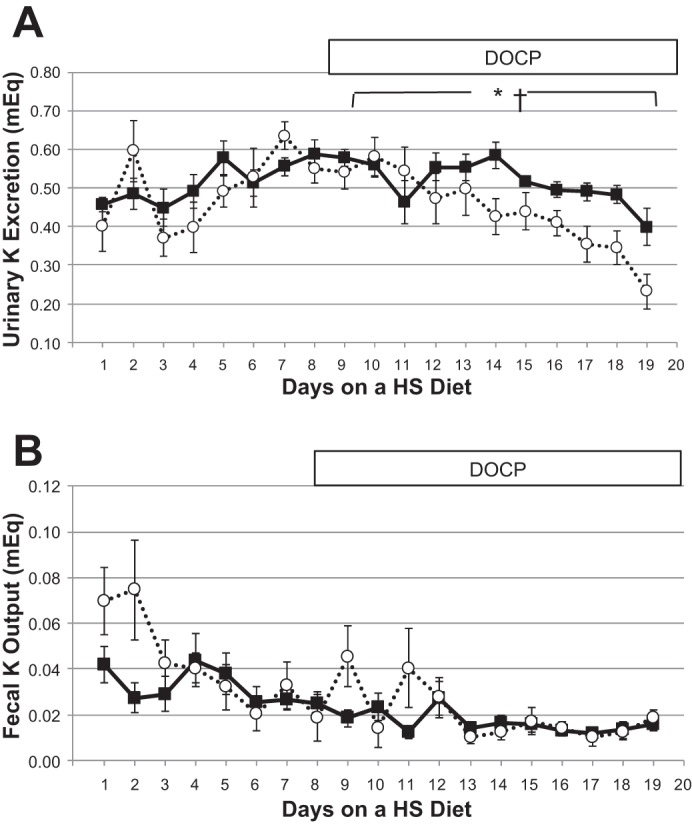
Urinary and fecal K excretion. A and B: urinary and fecal K excretion, respectively. Solid lines are controls, and dashed lines are CD ET-1 KO. Control, n = 9; CD ET-1 KO, n = 6. *P < 0.05, significant effect of genotype, repeated-measures ANOVA; †P < 0.05 significant effect of treatment in both genotypes, repeated-measures ANOVA.
DISCUSSION
Although aldosterone escape has been known for more than 50 years, its mechanism is not fully elucidated. Studies examining this mechanism have shown evidence that natriuretic peptides, prostaglandins, nitric oxide, specific transporters, and physical factors all participate in aldosterone escape (2, 23, 25, 27, 37, 41, 46, 47). However, the quantitative importance of these mechanisms, using the gold standard of complete metabolic balance, is quite limited. These studies represent one of the few experiments to quantify directly the importance of a mechanism to aldosterone escape. Our study implies that, under the conditions of these experiments, ET-1 expression by the CD is necessary for aldosterone escape. However, since many mechanisms may act in concert, the dependence of aldosterone escape on locally expressed ET-1 does not exclude the requirement of other factors or mechanisms. Purinergic signaling has been demonstrated to mediate aldosterone escape through downregulation of ENaC (37). There is evidence that purinergic and the ET systems may work in combination to elicit a diuretic and natriuretic response (35). ET-1 stimulates NO production (20). NO inhibits ENaC (17, 32), and the CD-specific KO of NO synthase 1 causes salt-sensitive hypertension (20). Aldosterone infusion in normal mice caused a marked increase in urinary microsomal prostaglandin E synthanse-1 (mPGES-1) and coinduction in proximal tubules, and the single-gene deletion of mPGES-1 caused impaired aldosterone escape by attenuating the downregulation of many Na transporters, including ENaC (23). The interaction between ET-1 and prostaglandins in the kidney requires further clarification.
The present study demonstrates that the absence of ET-1 expression principally in cells of the CD impairs the renal response to dietary Na loading. The physiological effects of a high-salt diet and DOCP treatment in CD ET-1 KO mice suggest a primary role for CD-expressed ET-1 to modify the effects of aldosterone on CD Na reabsorption. Control mice responded to DOCP treatment predictably by acutely decreasing urinary Na excretion and entering into a brief period of positive electrolyte balance (Figs. 4A and 5B). Control mice consequently developed modest positive fluid balance and body weight gain and exhibited acute adaptation of Na balance within 3–5 days, similar to previous reports. (23, 41, 47) In contrast, the CD ET-1 KO mice exhibited no acute response to DOCP but demonstrated progressive cumulative positive Na balance. At the end of the study, the CD ET-1 KO mice were severely hypernatremic.
