

Abstract
We tested the hypothesis that suppression of epoxyeicosatrienoic acid (EET) metabolism via genetic knockout of the gene for soluble epoxide hydrolase (sEH-KO), or female-specific downregulation of sEH expression, plays a role in the potentiation of pulmonary hypertension. We used male (M) and female (F) wild-type (WT) and sEH-KO mice; the latter have high pulmonary EETs. Right ventricular systolic pressure (RVSP) and mean arterial blood pressure (MABP) in control and in response to in vivo administration of U46619 (thromboxane analog), 14,15-EET, and 14,15-EEZE [14,15-epoxyeicosa-5(z)-enoic acid; antagonist of EETs] were recorded. Basal RVSP was comparable among all groups of mice, whereas MABP was significantly lower in F-WT than M-WT mice and further reduced predominantly in F-KO compared with M-KO mice. U46619 dose dependently increased RVSP and MABP in all groups of mice. The increase in RVSP was significantly greater and coincided with smaller increases in MABP in M-KO and F-WT mice compared with M-WT mice. In F-KO mice, the elevation of RVSP by U46619 was even higher than in M-KO and F-WT mice, associated with the least increase in MABP. 14,15-EEZE prevented the augmentation of U46619-induced elevation of RVSP in sEH-KO mice, whereas 14,15-EET-induced pulmonary vasoconstriction was comparable in all groups of mice. sEH expression in the lungs was reduced, paralleled with higher levels of EETs in F-WT compared with M-WT mice. In summary, EETs initiate pulmonary vasoconstriction but act as vasodilators systemically. High pulmonary EETs, as a function of downregulation or deletion of sEH, potentiate U46619-induced increases in RVSP in a female-susceptible manner.
Keywords: pulmonary hypertension, soluble epoxide hydrolase, epoxyeicosatrienoic acids, right ventricular systolic pressure, sex difference
pulmonary arterial hypertension (PAH) is a progressive disease with different etiologies that primarily affects small pulmonary arteries (28) and is associated with elevated pulmonary vasoconstriction, smooth muscle proliferation, endothelial dysfunction, and pulmonary remodeling (32). The two major factors that specifically contribute to the increase in pulmonary vascular resistance are vasoconstriction and vascular remodeling (9). Acute vasodilation resulting from the inhibition of Rho-kinase, an enzyme that causes sustained pulmonary vasoconstriction to generate pulmonary hypertension (33), significantly ameliorates pulmonary arterial pressure in patients with severe pulmonary hypertension (PH) (14, 17). Based on this evidence, recent studies arose to address the concern that specific roles of vasoconstriction in late stages of PH was underappreciated (39). The findings address an important issue that pulmonary artery tone and consequentially pulmonary resistance are controlled by factors released from the pulmonary vasculature. In this regard, any lung-sourced vasoconstrictors merit consideration as an endogenous trigger for PAH. For instance, endothelium-derived metabolites of arachidonic acid (AA) by lipoxygenase to form 15-HETE (35), or by cyclooxygenase to form thromboxane A2 (3, 5) initiate pulmonary vasoconstriction in mice and rabbits. Also, in vivo infusion of U46619, a thromboxane analog, acutely generates PAH in mice (44) and rats (10).
Idiopathic pulmonary arterial hypertension (IPAH) occurs more frequently in women (12) and is, therefore, categorized as a disease with female-specific prevalence (31). Based on data from across the U.S. published in 2010, of a total 2,525 patients with PAH, 2,007 (79.5%) were women (4). In this regard, the sex difference in the incidence and pathogenesis of pulmonary hypertension has encouraged research that is focused on the mechanisms involved. The change in female hormones was put forth as one possible explanation, based on studies showing that PAH was more prevalent in women who had taken oral contraceptives (16, 23) or received hormone replacement therapy (43). Nonetheless, some controversial findings refuted such an association (1) and a female-favorable advantage in IPAH survival has even been documented (18). Interestingly, a previous study reported a female-specific enhancement in pulmonary vasoconstriction to AA via an estrogen-dependent upregulation of lipoxygenase pathway in rabbits (34). The study, however, did not specifically correlate the increase in pulmonary vasoconstriction with in vivo changes in pulmonary artery pressure and instead focused mainly on the altered lipoxygenase pathway in isolated pulmonary arteries.
In addition to cyclooxygenase- and lipoxygenase-dependent pathways, the third pathway for AA metabolism is cytochrome P-450 (CYP)/epoxygenase-dependent production of epoxyeicosatrienoic acids (EETs). EETs are endothelial mediators that evoke systemic vasodilation to lower blood pressure (6) but elicit vasoconstriction in the pulmonary circulation (20, 21). Degradation of vascular EETs occurs rapidly by soluble epoxide hydrolase (sEH) to produce their corresponding non- or less vasoactive diols (dihydroxyeicosatrienoic acids, DHETs) (7), a response that contributes significantly to the angiotensin II-induced hypertension (19). In this context, the dual biological action of EETs in the sex-specific regulation of systemic and pulmonary circulations forms the basis of the present study. Given that IPAH correlates with a female-prevalent characteristic, and that compromising EET metabolism increases EET bioavailability, it is plausible to hypothesize that the suppression of EET hydrolysis via either genetic disruption of the sEH gene or the female-specific downregulation of sEH expression contributes significantly to potentiate pulmonary vasoconstriction. By using both male and female mice that have genetically deleted Ephx2 gene encoding sEH, along with their wild-type (WT) counterparts, we assessed dynamic changes in pulmonary (right ventricular systolic pressure, RVSP) and systemic blood pressure both under basal conditions and during stimulation with U46619 (a thromboxane analog) and EETs.
