

Abstract
Transendothelial hyperpermeability caused by numerous agonists is dependent on heat shock protein 90 (Hsp90) and leads to endothelial barrier dysfunction (EBD). Inhibition of Hsp90 protects and restores transendothelial permeability. Hyperacetylation of Hsp90, as by inhibitors of histone deacetylase (HDAC), suppresses its chaperone function and mimics the effects of Hsp90 inhibitors. In this study we assessed the role of HDAC in mediating lipopolysaccharide (LPS)-induced transendothelial hyperpermeability and acute lung injury (ALI). We demonstrate that HDAC inhibition protects against LPS-mediated EBD. Inhibition of multiple HDAC by the general inhibitors panobinostat or trichostatin provided protection against LPS-induced transendothelial hyperpermeability, acetylated and suppressed Hsp90 chaperone function, and attenuated RhoA activity and signaling crucial to endothelial barrier function. Treatment with the HDAC3-selective inhibitor RGFP-966 or the HDAC6-selective inhibitor tubastatin A provided partial protection against LPS-mediated transendothelial hyperpermeability. Similarly, knock down of HDAC3 and HDAC6 by specific small-interfering RNAs provided significant protection against LPS-induced EBD. Furthermore, combined pharmacological inhibition of both HDAC3 and -6 attenuated the inflammation, capillary permeability, and structural abnormalities associated with LPS-induced ALI in mice. Together these data indicate that HDAC mediate increased transendothelial hyperpermeability caused by LPS and that inhibition of HDAC protects against LPS-mediated EBD and ALI by suppressing Hsp90-dependent RhoA activity and signaling.
Keywords: histone deacetylases, endothelium, barrier function, r RhoA, myosin light chain, lipopolysaccharide, inflammation, acute lung injury
lipopolysaccharide (LPS), a major component of the bacterial wall cell membrane, is released in the bloodstream during infection after bacteria are lysed by white blood cells (8, 44). LPS binds to the Toll-like receptor 4 receptor complex on endothelial cells and induces multiple signaling pathways that lead to the production and release of proinflammatory cytokines and endothelial barrier dysfunction (EBD) (19, 45, 54). Inflammation and EBD are the primary causes of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) (3, 18, 44, 46).
The ATP-dependent molecular chaperone heat shock protein 90 (Hsp90) plays a central role in LPS-mediated EBD (4, 16). LPS potentiates Hsp90 chaperone function by inducing pp60Src-dependent Y-300 phosphorylation of Hsp90 (9). Inhibitors of Hsp90 block LPS-induced Y-300 phosphorylation in cultured human lung microvascular endothelial cells (HLMVEC) (9) and prolong survival, attenuate inflammation, and reduce lung injury in a murine model of LPS-induced ALI/ARDS (15). Long-term treatment (>8 h) with small molecule inhibitors of Hsp90 causes destabilization and ubiquitin-mediated degradation of numerous client proteins, such as protein kinase B (Akt) and pp60Src (10, 21, 24, 55). However, even short-term treatment (<4 h) with Hsp90 inhibitors attenuates LPS-induced RhoA activation and signaling and protects against LPS-mediated EBD in HLMVEC (31).
Histone deacetylases (HDAC) remove acetyl groups from the ε-amino group of lysine residues in numerous histone and nonhistone proteins such as Hsp90 (11, 61). In humans, there are 18 characterized HDAC distributed into four classes. HDAC3, a class I HDAC, is primarily a nuclear protein that shuttles to the cytoplasm when phosphorylated (53, 61), localizes to the plasma membrane, and is a substrate of Src (39). Conversely, HDAC6 is primarily a cytoplasmic protein and a unique member of class II HDAC (12). HDAC6 acetylation results in nuclear translocation and regulation of gene expression (38). HDAC inhibitors exhibit anti-inflammatory properties by suppressing proinflammatory cytokine production (6, 22, 30, 59) and reducing disease severity in animal models of inflammatory and autoimmune disease (6, 43, 48). In endothelial cells, HDAC inhibitors suppress thrombin-induced EBD (49). Importantly, inhibition of HDAC induces Hsp90 acetylation at lysine-294 and suppresses its chaperone function, thereby mimicking the effect of Hsp90 inhibitor (50). Both HDAC3 and -6 interact with and deacetylate Hsp90, and inhibition of HDAC3 and -6 hyperacetylates Hsp90 (29, 35, 50), suggesting a possible role of these two HDAC in EBD.