Increased fecal excretion of Na observed in CD ET-1 KO mice immediately following the start of the high-salt diet and DOCP treatment may reflect compensation to excessive renal Na retention (Fig. 4B). However, in the absence of CD ET-1, other adaptive mechanism(s) fail to adequately compensate and restore neutral Na balance in response to chronic DOCP treatment. Previous studies have shown that CD ET-1 KO mice exhibit salt-sensitive hypertension (1, 24, 26). The present data confirm this finding and support the hypothesis that CD ET-1 attenuates the renal action of aldosterone. Specifically, CD ET-1 KO mice have increased systolic BP during the day and night when placed on the high-salt diet, whereas the control mice do not significantly increase BP on a high-salt diet alone (Fig. 3A).
The degree of hypertension observed in this study in mice on a C57BL/6J genetic background was not as pronounced as experiments conducted on a C57BL6/CBA background but is consistent with the resistance of this strain to develop hypertension (1). This observation is also of interest because it is well known that there are major effects of the genetic background on the degree of hypertension exhibited by specific mice strains (14, 14, 16). Indeed, genetic differences between the Dahl salt-sensitive and salt-resistant strains of rat have been the subject of extensive interest and research (5–7, 22, 29).
The protocol used in these studies has been used extensively as an accepted model of desoxycorticosterone-salt hypertension to produce a model of hypertension similar to primary hyperaldosteronism (10, 28, 42, 44, 45). This protocol was used specifically in the present study because the C57BL/6J mouse strain is resistant to DOCA-induced increases in BP. It has been used to provide important information on the factors that contribute to or attenuate this form of hypertension. Mice were monitored daily for adequate food and fluid intake and changes in body weight and body condition scoring. Prolonged isolation in metabolic cages can be stressful to the mice and must be recognized. However, since both controls and CD-ET-1 KO were subjected to identical conditions, the difference in the response of the CD ET-1 KO and the controls demonstrates the importance of CD ET-1 expression in the response to mineralocorticoids.
The changes in heart rate reflect the opposing effects of volume-independent changes in blood pressure via baroreceptors and volume-dependent changes in cardiac venous return or hypovolemia. Although a reflex increase in blood pressure will activate baroreceptors and decrease heart rate, isotonic sodium loading might be expected to increase in blood volume, which would increase venous return to the heart and consequently heart rate.
Despite slightly smaller average body weight, CD ET-1 KO mice consumed a substantially larger volume of saline than controls. The propensity for the CD ET-1 KO mice to consume a substantially greater volume of fluid during high-salt and DOCP treatment (Fig. 1A) suggests a central nervous system stimulation of thirst. Since these mice do not have access to water without Na, apparently they cannot correct the hypernatremia, and their condition is exacerbated with DOCP treatment. Indeed, by the end of the study, the CD ET-1 mice are substantially hypernatremic. A formal comparison of the CD ET-1 KO mice preference for water vs. saline revealed no selection of saline over water on a normal Na diet (Fig. 1B).
Although the inner medullary CD produces the greatest amount of ET-1, principal cells from all regions of the CD produce the autocrine (24). The connecting segment (CNT) of the distal nephron has a large Na transport capacity and appears to be an important regulator of Na excretion (33). Furthermore, an electroneutral thiazide-sensitive Na reabsorption mechanism identified as the NaCl cotransporter in the distal convoluted tubule (DCT) is downregulated during primary hyperaldosteronism (9). Therefore, the effects of chronic mineralocorticoid treatment and possible inhibitory effects of ET-1 on Na transport mechanisms in the CNT and DCT may also contribute to the genotype effects.
A significant increase in urinary K excretion was not observed as a result of DOCP treatment in either genotype. In contrast, both genotypes progressively reduced urinary K excretion after DOCP treatment by about 20% in the controls and 50% in the CD ET-1 KO mice by the final days of the experiment (Fig. 6A). Furthermore, the CD ET-1 KO mice retained more K than controls after DOCP treatment. Despite positive K balance, DOCP treatment induced significant hypokalemia in both genotypes (Fig. 6D). In a recent review, Gumz et al. (15) discuss intracellular redistribution of K by aldosterone. Interestingly, chronic balance studies on the effect of mineralocorticoids in several species (dog, rabbit, pig, and mouse) have failed to find a significant effect of either desoxycorticosterone or aldosterone to produce a significant negative K balance (8, 13, 18, 19, 34). One study has demonstrated that desoxycorticosterone produced a positive K balance despite the development of hypokalemia (12). Previously, our laboratory has reported that mineralocorticoids increased HKα2 H,K-ATPase activity, and mice lacking this H-K pump exhibited negative K balance, which supports a role for this K reabsorptive mechanism as one mechanism to maintain K balance with chronic mineralocorticoid treatment or excess. The current findings are consistent with these observations (12).