MATERIALS AND METHODS
Animals
Twelve- to 15-wk-old male (M) and female (F) Ephx2−/− (sEH-KO) and Ephx2+/+ (WT) mice were used. As described previously (40), cryorecovered heterozygous (Ephx2+/−, B6.129X-Ephx2tm1Gonz/J) and WT (Ephx2+/+) mice were received from the Jackson Laboratory (Bar Harbor, ME) and the homozygous (Ephx2−/−) mice were bred in the Department of Comparative Medicine, New York Medical College.
All protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College and conform to the guidelines of the National Institutes of Health and the American Physiological Society for the use and care of laboratory animals.
Experimental Protocols
Surgery.
The procedure was described in detail previously (20). Briefly, mice were anesthetized with inhalation of Isothesia (isoflurane). Heart rate (HR) and systemic arterial pressure (BP) were recorded through a left carotid artery catheterization. The left jugular vein catheterization was used for infusion of U46619, 14,15-EET, and 14,15-EEZE. The third catheter (20) was placed in the right jugular vein and advanced into the right ventricle for monitoring RVSP. The pressure catheters were filled with heparinized saline and connected with pressure transducers (TRN050; Kent Scientific, Torrington, CT) and transducer amplifiers (Kent Scientific). Changes in systemic arterial pressure, HR, and RVSP were recorded on PowerLab (ADInstruments, Colorado Springs, CO) and analyzed with Chart V8 software (ADInstruments).
Administration of U46619.
Mice were allowed to recover from the surgery for 10 min, during which HR was controlled at ∼500 beats/min by adjusting the depth of anesthesia. RVSP, BP, and HR were recorded to obtain stable baselines. Subsequently, U46619 was administered through the left jugular vein catheter. Four doses of U46619 (0.01, 0.05, 0.1 and 0.2 nmol/g body wt) were used. A final volume of 20 μl for each dose of U46619 was injected via syringe pump at a delivery rate of 10 μl/min. A 15-min interval was given between injections. Changes in RVSP and BP were continuously recorded and the vehicle for U46619 was tested and no hemodynamic effects were observed.
In separate experiments, after control responses to 0.2 nmol/g body wt U46619 were recorded, 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE, 10 μM; a putative EET antagonist) was administered. After 10 min, hemodynamic changes to the same dose of U46619 were once more recorded.
Administration of EET.
In this protocol, EET-induced changes in RVSP and BP were measured via infusion of 14,15-EET (1, 2, 5 and 10 ng/g body wt in a final volume of 20 μl) through the left jugular vein catheter (20). Each dose of 14,15-EET was injected over 2 min, and a 15-min interval was given between injections. Changes in RVSP and BP were continuously recorded.
Echocardiography.
In separate echocardiograph experiments, mouse cardiac output was determined to clarify that the EET-induced increases in RVSP did not significantly affect the cardiac output. As described previously (2), mice were anesthetized with isoflurane, and the left jugular vein was catheterized for EET infusion. The mouse was then placed onto a heated echo platform in the supine position with the limbs taped onto the EKG leads. Transthoracic echocardiography was performed by placing the 30-MHz transducer (Vevo 770, Visualsonics, Toronto, Ontario, Canada) along the long axis of the left ventricle (LV) in a direction toward the right side of the neck. After recording LV long-axis images, the transducer was rotated clockwise by 90° and short-axis views were recorded. Parasternal long-axis views and short-axis views at the papillary muscle level and 2D guided M-mode images were obtained and used to calculate the cardiac output before and after administration of 14,15-EET (2 ng/g body wt). The saline containing 0.1% ethanol was used as the vehicle control before injection of the EET.
LC/MS/MS-based measurements for lung EETs and DHETs.
At the end of experiments, the lungs were excised for EET measurements. As described in detail (36, 40), esterified EETs and DHETs from tissue extracts were quantified via LC/MS/MS (AB ScieX, Qtrap 3200). Protein concentration of samples, determined by the Bradford method (Bio-Rad, Hercules, CA), was used to normalize the detected lipids, such that each regioisomer of EETs and DHETs was expressed as picograms per microgram protein. Total EETs and DHETs were the sum of all four isoforms of EET and DHET.
Western blot analysis.
We separated 25 μg protein extracted from lungs by a 10% SDS-PAGE gel and transferred it to a PVDF membrane. The membrane was probed with specific primary antibodies for sEH (Santa Cruz Biotechnology, Santa Cruz, CA), CYP2C29 (Biodesign, Maco, ME) and thromboxane A2 receptor (TXA2R; Cayman Chemical, Ann Arbor, MI), respectively, and appropriate secondary antibodies were conjugated with horseradish peroxidase. The specific bands were visualized with an enhanced chemiluminescent reaction and normalized to β-actin or GAPDH.
Statistics
Data are represented as means ± SE, and n refers to the number of mice. Statistical analysis was performed with GraphPad Prism 5 software. Repeated-measures ANOVA followed by the Tukey-Kramer post hoc test were used to compare the difference among multiple groups. Student's t-test was used to compare the difference between two groups. Statistical significance was accepted at a level of P < 0.05.