In this study, we demonstrate that inhibition of HDAC protects against LPS-mediated EBD. Particularly, we show that selective pharmacological or genetic inhibition of HDAC3 and/or HDAC6 provides partial protection against LPS-mediated endothelial hyperpermeability. Finally, we demonstrate that HDAC acts upstream of Hsp90 and that inhibition of HDAC acetylates Hsp90, suppressing its chaperone function. Together, our data suggest a central role for basal and induced HDAC activity in LPS-mediated endothelial hyperpermeability and EBD.
MATERIALS AND METHODS
Antibodies and reagents.
Antibodies against acetyl-lysine, phospho-Akt, Akt, myosin light chain (MLC), and di-phospho-MLC were purchased from Cell Signaling. Anti-Hsp90 antibodies were purchased from BD Biosciences (610418) and Enzo Life Sciences (ADI-SPA-840). Anti-β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO), as were antibodies against S424-HDAC3, HDAC3, and HDAC6. PY99 antibody was purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). The HDAC inhibitors panobinostat (Pan) and trichostatin (TSA) and the Hsp90 inhibitor AUY-922 (S1069) were obtained from Selleck Chemicals. Selective HDAC3 inhibitor RGFP-966 and selective HDAC6 inhibitor tubastatin were purchased from Selleck Chemicals. HDAC3 (sc-35538), HDAC6 (sc-35544), and control small-interfering RNA (siRNA, sc-37007) were purchased from Santa Cruz Biotechnology. Oligofectamine was obtained from Fisher Scientific (Pittsburg, PA).
Cell culture.
HLMVEC were isolated and cultured in-house as described previously (14).
HDAC activity assay.
HDAC activity was measured using the fluor-de-lys HDAC activity assay kit in accordance with the manufacturer's instructions (Enzo Life Sciences). After treatment, HLMVEC were lysed, and cell lysates were used to measure HDAC activity. Results were normalized to protein levels measured using the bicinchonic acid assay reagent (Bio-Rad).
Transendothelial permeability.
Transendothelial resistance (TER) measurements were performed as described previously (16). Briefly, ∼60,000 cells were seeded on each well of a 8W10E+ ECIS array. After 24 h, the media were changed, and treatments began at 48 h when the resistance was stable between 900 and 1,200 Ω at a frequency of 4,000 Hz, and the capacitance was between 22 and 29 nF. Each experiment was performed at least in triplicate and repeated at least three times (n = 3). Resistance was measured using the ECIS model Zθ and normalized to each well's value at time = 0 h.
RhoA activity assay.
RhoA activity was determined using a Rho G-LISA assay kit in accordance with the manufacturer's instructions (Cytoskeleton) using HLMVEC cell lysates. Results were normalized to protein levels, measured by the Precision Red protein assay reagent.
Cell fractionation.
Cytoplasmic and nuclear extract were prepared using the nuclear kit from Active Motif.
Western blotting and immunoprecipitation.
Western blot analyses and immunoprecipitation experiments were performed as described previously (9, 31). Densitometry was performed using Image Studio version 3.1 from Licor and plotted as fold change from vehicle.
Animal experiments.
Mice (C57/6; Harlan) were injected intraperitoneally with 10 mg/kg each of RGFP-966 and tubastatin. After 24 h, the animals were anesthetized (ketamine/xylaxine), and 1.5 mg/kg LPS were instilled through the trachea. Later (24 h), the animals were similarly anesthetized, and bronchoalveolar lavage (BAL) was performed with 1 ml saline. BAL fluid (BALF) was collected and used for measurements of cellularity and protein concentration.
siRNA transfection.
siRNAs against human HDAC3 and HDAC6 were used to knock down the expression of the respective proteins in HLMVEC. siRNA, which does not lead to the degradation of any known cellular mRNA, was used as control. siRNAs were diluted in Opti-MEM I Reduced Serum Medium, and oligofectamine was diluted in an equal volume of Opti-MEM I and incubated for 30 min at room temperature. The oligomer-Lipofectamine complexes were added to cells, which were cultured in media free of antibiotics. The medium was changed 8 h after transfection. Cells were incubated at 37°C in an atmosphere of 5% CO2 and 95% air for 48 h after transfection and then assayed by Western blotting or used in ECIS experiments.