In summary, the absence of ET-1 expression in cells of the CD alters the renal response to aldosterone. When fed a high-salt diet, the CD ET-1 KO mice have increased blood pressure with fluid retention and body weight gain. DOCP treatment combined with the high-salt diet resulted in chronic electrolyte retention and substantial hypertension. Thus, disruption of a renal aldosterone-endothelin feedback mechanism prevented renal compensation for the effects of a mineralocorticoid-salt regiment. Such a locally acting system appears to be of primary importance in the strict regulation of Na and fluid balance.
The local autocrine-paracrine action of ET-1 to modulate Na transport in the CD involves multiple steps that are important for proper operation of this feedback control system, as demonstrated by others (20, 21), and may also act in concert with humoral or physical factors (25, 37, 41, 47). Further investigation is necessary to determine to what degree defects in other proteins or signaling mechanisms contribute to this impairment of fluid and electrolyte excretion and balance.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-82680 to B. D. Cain and C. S. Wingo, National Institutes of Health postdoctoral fellowship 2T32-HL-083810 to A. K. Welch, and funds from the Department of Veterans Affairs.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.J.L., A.K.W., M.L.G., D.E.K., B.D.C., and C.S.W. conception and design of research; I.J.L. performed experiments; I.J.L. analyzed data; I.J.L., A.K.W., M.L.G., D.E.K., B.D.C., and C.S.W. interpreted results of experiments; I.J.L. prepared figures; I.J.L. drafted manuscript; I.J.L., A.K.W., M.L.G., D.E.K., B.D.C., and C.S.W. edited and revised manuscript; I.J.L., A.K.W., M.L.G., D.E.K., B.D.C., and C.S.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. David Weiner for the kind gift of the AQP-2 Cre transgenic mice and Dr. Chris Baylis for review of the manuscript. We also thank Jennifer Prevot and John Collins for superb technical assistance.
Footnotes
Aldosterone escape should not be confused with “aldosterone breakthrough,” a response to therapy that may occur in patients treated with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) to reduce plasma aldosterone levels. In this condition, after initial reduction, plasma aldosterone increases to pretreatment levels. These patients tend to have a worse clinical prognosis than those that do not exhibit breakthrough (36).
REFERENCES
- 1.Ahn D, Ge Y, Stricklett PK, Gill P, Taylor D, Hughes AK, Yanagisawa M, Miller L, Nelson RD, Kohan DE. Collecting duct-specific knockout of endothelin-1 causes hypertension and sodium retention. J Clin Invest 114: 504–511, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballermann BJ, Bloch KD, Seidman JG, Brenner BM. Atrial natriuretic peptide transcription, secretion, and glomerular receptor activity during mineralocorticoid escape in the rat. J Clin Invest 78: 840–843, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bugaj V, Mironova E, Kohan DE, Stockand JD. Collecting duct-specific endothelin B receptor knockout increases ENaC activity. Am J Physiol Cell Physiol 302: C188–C194, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bugaj V, Pochynyuk O, Mironova E, Vandewalle A, Medina JL, Stockand JD. Regulation of the epithelial Na+ channel by endothelin-1 in rat collecting duct. Am J Physiol Renal Physiol 295: F1063–F1070, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cowley AW., Jr The genetic dissection of essential hypertension. Nat Rev Genet 7: 829–840, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Cowley AW Jr, Stoll M, Greene AS, Kaldunski ML, Roman RJ, Tonellato PJ, Schork NJ, Dumas P, Jacob HJ. Genetically defined risk of salt sensitivity in an intercross of Brown Norway and Dahl S rats. Physiol Genomics 2: 107–115, 2000. [DOI] [PubMed] [Google Scholar]
- 7.Dahl LK, Heine M, Thompson K. Genetic influence of renal homografts on the blood pressure of rats from different strains. Proc Soc Exp Biol Med 140: 852–856, 1972. [DOI] [PubMed] [Google Scholar]
- 8.Dawborn JK, Ross EJ. The effect of prolonged administration of aldosterone on sodium and potassium turnover in the rabbit. Clin Sci 32: 559–570, 1967. [PubMed] [Google Scholar]