RESULTS
Changes in RVSP
Figure 1 shows an original tracing (A) recorded under basal conditions and in response to administration of U46619 (0.1 nmol/g body wt). Additionally, summarized data of changes in RVSP are included, as a function of U46619 (B) and EEZE (C), respectively, in the four groups of mice: abbreviated as M-WT (male wild type), F-WT (female wild type), M-KO (male sEH-KO) and F-KO (female sEH-KO). As shown in Fig. 1B, all groups of mice, which had comparable basal RVSP (M-WT: 28.1 ± 0.7; F-WT: 29.1 ± 1.2; M-KO: 29.2 ± 0.7; F-KO: 29.5 ± 0.5 mmHg), exhibited dose-dependent increases in RVSP in response to stimulation with U46619, but with significant difference in their increments. Specifically, F-WT mice displayed a significant elevation of U46619-induced increase in RVSP compared with M-WT, revealing a sex-different response. Disruption of the sEH gene dramatically enhanced U46619-induced increases in RVSP in both sexes of mice, as indicated by the significantly upward-shifted curves of M-KO and F-KO mice from their WT controls, suggesting that sEH may serve as a modulator in the pulmonary circulation. Moreover, sEH deficiency-dependent augmentation of U46619-induced increases in RVSP was significantly greater in F-KO compared with M-KO, suggesting an additive/or synergistic effect between the female sex and sEH deficiency. It is worth noting that RVSP of M-KO mice was close to that of F-WT mice (manifested as comparable curves between the two groups), which alludes to the presence of female-specific properties that may mimic actions of sEH deficiency in males.
Fig. 1.

A: original tracing for changes in right ventricular systolic pressure (RVSP) in response to U46619. B: dose-dependent increases in RVSP in response to U46619 in male (M) and female (F) wild-type (WT) mice and mice with genetic knockout (KO) of the gene for soluble epoxide hydrolase (sEH) (n = 6–9 in each group). *Curve of M-KO or F-WT mice shows significant enhancement compared with that of M-WT mice. #Curve of F-KO mice shows significant enhancement compared with those of other 3 groups of mice. C: effects of 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE) on U46619 (0.2 nmol/g body wt)-induced increases in RVSP in the 4 groups of mice (n = 6–9 in each group). *Significant difference from M-KO mice. #Significant difference from F-KO mice. BW, body weight.
Next, roles of EETs in the mediation of U46619-elicited augmentation of RVSP in sEH-KO mice were evaluated, and results are shown in Fig. 1C. We found that 14,15-EEZE reversed the augmented response to U46619 (0.2 nmol/g body wt) in both M-KO and F-KO mice back down to a level similar to their WT controls but had no effects on the responses in WT mice. This specifies that the sEH-deficient augmentation of U46619-induced increases in RVSP is indeed EET-dependent in nature.
Values for each regioisomer of EETs and total EETs in the lungs are summarized in Table 1 and Fig. 2. There was a significantly higher total level of EETs (Fig. 2A) and a greater EETs/DHETs ratio (Fig. 2B) in F-WT than M-WT mice, revealing a sex difference in pulmonary EET metabolism. In response to deletion of the sEH gene, total EETs, as well as the ratio of EETs/DHETs were significantly increased in M-KO mice. The increment was non-statistically significant in F-KO compared with F-WT mice. Moreover, Table 1 shows that 14,15-EET, 11,12-EET, and 8,9-EET were elevated, associated with reductions in corresponding DHETs, resulting in a significantly greater EETs/DHETs ratio in both sexes of sEH-KO mice, as well as in F-WT mice. This finding implies that the female sex may possess a specific regulatory mechanism that mimics the Ephx2 gene deletion. Also, an unchanged 5,6-EET/DHET in response to disruption of the Ephx2 gene indicates that this isoform of EET is a poor substrate for sEH (36, 46). Importantly, administration of 14,15-EET dose dependently elicited comparable increases in RVSP in all groups of mice (Fig. 2C), suggesting that their vascular reactivity to EETs remains unchanged.
Table 1.
Pulmonary cytochrome P-450 metabolites
| M-WT (n = 8) | M-KO (n = 6) | F-WT (n = 6) | F-KO (n = 6) | |
|---|---|---|---|---|
| 14,15-EET | 0.98 ± 0.14 | 2.7 ± 0.6* | 1.78 ± 0.2* | 3.11 ± 0.11† |
| 11,12-EET | 0.85 ± 0.2 | 2.8 ± 0.7* | 2.16 ± 0.4* | 2.4 ± 0.3 |
| 8,9-EET | 1.14 ± 0.18 | 2.3 ± 0.3* | 1.9 ± 0.6 | 3.4 ± 0.4† |
| 5,6-EET | 0.97 ± 0.1 | 1.2 ± 0.2 | 1.7 ± 0.2 | 1.3 ± 0.2 |
| 14,15-DHET | 0.12 ± 0.02 | 0.05 ± 0.009* | 0.09 ± 0.015 | 0.08 ± 0.008 |
| 11,12-DHET | 0.17 ± 0.3 | 0.07 ± 0.02* | 0.12 ± 0.15* | 0.06 ± 0.012† |
| 8,9-DHET | 0.24 ± 0.02 | 0.09 ± 0.03* | 0.18 ± 0.04 | 0.084 ± 0.03† |
| 5,6-DHET | 0.25 ± 0.04 | 0.21 ± 0.03 | 0.27 ± 0.03 | 0.15 ± 0.04† |
| 14,15EET/DHET | 7.5 ± 1.4 | 35.3 ± 6.7* | 19.8 ± 4.9* | 35.54 ± 5.4† |
| 11,12-EET/DHET | 5.6 ± 1.3 | 46.43 ± 16.4* | 18.4 ± 3.6* | 40.32 ± 7.3*† |
| 8,9-EET/DHET | 9.1 ± 3.3 | 35.77 ± 8.7* | 10.94 ± 4.01 | 33.14 ± 9.1† |
| 5,6-EET/DHET | 5.1 ± 1.3 | 7.0 ± 1.0 | 6.3 ± 1.1 | 11.1 ± 4.3 |
M, male; F, female; WT, wild type; KO, knockout; EET, epoxyeicosatrienoic acid; DHET, dihydroxyeicosatrienoic acid.