Statistical analyses.
Data are presented as mean values ± SE. Comparisons among groups were performed using either paired t-test, one-way or two-way ANOVA with Bonferroni posttest, as appropriate. Differences were considered significant at P < 0.05, and n represents the number of experimental repeats.
RESULTS
HLMVEC grown to confluence were exposed to either PBS or LPS [0.2, 1, or 5 endotoxin units (EU)/ml] for 2 h. After cell lysis, HDAC activity was measured as described in materials and methods. Compared with PBS-treated cells, LPS-exposed cells exhibited a modest but consistent and significant increase in HDAC activity in whole cell lysates (111 ± 3, 122 ± 3, and 117 ± 2%, respectively, for 0.2, 1, and 5 EU/ml LPS), indicating that LPS signaling activates cellular HDAC function. The LPS-induced HDAC activity increased as early as 1 h (122 ± 2%) and remained elevated at 2 h (130 ± 7%). Because HDAC are found in both the cytoplasm and the nucleus, we isolated the cytoplasmic and the nuclear extract and measured their HDAC activity. LPS induced HDAC activity in the cytoplasm (100 ± 4, 112 ± 1, and 115 ± 1%) and nucleus (100 ± 1, 118 ± 1, and 115 ± 1%) for 0.2, 1, and 5 h, respectively.
HDAC inhibition protects against the LPS-mediated decrease in TER.
Pan-HDAC inhibitors target multiple class I and class II HDAC and have a broad effect on HDAC activity and function. Therefore, we hypothesized that treatment with pan-HDAC inhibitors would attenuate LPS-mediated endothelial hyperpermeability. ECIS arrays were used to measure TER across the endothelial cell monolayer. HLMVEC were grown on gold electrode arrays, and TER values were monitored continuously until a constant value was attained, suggesting a confluent monolayer. Cells were then exposed to vehicle or the HDAC inhibitor Pan (1 μM) or TSA (2 μM) for 2 h followed by PBS or LPS (1 EU/ml). These concentrations are higher than those frequently used in tumor cells because it is well known that nontumor normal cells are less responsive to HDAC inhibitors (56). LPS decreased TER values, suggesting an increase in monolayer permeability. Pretreatment with either Pan (Fig. 1A) or TSA (Fig. 1B) prevented the LPS-mediated decrease in HLMVEC monolayer TER, suggesting that HDAC activity and function is necessary for LPS-induced endothelial hyperpermeability.
Fig. 1.
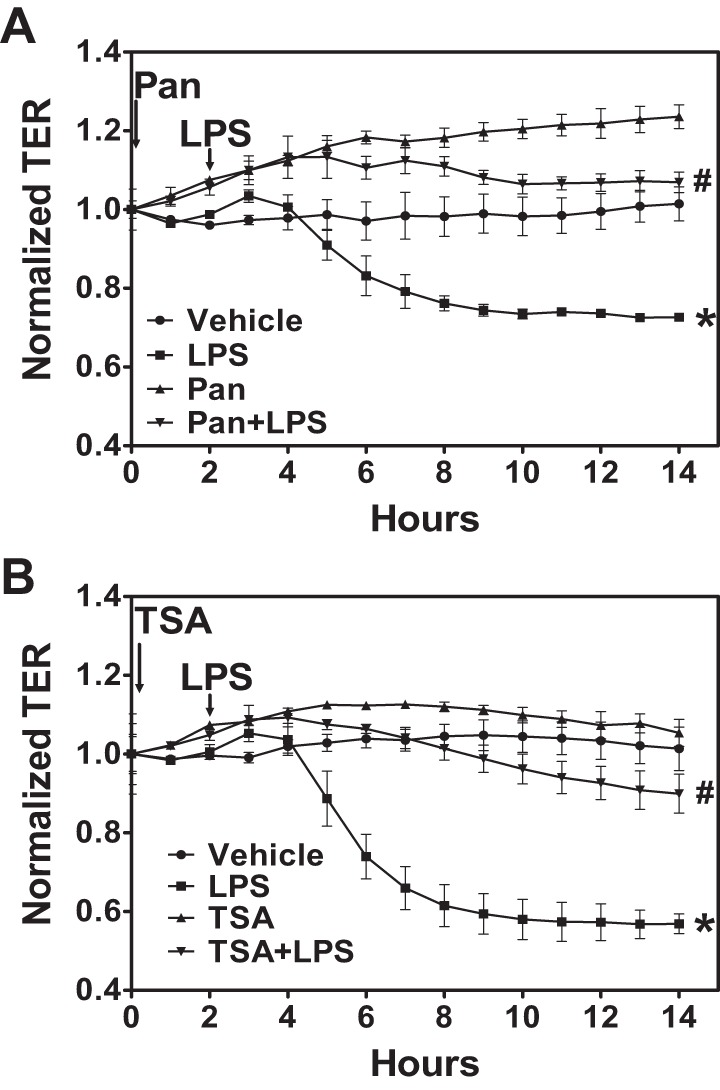