- 9.Eladari D, Chambrey R, Picard N, Hadchouel J. Electroneutral absorption of NaCl by the aldosterone-sensitive distal nephron: implication for normal electrolytes homeostasis and blood pressure regulation. Cell Mol Life Sci 71: 2879–2895, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elijovich F, Zhao HW, Laffer CL, Du Y, DiPette DJ, Inagami T, Wang DH. Regulation of growth of the adrenal gland in DOC-salt hypertension. Role of angiotensin II receptor subtypes. Hypertension 29: 408–413, 1997. [DOI] [PubMed] [Google Scholar]
- 11.Ge Y, Huang Y, Kohan DE. Role of the renin-angiotensin-aldosterone system in collecting duct-derived endothelin-1 regulation of blood pressure. Can J Physiol Pharmacol 86: 329–336, 2008. [DOI] [PubMed] [Google Scholar]
- 12.Greenlee MM, Lynch IJ, Gumz ML, Cain BD, Wingo CS. Mineralocorticoids stimulate the activity and expression of renal H+,K+-ATPases. J Am Soc Nephrol 22: 49–58, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grekin RJ, Terris JM, Bohr DF. Electrolyte and hormonal effects of deoxycorticosterone acetate in young pigs. Hypertension 2: 326–332, 1980. [DOI] [PubMed] [Google Scholar]
- 14.Gumz ML, Popp MP, Wingo CS, Cain BD. Early transcriptional effects of aldosterone in a mouse inner medullary collecting duct cell line. Am J Physiol Renal Physiol 285: F664–F673, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Gumz ML, Rabinowitz L, Wingo CS. An Integrated View of Potassium Homeostasis. N Engl J Med 373: 60–72, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartner A, Cordasic N, Klanke B, Veelken R, Hilgers KF. Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol Dial Transplant 18: 1999–2004, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Helms MN, Yu L, Malik B, Kleinhenz DJ, Hart CM, Eaton DC. Role of SGK1 in nitric oxide inhibition of ENaC in Na+-transporting epithelia. Am J Physiol Cell Physiol 289: C717–C726, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Hulter HN, Licht JH, Sebastian A. K+ deprivation potentiates the renal acid excretory effect of mineralocorticoid: obliteration by amiloride. Am J Physiol Renal Fluid Electrolyte Physiol 236: F48–F57, 1979. [DOI] [PubMed] [Google Scholar]
- 19.Hulter HN, Sigala JF, Sebastian A. K+ deprivation potentiates the renal alkalosis-producing effect of mineralocorticoid. Am J Physiol Renal Fluid Electrolyte Physiol 235: F298–F309, 1978. [DOI] [PubMed] [Google Scholar]
- 20.Hyndman KA, Boesen EI, Elmarakby AA, Brands MW, Huang P, Kohan DE, Pollock DM, Pollock JS. Renal collecting duct NOS1 maintains fluid-electrolyte homeostasis and blood pressure. Hypertension 62: 91–98, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyndman KA, Bugaj V, Mironova E, Stockand JD, Pollock JS. NOS1-dependent negative feedback regulation of the epithelial sodium channel in the collecting duct. Am J Physiol Renal Physiol 308: F244–F251, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwai J, Knudsen KD, Dahl LK. Genetic influence on the renin-angiotensin system. Evidence for a renin inhibitor in hypertension-prone rats. J Exp Med 131: 543–557, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia Z, Aoyagi T, Kohan DE, Yang T. mPGES-1 deletion impairs aldosterone escape and enhances sodium appetite. Am J Physiol Renal Physiol 299: F155–F166, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kohan DE. Role of collecting duct endothelin in control of renal function and blood pressure. Am J Physiol Regul Integr Comp Physiol 305: R659–R668, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohan DE, Knox FG. Localization of the nephron sites responsible for mineralocorticoid escape in rats. Am J Physiol Renal Fluid Electrolyte Physiol 239: F149–F153, 1980. [DOI] [PubMed] [Google Scholar]
- 26.Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev 91: 1–77, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee YJ, Shin SJ, Tan MS, Hsieh TJ, Tsai JH. Increased renal atrial natriuretic peptide synthesis in rats with deoxycorticosterone acetate-salt treatment. Am J Physiol Renal Fluid Electrolyte Physiol 271: F779–F789, 1996. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Crockett E, Wang DH, Galligan JJ, Fink GD, Chen AF. Gene transfer of endothelial NO synthase and manganese superoxide dismutase on arterial vascular cell adhesion molecule-1 expression and superoxide production in deoxycorticosterone acetate-salt hypertension. Arterioscler Thromb Vasc Biol 22: 249–255, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Liang M, Lee NH, Wang H, Greene AS, Kwitek AE, Kaldunski ML, Luu TV, Frank BC, Bugenhagen S, Jacob HJ, Cowley AW Jr. Molecular networks in Dahl salt-sensitive hypertension based on transcriptome analysis of a panel of consomic rats. Physiol Genomics 34: 54–64, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Lynch IJ, Welch AK, Kohan DE, Cain BD, Wingo CS. Endothelin-1 inhibits sodium reabsorption by ETA and ETB receptors in the mouse cortical collecting duct. Am J Physiol Renal Physiol 305: F568–F573, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ni W, Zhou H, Diaz J, Murphy DL, Haywood JR, Watts SW. Lack of the serotonin transporter does not prevent mineralocorticoid hypertension in rat and mouse. Eur J Pharmacol 589: 225–227, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ortiz PA, Garvin JL. Role of nitric oxide in the regulation of nephron transport. Am J Physiol Renal Physiol 282: F777–F784, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Palmer LG, Frindt G. Na+ and K+ transport by the renal connecting tubule. Curr Opin Nephrol Hypertens 16: 477–483, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Pan YJ, Young DB. Experimental aldosterone hypertension in the dog. Hypertension 4: 279–287, 1982. [DOI] [PubMed] [Google Scholar]
- 35.Pandit MM, Inscho EW, Zhang S, Seki T, Rohatgi R, Gusella L, Kishore B, Kohan DE. Flow regulation of endothelin-1 production in the inner medullary collecting duct. Am J Physiol Renal Physiol 308: F541–F552, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schrier RW. Aldosterone ‘escape’ vs ‘breakthrough’. Nat Rev Nephrol 6: 61, 2010. [DOI] [PubMed] [Google Scholar]
- 37.Stockand JD, Mironova E, Bugaj V, Rieg T, Insel PA, Vallon V, Peti-Peterdi J, Pochynyuk O. Purinergic inhibition of ENaC produces aldosterone escape. J Am Soc Nephrol 21: 1903–1911, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stow LR, Gumz ML, Lynch IJ, Greenlee MM, Rudin A, Cain BD, Wingo CS. Aldosterone modulates steroid receptor binding to the endothelin-1 gene (edn1). J Biol Chem 284: 30087–30096, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sullivan JC, Goodchild TT, Cai Z, Pollock DM, Pollock JS. Endothelin(A) [ET(A)] and ET(B) receptor-mediated regulation of nitric oxide synthase 1 (NOS1) and NOS3 isoforms in the renal inner medulla. Acta Physiol (Oxf) 191: 329–336, 2007. [DOI] [PubMed] [Google Scholar]
- 40.Tomita K, Nonoguchi H, Terada Y, Marumo F. Effects of ET-1 on water and chloride transport in cortical collecting ducts of the rat. Am J Physiol Renal Fluid Electrolyte Physiol 264: F690–F696, 1993. [DOI] [PubMed] [Google Scholar]
- 41.Wang XY, Masilamani S, Nielsen J, Kwon TH, Brooks HL, Nielsen S, Knepper MA. The renal thiazide-sensitive Na-Cl cotransporter as mediator of the aldosterone-escape phenomenon. J Clin Invest 108: 215–222, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Babankova D, Huang J, Swain GM, Wang DH. Deletion of transient receptor potential vanilloid type 1 receptors exaggerates renal damage in deoxycorticosterone acetate-salt hypertension. Hypertension 52: 264–270, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Wang DH. Aggravated renal inflammatory responses in TRPV1 gene knockout mice subjected to DOCA-salt hypertension. Am J Physiol Renal Physiol 297: F1550–F1559, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Wang DH. Protective effect of TRPV1 against renal fibrosis via inhibition of TGF-beta/Smad signaling in DOCA-salt hypertension. Mol Med 17: 1204–1212, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Wang DH. Role of substance P in renal injury during DOCA-salt hypertension. Endocrinology 153: 5972–5979, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wright FS, Knox FG, Howards SS, Berliner RW. Reduced sodium reabsorption by the proximal tubule of Doca-escaped dogs. Am J Physiol 216: 869–875, 1969. [DOI] [PubMed] [Google Scholar]
- 47.Yokota N, Bruneau BG, Kuroski de Bold ML, de Bold AJ. Atrial natriuretic factor significantly contributes to the mineralocorticoid escape phenomenon. Evidence for a guanylate cyclase-mediated pathway. J Clin Invest 94: 1938–1946, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeidel ML, Brady HR, Kone BC, Gullans SR, Brenner BM. Endothelin, a peptide inhibitor of Na(+)-K(+)-ATPase in intact renaltubular epithelial cells. Am J Physiol Cell Physiol 257: C1101–C1107, 1989. [DOI] [PubMed] [Google Scholar]