Significant difference from M-WT;
significant difference from F-WT.
Fig. 2.

Total epoxyeicosatrienoic acids (EETs) (A) and the ratio of EETs to dihydroxyeicosatrienoic acids (DHETs) (B) in lungs taken from male and female WT and sEH-KO mice (n = 6–8 in each group). *Significant difference from M-WT mice. #Significant difference from F-WT mice. C: 14,15-EET-induced increases in RVSP in the 4 groups of mice (n = 5 in each group).
Changes in Systemic Blood Pressure
Figure 3 shows an original tracing (A) and summarized data of changes in mean arterial blood pressure (MABP) of mice, as a function of U46619 (B) and EEZE (C). Unlike pulmonary circulation, which displayed comparable basal RVSP among the four groups, systemic circulation revealed a sex difference in MABP among both WT and sEH-KO mice, showing that female mice had significantly lower blood pressure (F-WT: 84.7 ± 0.78 and F-KO: 80.6 ± 0.9 mmHg) than their male counterparts (M-WT: 90.3 ± 0.7 and M-KO: 83.9 ± 0.58 mmHg). Deficiency in sEH initiated a parallel reduction of MABP in both sexes, leading to the lowest basal MABP in F-KO mice. In response to U46619, Fig. 3B shows that all groups of mice displayed a dose-dependent increase in MABP. This increase in MABP also presented a sex-specific responsive phenotype, as evidenced by the fact that F-WT exhibited a significantly less increase in MABP than M-WT. Deletion of the sEH gene led to an even smaller increase in blood pressure in both sexes of mice, but once again the female phenotype (F-KO mice) exhibited the lowest incremental increase in MABP. Direct infusion of 14,15-EET initiated a significant reduction of MABP in all groups of mice (Fig. 3C), further confirming that EETs regulate pulmonary and systemic vascular tone via opposable means. F-KO mice also displayed significant decrements at each selected dose-point compared with the other three groups of mice, due perhaps, to having the lowest MABP.
Fig. 3.

A: original tracing for changes in arterial blood pressure in response to U46619. B: dose-dependent increases in MABP in response to U46619 in male and female WT and sEH-KO mice (n = 6–9 in each group). *Curve of M-KO or F-WT mice shows significant reduction compared with that of M-WT mice. #Curve of F-KO mice shows significant reduction compared with those of other 3 groups of mice. C: 14,15-EET-induced reduction in MABP in the 4 groups of mice (n = 5 in each group). *Significant reduction at each dose of 14,15-EET in 4 groups of mice compared with their basal line.
Collectively, the female sex and sEH-KO are engaged synergistically in the regulation of pulmonary and systemic circulations, such that RVSP is increased and MABP is reduced.
Characteristics of EET-Induced Pulmonary and Systemic Responses
To exclude the possibility that EET-induced increases in RVSP (Fig. 2C) may result from increased systemic venous return as a function of EET-induced systemic vasodilation, rather than EET-induced pulmonary vasoconstriction, we performed specific time-course experiments by administering a total volume of 20 μl of 14,15-EET in a final concentration of 2 ng/g body wt over 1 and 2 min, respectively (Fig. 4). Figure 4A shows that the elevation of RVSP accelerated more rapidly and dramatically when 14,15-EET was delivered over 1 min compared with the same amount of 14,15-EET delivered over 2 min, suggesting that 14,15-EET-induced pulmonary vasoconstriction is proportional to its local/pulmonary concentration. This difference, however, was not present in the systemic circulation, as evidenced by comparable MABP curves between the groups (Fig. 4B). Moreover, 14,15-EET-induced increases in RVSP took precedence over its reduction in MABP, and HR was not significantly affected by either injection rate of EET (Fig. 4C). Together, these results indicate that the increase in RVSP shown in Fig. 4A is independent of EET-induced systemic vasodilation. The enhanced venous return, if any, is not a primary determinant in the EET-induced elevation of RVSP presented in this study. Moreover, Fig. 4D aimed to evaluate effects of EET on the cardiac output and shows that neither vehicle nor 14,15-EET injections affected the cardiac output. The cardiac output measured at 1 and 3 min after EET injection was correlated to the time points at which peak changes in RVSP and MABP were recorded. These results suggested that neither the increased RVSP nor the reduced MABP was secondary to EET-induced changes in cardiac output.
Fig. 4.