Histone deacetylase (HDAC) inhibition protects against the lipopolysaccharide (LPS)-mediated decrease in transendothelial resistance (TER). Human lung microvascular endothelial cells (HLMVEC) were grown to confluence on ECIS arrays (8W10E+). Once a constant resistance was attained, the cells were treated with either vehicle (0.1% DMSO) or 1 μM panobinostat (Pan; A) or 2 μM trichostatin (TSA; B) for 2 h. LPS [1 endotoxin unit (EU)/ml] was then added as indicated, and TER values were measured using ECIS Zθ. Normalized resistance was calculated and plotted as a function of time. P < 0.01 from vehicle (*) and LPS (#); n ≥ 4 experiments.
HDAC inhibition attenuates LPS-induced Hsp90 activation and chaperone function.
Hsp90 plays a central role in LPS-mediated EBD. Therefore, we hypothesized that the HDAC effects on EBD may involve Hsp90. Confluent HLMVEC were pretreated with vehicle or the specific Hsp90 inhibitor AUY-922 followed by exposure to LPS (1 EU/ml). At the end of treatment, cells were lysed, and total cellular HDAC activity was measured, as described in materials and methods. LPS alone increased HDAC activity. AUY-922 did not affect either the basal or the LPS-induced HDAC activity (Fig. 2A), suggesting that Hsp90 functions downstream of HDAC. To prove this, we then examined the effects of HDAC inhibitors on Hsp90 activation and function.
Fig. 2.

HDAC inhibition attenuates LPS-induced heat shock protein 90 (Hsp90) chaperone function. A: confluent monolayers were treated with vehicle or 2 μM AUY-922 (Hsp90 inhibitor) for 4 h followed by exposure to LPS (1 EU/ml) for 2 h. Cells were then lysed, and HDAC activity was measured using the Flour-de-lys HDAC activity assay. *P < 0.05 from vehicle or AUY. B: confluent monolayers were treated with vehicle, LPS (1 EU/ml), 1 μM TSA for 2 h followed by exposure to LPS for 2 h, or 1 μM Pan for 2 h followed by exposure to LPS for 2 h. Cells were lysed, and Hsp90 was immunoprecipitated and immunoblotted for phoshotyrosine and Hsp90. P < 0.05 from vehicle (*) and LPS (#); n = 3. C: HLMVEC were treated with vehicle (VEH) or 1 μM Pan; cells were then lysed, and Hsp90 was immunoprecipitated and immunoblotted for acetyl-lysine and Hsp90. *P < 0.05; n = 3. D: HLMVEC were treated with vehicle, LPS (1 EU/ml), 1 μM panobinostat, or 1 μM panobinostat for 2 h followed by exposure to LPS (1 EU/ml) for 2 h. Cells were then lysed and immunoblotted for phosho-protein kinase B (Akt, S473) and Akt. Densitometric analysis was carried out, and the ratio of pAkt to Akt was plotted. *P < 0.01 vs. vehicle.