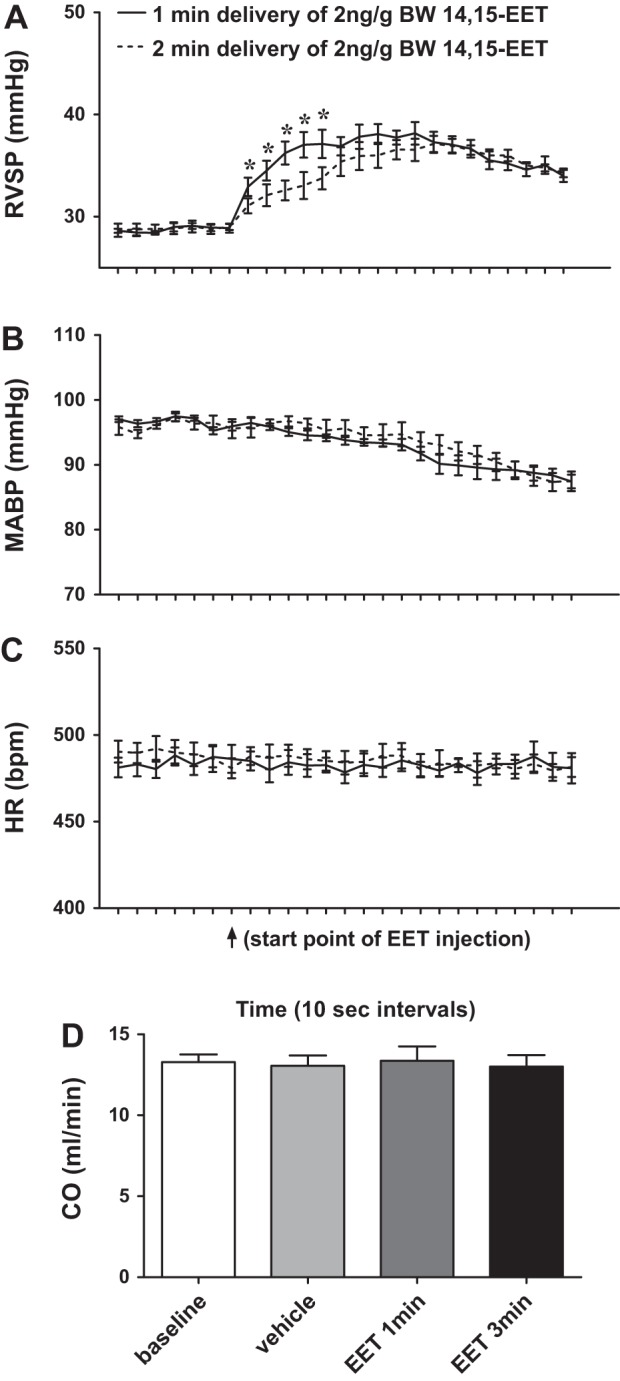
Time-dependent changes in RVSP (A), mean arterial blood pressure (MABP; B), and heart rate (HR; C) of male WT mice in response to a single dose of 14,15-EET (2 ng/g body wt), injected in a volume of 20 μl within 1 and 2 min, respectively (n = 5). *Significant difference from 2-min delivery group at the same time points. D: cardiac output (CO) of male WT mice in control conditions (baseline and vehicle) and after injection of 14,15-EET (2 ng/g body wt) for 1 and 3 min, respectively (n = 5).
Influence of Sex on Protein Expression of sEH
Western blot analysis (Fig. 5) shows that pulmonary expression of sEH (A and B) was significantly reduced by approximately fivefold in F-WT compared with M-WT mice and was undetectable in sEH-KO mice (data not shown). Figure 5C shows the comparable expression of CYP2c29 (EET synthase) and TXA2R among the four groups of mice.
Fig. 5.

Protein expression of sEH (A and B), CYP2C29, and thromboxane A2 receptor (TXA2R) (C) in the lungs of male and female WT and sEH-KO mice. *Significant difference from M-WT mice (n = 3 blots from 10 mice in each group).
DISCUSSION
The salient findings of our studies are as follows: 1) In vivo administration of U46619 and 14,15-EET dose dependently increases RVSP. 2) There is a sex difference in U46619-induced pulmonary hypertension, characterized as a higher RVSP in female mice than in their male controls under an identical basal RVSP. 3) Deletion of the gene for sEH significantly enhances U46619-induced increases in RVSP in both sexes of mice with greater predominance in females. 4) Disruption of the sEH gene in males evokes the same changes in RVSP (Fig. 1B) and pulmonary EET metabolism (Fig. 2 and Table 1) as downregulation of sEH expression in F-WT mice. In the latter, however, an additional sEH knockout (F-KO) further promotes augmented RVSP in a sEH-independent manner. 5) High pulmonary levels of EETs, as a consequence of sEH deficiency, contribute significantly to the augmented U46619-induced increases in RVSP, since an EET antagonist prevents the response. 6) In the systemic circulation, the female phenotype of lower blood pressure exists in both basal and U46619-stimulated conditions, as a function of EET-induced systemic vasodilation. Thus we provided a mechanistically based explanation, at least in part, for the female susceptibility to PAH, characterized as a female-specific downregulation of sEH expression to promote EET-induced pulmonary vasoconstriction and potentiate U46619-induced pulmonary hypertension. A recent study has demonstrated an estrogen-dependent improvement of right ventricular function in rats with established PH (13), which provides clinical significance relevant to the prognosis of the disease, as a function of sex. Our studies conducted on normal mice, further reveal a sex difference in the incidence of PAH. However, it is important to note that the deletion of the sEH gene was unable to eliminate the sex-specific variances in RVSP and MABP, suggesting that other mechanisms that are independent of sEH may also participate in the responses.
The recognition that the female sex and/or estrogen favors the contribution of EETs has been documented (38). We also demonstrated that, in resistance arteries, actions of EETs become discernable when EET degradation is compromised (40). Thus, via genetic disruption of the sEH gene to increase intracellular EET levels and prolong their vascular actions, we were able to evaluate the pathophysiological significance of sEH/EETs in the regulation of pulmonary and systemic circulations, as a function of sex.