LPS induces Hsp90-Y-300 phosphorylation via pp60Src to activate Hsp90 and potentiate its chaperone function. If HDACs act upstream of Hsp90 then inhibition of HDAC function would affect LPS-induced Hsp90-Y-300 phosphorylation. To test this, we pretreated HLMVEC with Pan or TSA followed by exposure to LPS. Both Pan and TSA completely blocked LPS-induced Y-300 phosphorylation of Hsp90 (Fig. 2B).
Acetylation of Hsp90 suppresses its chaperone function. Therefore, we determined whether HDAC inhibition acetylates Hsp90 in HLMVEC. Treatment with Pan for 2 h increased Hsp90 acetylation (Fig. 2C). Furthermore, Pan completely abolished the LPS-induced and Hsp90-dependent phosphorylation of Akt, an Hsp90 client protein (Fig. 2D). Taken together, the data in Fig. 2 suggest that HDAC inhibition prevents Hsp90 activation and suppresses its chaperone function.
HDAC inhibition suppresses LPS-mediated RhoA activity and signaling.
The RhoA-Rho kinase (ROCK) pathway is an important mediator of the cytoskeletal changes associated with HLMVEC exposure to LPS (13, 25). ROCK inhibitors block LPS-induced MLC phosphorylation and protect against LPS-induced EBD (23, 37). Recently, we reported that Hsp90 inhibition attenuates LPS-induced RhoA activation and MLC phosphorylation in HLMVEC (31). Therefore, we hypothesized that HDAC inhibition would block the LPS-triggered and Hsp90-dependent RhoA-mediated signaling. LPS increased RhoA activity, and Pan pretreatment for 2 h attenuated this LPS-induced RhoA activity (Fig. 3A). Furthermore, LPS induced diphosphorylation of MLC2 in HLMVEC, and this induction was completely blocked by either Pan or TSA (Fig. 3, B and C). These data suggest that HDAC inhibition blocks RhoA activity and signaling, most likely by attenuating Hsp90 chaperone function.
Fig. 3.

HDAC inhibition suppresses LPS-induced RhoA activity and signaling. A: confluent HLMVEC were treated with vehicle, LPS (1 EU/ml), 1 μM Pan, or Pan for 2 h followed by exposure to LPS for 2 h (Pan + LPS). Cells were then lysed, and RhoA activity was measured using the G-LISA RhoA activation assay. P < 0.05 from vehicle (*) and LPS (#); n = 5. B: HLMVEC were grown in 6-well plates. Confluent monolayers were treated with vehicle, LPS (1 EU/ml), 1 μM panobinostat, or panobinostat for 2 h followed by exposure to LPS for 2 h. Cells were then lysed and immunoblotted with antibodies against pp-myosn light chain (MLC)2, MLC2, and β-actin. *P < 0.01 vs. vehicle. *P < 0.05 from LPS. C: cells were treated as in B except instead of Pan 1 μM TSA was added. P < 0.05 from vehicle (*) and LPS (#); n = 5.
HDAC3 is involved in LPS-induced EBD.
HDAC3 and HDAC6 have been reported to interact with Hsp90. Therefore, we hypothesized that they may be selectively involved in LPS-induced EBD. Confluent HLMVEC were exposed to vehicle or LPS (1 EU/ml) for 2 h and lysed, HDAC3 was immunoprecipitated, and HDAC activity was measured as described in materials and methods. LPS significantly increased HDAC3 activity above basal levels observed in vehicle-treated cells (Fig. 4A). Immunoprecipitated HDAC3 exhibited an LPS-induced increase in S424 phosphorylation (Fig. 4B), which is consistent with studies showing increased activity of S424-phosphorylated HDAC3 (62).
Fig. 4.

HDAC3 inhibition protects from LPS-induced endothelial barrier dysfunction (EBD). A: HLMVEC were grown in 100-mm dishes. When confluent, the cells were treated with either PBS or LPS (1 EU/ml) for 2 h. Cells were then lysed, HDAC3 was immunoprecipitated, and HDAC activity was measured using the Flour-de-lys HDAC activity assay. *P < 0.05; n = 4. B: HLMVEC were treated as above. Cells were lysed, and HDAC3 was immunoprecipitated and then immunoblotted for phospho (p)-serine-424 and HDAC3. *P < 0.05; n = 3. C and D: HLMVEC were grown in 100-mm dishes. When confluent, the cells were treated with the indicated concentrations of the HDAC3-selective inhibitor RGFP-966 for 2 h, and total HDAC (C) and HDAC3 (D) activity was measured, as described previously. *P < 0.05 from vehicle; n = 4. E: HLMVEC were grown to confluence on ECIS arrays (8W10E+). Once a constant resistance was attained, the cells were treated with either vehicle (0.1% DMSO) or 5 μM RGFP-966 (RGF) for 2 h. LPS (1 EU/ml) was then added as indicated, and TER values were measured using an ECIS Zθ. *P < 0.05 from vehicle. #P < 0.01 from LPS.
We then determined whether inhibition of HDAC3 blocks the LPS-mediated decrease in TER. To that end, we employed the HDAC3-selective inhibitor RGFP-966. Pretreatment with RGFP-966, at a concentration similar to those reported in the literature without apparent toxic effects (57), moderately but significantly reduced total HDAC activity (Fig. 4C) in a concentration-dependent manner but nearly abolished HDAC3 activity, also in a concentration-dependent manner (Fig. 4D). Importantly, pretreatment with RGFP-966 significantly attenuated the LPS-induced decrease in TER (Fig. 4E). These data suggest that HDAC3 plays a significant role in LPS-mediated EBD.
HDAC6 inhibition attenuates the LPS-mediated EBD.
HDAC6 interacts with and regulates Hsp90 chaperone function (7, 50). Inhibition of HDAC6 blocks endothelial hyperpermeability induced by thrombin (49). Therefore, we hypothesized that LPS modulates HDAC6 activity and that inhibition of HDAC6 may prevent the LPS-induced endothelial hyperpermeability. Confluent HLMVEC were exposed to vehicle or LPS (1 EU/ml) for 1 or 2 h and lysed, HDAC6 was immunoprecipitated, and HDAC activity was measured as described in materials and methods. LPS did not affect basal HDAC6 activity (Fig. 6A). Similarly, LPS did not affect the expression of either HDAC3 or HDAC6 proteins (data not shown). We then determined whether HDAC6 is involved in the LPS-mediated decrease in TER. Pretreatment with the HDAC6-selective inhibitor tubastatin A for 2 h and at a concentration similar to those reported in the literature without apparent toxic effects (17) moderately but significantly reduced total HDAC activity (Fig. 5B) in a concentration-dependent manner but nearly abolished HDAC6 activity, also in a concentration-dependent manner (Fig. 5C). Furthermore, pretreatment with tubastatin A blocked the LPS-induced decrease in TER (Fig. 5D). These data suggest that, even though HDAC6 activity is not affected by LPS, HDAC6 plays an important part in LPS-mediated EBD.
Fig. 6.