Female-Specific Potentiation of RVSP in an EET-Dependent Manner
Although the pulmonary level of EETs was significantly higher in females than males (Fig. 2), their basal RVSP were comparable (Fig. 1). These results suggest that EETs may not be the major contributors to the physiological control of pulmonary vascular tone but instead may play a role in the mediation of the pathological processes. Indeed, U46619-induced elevation of RVSP increased significantly more in female mice (Fig. 1B), results that are consistent with those observed in isolated perfused lungs that show an increase in vasoconstriction to U46619 in an estrogen-dependent manner (10). Further increases in RVSP caused by sEH deficiency were also characterized as a female-predominant phenotype (F-KO > M-KO; Fig. 1B), suggesting the presence of an additional female-specific mechanism(s) that is independent of sEH. These findings fit well with the categorization of PAH as a disease with sex-specific prevalence (30), since CYP/epoxygenase, as well as sEH are favorable targets for female hormones/estrogens (41, 42). In the present study, pulmonary expression of CYP2C29 (an endothelial EET synthase) and TXA2R were comparable between WT and sEH-KO mice, suggesting that it is neither the upregulation of EET synthase nor the changes in the receptor density, but rather the reduction of EET degradation to increase EET bioavailability, that sensitizes vasoconstrictor responses to U46619. On the other hand, compared with M-KO mice, the sEH-independent component of augmented U46619-induced increase in RVSP in F-KO mice (Fig. 1B) could be attributed to a female-favorable increase in activity of EET synthase or downregulation of CYP4A to reset the balance between EETs and 20-hydroxyeicosatetraenoic acid (20-HETE). Additional studies are needed to explore specific signaling molecules in the pulmonary vasculature, such as CYP-ω-hydroxylation of arachidonic acid to produce 20-HETE and endothelial nitric oxide synthase to produce NO, both pathways that interact with each other and are regulated by actions of epoxygenase/EETs/sEH in a sex-specific manner. It is important to note that in alignment with the EET metabolic pattern (Fig. 2 and Table 1), the U46619-RVSP curve of M-KO mice (Fig. 1B) profiled a significant upward shift from their WT controls, leading to a statistically overlapped curve with that of F-WT mice. Thus a question was raised as to whether Ephx2 gene knockout in males imitates outcomes that are similar to a lower expression of sEH in females. Figure 5A provides molecular evidence supporting our hypothesis that the drastically suppressed expression of sEH in the lungs of F-WT mice is responsible for their significantly higher RVSP compared with M-WT mice. This conclusion also serves as an explanation for the phenomenon that deletion of the sEH gene in M-WT mice eliminated their difference from F-WT mice. Thus the functional assessment of RVSP (Fig. 1) and biochemical measurement of pulmonary EETs (Fig. 2 and Table 1) have provided strong evidence of an EET-dependent promotion of pulmonary hypertension in a female-susceptible manner. Alternatively, conflicting results have also shown an estrogen-dependent prevention of pulmonary pressor response to phenylephrine and hypoxia in isolated pulmonary artery rings that are challenged with superphysiological concentrations of estradiol (24, 25). In such cases, however, contributing responses are mainly due to nongenomic effects of estrogen on the activation of NO and prostacyclin synthases or direct vasodilation induced by high doses of estradiol (26), mechanisms that are different from those identified in the present study. Moreover, sEH inhibitors attenuated monocrotaline-induced PH in rats (37), but in this instance the underlying mechanism might refer either to an intrinsic cardioprotective property of sEH inhibitors used or to a direct EET-mediated protection against monocrotaline-induced inflammation. Since the direct responses to 14,15-EET were comparable among the groups (Fig. 2C), whereas EEZE-treatment reversed the augmented U46619-induced increases in RVSP in sEH-KO mice without impacts on those of WT controls (Fig. 1C), it can be speculated that the elevated pulmonary EETs intensified the vasoconstrictor effect of U46619. Although the specific mechanism(s) responsible for the augmented RVSP response to U46619 are currently unknown, an alteration in the metabolism of AA seems to be a promising explanation, since all three major pathways responsible for AA metabolism via lipoxygenase (34, 35), cyclooxygenase (3, 5), and cytochrome P-450/exoxygenase (20–22) are reported to be involved in potentiating pulmonary arterial pressure. Particularly, activation of Rho-kinase activity by EETs (21) has been considered as a primary determinant in human and animal models of PAH with different etiologies, including hypoxia-induced PH (8, 15). Moreover, the expression and activity of EET synthase is demonstrated to be sensitive to changes in oxygen tension (11). Such a phenomenon could explain our present findings indicating that hypoxia per se directly augments EET-induced increases in RVSP (20), and, vice versa, RVSP in sEH-KO mice exposed to hypoxia is increased by more than twofold compared with WT controls (unpublished results). Owing to the female-specific prevalence of PAH, sex may have a major influence on the converging actions of Rho-kinase, hypoxia, and sEH/EETs to trigger pathogenesis of PAH. During this process, an altered cyclooxygenase/thromboxane signaling may play a role in the EET-dependent augmentation of U46619-induced increases in RVSP.
Female-Specific Prevention of U46619-Induced Increases in MABP
Enhanced EET bioavailability as the consequence of sEH downregulation (F-WT) and deletion of sEH to promote systemic vasodilation and lower blood pressure was confirmed in Fig. 3. Both female WT and sEH-KO mice had lower blood pressure, as a function of desensitizing U46619-induced systemic vasoconstriction and potentiating EET-dependent systemic vasodilation, compared with their male controls. Blood pressure is indeed regulated through different mechanisms in physiological conditions. For instance, the arteriolar tone is controlled by interactions between vasoconstrictor (such as myogenic response) and dilator (such as shear stress-induced vasodilation) mechanisms to establish a stable resistance. It is plausible to speculate that the balance between these two mechanisms can be reset to a new level as a consequence of the increase in EET-induced vasodilation, leading to lower vascular resistance and blood pressure.
Perspective and Significance
The female-specific sensitization to pulmonary vasoconstriction as a result of compromising EET degradation was explored. EETs augmented U46619-induced increases in RVSP in both male and female mice, but to a greater extent in females. The potentiated increase in RVSP in female mice can be attributed, at least in part, to the female-specific downregulation of sEH in the pulmonary vasculature. To date, there is a lack of congruency regarding the role of estrogen in the predominance of IPAH in females, due in part to the presence of the “estrogen paradox” that exists between outcomes from experimental (animal) studies and those from clinical (human) studies (27). As indicated, findings of better prognoses in female pulmonary hypertensive animals, exacerbation of the diseases by ovariectomy, and general evidence for the vasoprotective properties of estrogens are documented from experimental studies (8, 45). In contrast, clinical and epidemiological studies provide a strong correlation between female/estrogen and increased risk/incidence of PAH (4, 29). In this regard, the etiological difference and environmental influence in the pathobiology of experimental PAH models compared with human IPAH may bear the responsibility for the discrepancy. In the present study, the female-specific downregulation of sEH, as well as the EET-dependent sensitization of the pulmonary vasopressor response to U46619, is observed physiologically, in in vivo conditions. As such, the detrimental action of EETs in the pulmonary circulation actually reflects a net activity of estrogen/EETs converging from pulmonary vasoconstriction, systemic vasodilation, and anti-inflammatory properties, etc. We believe that the knowledge obtained from our studies is of significance, because it can deepen our understanding of the mechanisms behind the higher susceptibility and incidence of PAH in women.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL070653, HL115124, and HL34300.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.K., J.Q., G.F., H.J., A.H., and D.S. performed experiments; S.K., J.Q., H.J., A.H., and D.S. analyzed data; S.K., J.Q., A.H., and D.S. interpreted results of experiments; S.K., J.Q., A.H., and D.S. prepared figures; S.K., A.H., and D.S. drafted manuscript; S.K., J.Q., G.F., H.J., M.L., M.S.W., A.H., and D.S. approved final version of manuscript; G.F., M.L., M.S.W., A.H., and D.S. edited and revised manuscript; A.H. and D.S. conception and design of research.
ACKNOWLEDGMENTS
We thank the New York Medical College/Westchester Medical Center Stem Cell Laboratory and its Core Facility for assistance with the echocardiography equipment.
REFERENCES
- 1.Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, Higenbottam T, Oakley C, Wouters E, Aubier M, Simonneau G, Begaud B. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med 335: 609–616, 1996. [DOI] [PubMed] [Google Scholar]
- 2.Alhawaj R, Patel D, Kelly MR, Sun D, Wolin MS. Heme biosynthesis modulation via δ-aminolevulinic acid administration attenuates chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L719–L728, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baber SR, Deng W, Rodriguez J, Master RG, Bivalacqua TJ, Hyman AL, Kadowitz PJ. Vasoactive prostanoids are generated from arachidonic acid by COX-1 and COX-2 in the mouse. Am J Physiol Heart Circ Physiol 289: H1476–H1487, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, Giles S, Feldkircher K, Miller DP, McGoon MD. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 137: 376–387, 2010. [DOI] [PubMed] [Google Scholar]
- 5.Buzzard CJ, Pfister SL, Campbell WB. Endothelium-dependent contractions in rabbit pulmonary artery are mediated by thromboxane A2. Circ Res 72: 1023–1034, 1993. [DOI] [PubMed] [Google Scholar]
- 6.Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflügers Arch 459: 881–895, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng Y, Theken KN, Lee CR. Cytochrome P450 epoxygenases, soluble epoxide hydrolase, and the regulation of cardiovascular inflammation. J Mol Cell Cardiol 48: 331–341, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Do.e Z, Fukumoto Y, Takaki A, Tawara S, Ohashi J, Nakano M, Tada T, Saji K, Sugimura K, Fujita H, Hoshikawa Y, Nawata J, Kondo T, Shimokawa H. Evidence for Rho-kinase activation in patients with pulmonary arterial hypertension. Circ J 73: 1731–1739, 2009. [DOI] [PubMed] [Google Scholar]
- 9.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med 351: 1655–1665, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Farhat MY, Ramwell PW. Estradiol potentiates the vasopressor response of the isolated perfused rat lung to the thromboxane mimic U-46619. J Pharmacol Exp Ther 261: 686–691, 1992. [PubMed] [Google Scholar]
- 11.Fradette C, du Souich P. Effect of hypoxia on cytochrome P450 activity and expression. Curr Drug Metab 5: 257–271, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Frost AE, Badesch DB, Barst RJ, Benza RL, Elliott CG, Farber HW, Krichman A, Liou TG, Raskob GE, Wason P, Feldkircher K, Turner M, McGoon MD. The changing picture of patients with pulmonary arterial hypertension in the United States: how REVEAL differs from historic and non-US contemporary registries. Chest 139: 128–137, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Frump AL, Goss KN, Vayl A, Albrecht M, Fisher A, Tursunova R, Fierst J, Whitson J, Cucci AR, Brown MB, Lahm T. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol 308: L873–L890, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, Abe K, Takeshita A, Shimokawa H. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 91: 391–392, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukumoto Y, Tawara S, Shimokawa H. Recent progress in the treatment of pulmonary arterial hypertension: expectation for rho-kinase inhibitors. Tohoku J Exp Med 211: 309–320, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Irey NS, Norris HJ. Intimal vascular lesions associated with female reproductive steroids. Arch Pathol 96: 227–234, 1973. [PubMed] [Google Scholar]