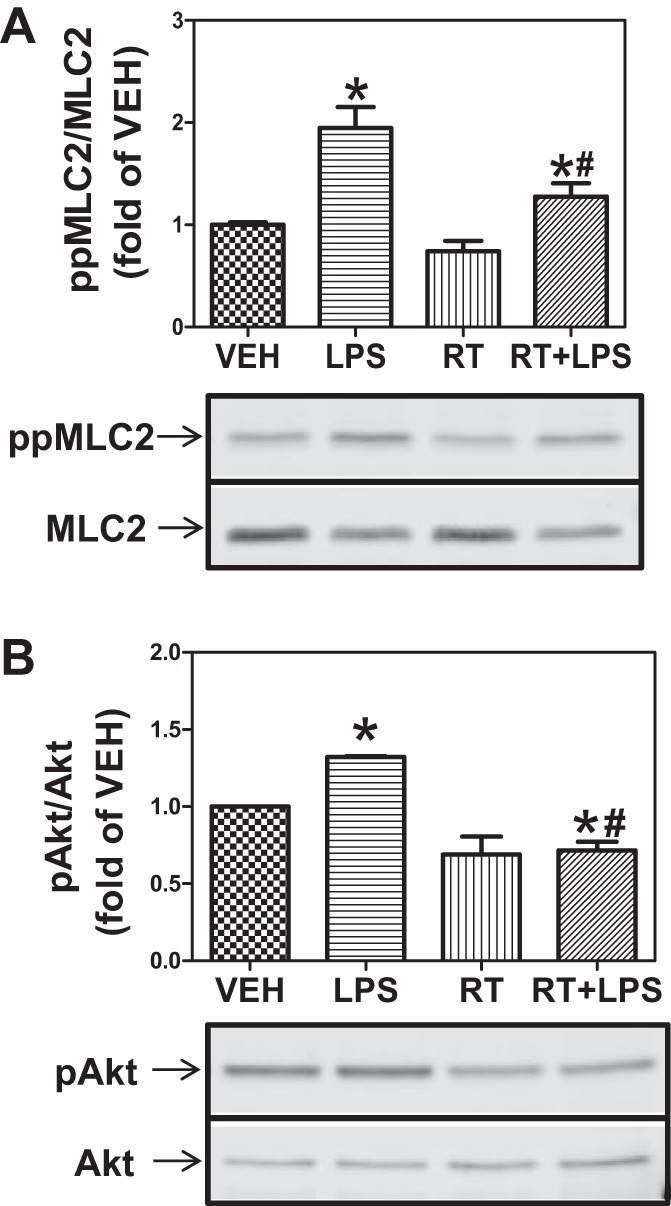
Combined inhibition of HDAC3 and -6 prevents MLC phosphorylation and abrogates Hsp90 chaperone function. A: HLMVEC were grown in 6-well plates. Confluent monolayers were treated with vehicle, LPS, 5 μM RGFP-966 + 5 μM tubastatin (RT), or RT for 2 h followed by exposure to LPS (1 EU/ml) for 2 h. Cells were then lysed and immunoblotted with antibodies against ppMLC2 and MLC2. P < 0.05 from vehicle (*) and LPS (#); n = 4. B: cells were treated as in A and immunoblotted with antibodies against pAkt and Akt. P < 0.05 from vehicle (*) and LPS (#); n = 4.
Fig. 5.

HDAC6 inhibition protects against LPS-induced EBD. A: HLMVEC were grown in 100-mm dishes. When confluent, the cells were treated with either PBS or LPS (1 EU/ml) for 2 h. Cells were then lysed, HDAC6 was immunoprecipitated, and HDAC activity was measured using the Flour-de-lys HDAC activity assay. B and C: HLMVEC were grown in 100-mm dishes. When confluent, the cells were treated with the indicated concentrations of the HDAC6-selective inhibitor tubastatin for 2 h, and total HDAC (B) and HDAC6 (C) activity were measured, as described previously. *P < 0.05 from vehicle; n = 4. D: HLMVEC were grown to confluence on ECIS arrays (8W10E+). Once a constant resistance was attained, the cells were treated with either vehicle (0.1% DMSO) or 5 μM tubastatin (Tub) for 2 h. LPS (1 EU/ml) was then added as indicated, and TER values were measured using ECIS Zθ. *P < 0.01 from vehicle. #P < 0.05 from LPS.
Combined inhibition of HDAC3 and -6 attenuates LPS-mediated EBD.
As shown in Figs. 4 and 5, selective inhibition of either HDAC3 or HDAC6 provides partial but significant protection against LPS-mediated EBD. Therefore, we investigated the effect of combined inhibition of HDAC3 and -6. HLMVEC were exposed to 5 μM each of RGFP-966 and tubastatin for 2 h, followed by LPS (1 EU/ml). This treatment essentially abolished the LPS-induced MLC2 phosphorylation (Fig. 6A) and Hsp90 chaperone function, as reflected in Akt phosphorylation (Fig. 6B). To further assess the combined role of HDAC3 and -6 in LPS-mediated EBD, HLMVEC were transfected with siRNAs against HDAC3 and -6. The siRNA produced >80% inhibition of HDAC3 and HDAC6 expression (Fig. 7, A and B). Subsequently, the two siRNAs were added to HLMVEC freshly plated on ECIS arrays. TER was monitored starting 24 h later, and, for comparison purposes, data are presented normalized to the 24-h values. Over the next 20 h HLMVEC lacking most of HDAC3 and -6 exhibited faster proliferation (Fig. 7C). At 48 h postplating, LPS (1 EU/ml) was added to the arrays, and, for comparison purposes, TER data were again normalized to the value just before LPS addition. Cells lacking HDAC3 and -6 were significantly more resistant to LPS than cells transfected with scrambled siRNA (Fig. 7D).