- 17.Ishikura K, Yamada N, Ito M, Ota S, Nakamura M, Isaka N, Nakano T. Beneficial acute effects of rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circ J 70: 174–178, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Jacobs W, van de Veerdonk MC, Trip P, de Man F, Heymans MW, Marcus JT, Kawut SM, Bogaard HJ, Boonstra A, Vonk NA. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 145: 1230–1236, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension 45: 759–765, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Kandhi S, Froogh G, Qin J, Luo M, Wolin MS, Huang A, Sun D. EETs elicit direct increases in pulmonary arterial pressure in mice. Am J Hypertens. 2015. Aug 24. pii: hpv148 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keseru B, Barbosa-Sicard E, Popp R, Fisslthaler B, Dietrich A, Gudermann T, Hammock BD, Falck JR, Weissmann N, Busse R, Fleming I. Epoxyeicosatrienoic acids and the soluble epoxide hydrolase are determinants of pulmonary artery pressure and the acute hypoxic pulmonary vasoconstrictor response. FASEB J 22: 4306–4315, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keseru B, Barbosa-Sicard E, Schermuly RT, Tanaka H, Hammock BD, Weissmann N, Fisslthaler B, Fleming I. Hypoxia-induced pulmonary hypertension: comparison of soluble epoxide hydrolase deletion vs. inhibition. Cardiovasc Res 85: 232–240, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleiger RE, Boxer M, Ingham RE, Harrison DC. Pulmonary hypertension in patients using oral contraceptives. A report of six cases. Chest 69: 143–147, 1976. [DOI] [PubMed] [Google Scholar]
- 24.Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Tan J, Meldrum DR. Selective estrogen receptor-α and estrogen receptor-β agonists rapidly decrease pulmonary artery vasoconstriction by a nitric oxide-dependent mechanism. Am J Physiol Regul Integr Comp Physiol 295: R1486–R1493, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Weil B, Meldrum DR. Exogenous estrogen rapidly attenuates pulmonary artery vasoreactivity and acute hypoxic pulmonary vasoconstriction. Shock 30: 660–667, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Lahm T, Crisostomo PR, Markel TA, Wang M, Weil BR, Novotny NM, Meldrum DR. The effects of estrogen on pulmonary artery vasoreactivity and hypoxic pulmonary vasoconstriction: potential new clinical implications for an old hormone. Crit Care Med 36: 2174–2183, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Lahm T, Tuder RM, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307: L7–L26, 2014. [DOI] [PubMed] [Google Scholar]
- 28.Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res 115: 115–130, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, Howard LS, Pepke-Zaba J, Sheares KK, Corris PA, Fisher AJ, Lordan JL, Gaine S, Coghlan JG, Wort SJ, Gatzoulis MA, Peacock AJ. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 186: 790–796, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Miller VM. In pursuit of scientific excellence: sex matters. Am J Physiol Heart Circ Physiol 302: H1771–H1772, 2012. [DOI] [PubMed] [Google Scholar]
- 31.Miller VM. Why are sex and gender important to basic physiology and translational and individualized medicine? Am J Physiol Heart Circ Physiol 306: H781–H788, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 54: S20–S31, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923–929, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Pfister SL. Role of lipoxygenase metabolites of arachidonic acid in enhanced pulmonary artery contractions of female rabbits. Hypertension 57: 825–832, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfister SL, Campbell WB. Role of endothelium-derived metabolites of arachidonic acid in enhanced pulmonary artery contractions in female rabbits. Hypertension 27: 43–48, 1996. [DOI] [PubMed] [Google Scholar]
- 36.Qin J, Sun D, Jiang H, Kandhi S, Froogh G, Hwang SH, Hammock BD, Wolin MS, Thompson CI, Hintze TH, Huang A. Inhibition of soluble epoxide hydrolase increases coronary perfusion in mice. Physiol Rep 3: e12427, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Revermann M, Barbosa-Sicard E, Dony E, Schermuly RT, Morisseau C, Geisslinger G, Fleming I, Hammock BD, Brandes RP. Inhibition of the soluble epoxide hydrolase attenuates monocrotaline-induced pulmonary hypertension in rats. J Hypertens 27: 322–331, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scotland RS, Madhani M, Chauhan S, Moncada S, Andresen J, Nilsson H, Hobbs AJ, Ahluwalia A. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation 111: 796–803, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Stenmark KR, McMurtry IF. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension: a time for reappraisal? Circ Res 97: 95–98, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Sun D, Cuevas AJ, Gotlinger K, Hwang SH, Hammock BD, Schwartzman ML, Huang A. Soluble epoxide hydrolase-dependent regulation of myogenic response and blood pressure. Am J Physiol Heart Circ Physiol 306: H1146–H1153, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun D, Jiang H, Wu H, Yang Y, Kaley G, Huang A. A novel vascular EET synthase: role of CYP2C7. Am J Physiol Regul Integr Comp Physiol 301: R1723–R1730, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun D, Yang YM, Jiang H, Wu H, Ojaimi C, Kaley G, Huang A. Roles of CYP2C29 and RXRγ in vascular EET synthesis of female mice. Am J Physiol Regul Integr Comp Physiol 298: R862–R869, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sweeney L, Voelkel NF. Estrogen exposure, obesity and thyroid disease in women with severe pulmonary hypertension. Eur J Med Res 14: 433–442, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thibault HB, Kurtz B, Raher MJ, Shaik RS, Waxman A, Derumeaux G, Halpern EF, Bloch KD, Scherrer-Crosbie M. Noninvasive assessment of murine pulmonary arterial pressure: validation and application to models of pulmonary hypertension. Circ Cardiovasc Imaging 3: 157–163, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Umar S, Rabinovitch M, Eghbali M. Estrogen paradox in pulmonary hypertension: current controversies and future perspectives. Am J Respir Crit Care Med 186: 125–131, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeldin DC, Wei S, Falck JR, Hammock BD, Snapper JR, Capdevila JH. Metabolism of epoxyeicosatrienoic acids by cytosolic epoxide hydrolase: substrate structural determinants of asymmetric catalysis. Arch Biochem Biophys 316: 443–451, 1995. [DOI] [PubMed] [Google Scholar]