Fig. 7.

Small-interfering RNA (siRNA)-induced downregulation of both HDAC3 and HDAC6 protects against LPS-induced EBD. A and B: HLMVEC were transfected with either scrambled siRNA or siRNAs against both HDAC3 and -6. Later (48 h) transfected cells were lysed and immunoblotted with antibodies against HDAC3 (A) and HDAC6 (B). *P < 0.05 and ***P < 0.001; n = 4. C: HLMVEC were transfected with either scrambled siRNA or siRNAs against both HDAC3 and -6. Later (24 h) transfected cells were added to ECIS arrays (8W10E+) and allowed to grow to confluence. *P < 0.01 from scrambled siRNA; n = 4. D: the same cells and arrays as in C. Following 24 h exposure to siRNA, TER was normalized and is presented as time = 0. The cells were then treated with either vehicle (PBS) or LPS (1 EU/ml) as indicated, and TER values were monitored. P < 0.05 from vehicle (*) and LPS (#); n = 4.
Combined inhibition of HDAC3 and -6 attenuates LPS-mediated ALI.
To investigate whether the EBD-protective effects of HDAC3 and -6 inhibitors conferred similar protection against LPS-induced ALI, mice were injected intraperitoneally with 10 mg/kg each of RGFP-966 and tubastatin. After 24 h, 1.5 mg/kg LPS were given intratracheally to mice under anesthesia, and BALF was collected 24 h post-LPS instillation. Cellular infiltration into alveoli was quantified by the counting number of cells in BALF. LPS induced a profound and significant increase in leukocyte infiltration compared with vehicle treatment; however, BALF from mice pretreated with RGFP-966 and tubastatin exhibited significantly less cellular infiltration (Fig. 8A). Similarly, a partial but significant attenuation of BALF protein concentration was observed in mice pretreated with RGFP-966 and tubastatin compared with mice treated with LPS (Fig. 8B). Histological examination of lung tissue stained with hematoxylin and eosin revealed significant protection in lungs of mice treated with RGFP-966 and tubastatin from the LPS-induced structural changes, reflected in septal thickness and cellular infiltration (Fig. 8C), whereas MPO staining demonstrated the granulocytic nature of the invading cells (Fig. 8D). When quantified with respect to lung injury index (42), the combination of HDAC3 and -6 inhibitor treatment effectively protected against LPS-induced lung pathology (Fig. 8E). Together, data in Fig. 8 indicate that combined inhibition of HDAC3 and -6 protects against LPS-mediated ALI.
Fig. 8.

Combined pharmacological inhibition of HDAC3 and -6 protects against LPS-mediated acute lung injury (ALI). Mice were injected ip with 10 mg/kg each of RGFP-966 and tubastatin. After 24 h, 1.5 mg/kg LPS were given it, and bronchoalveolar lavage fluid (BALF) was collected 24 h later. A: cellular infiltration into the alveolar spaces was quantified by counting the no. of cells in BALF. P < 0.05 from vehicle (*) and LPS (#); n = 7. B: capillary permeability was estimated by measuring protein concentration in BALF using the bicinchonic acid method. P < 0.05 from vehicle (*), LPS (#), and RT ($); n = 7. Histological evaluation of lung samples from treated mice was also performed. C and D: sections were stained with hematoxylin and eosin (C) or immunocytostained against the granulocyte marker myeloperoxidase (MPO, D). Arrows point to thickened septa and cellular infiltrates. Representative samples of 3 specimens/group. E: estimation of lung injury index as described previously (42). P < 0.05 from vehicle (*) and LPS (#); n = 4.
DISCUSSION
Lysine acetylation is fine-tuned by the opposing actions of two families of enzymes, histone acetyltransferases (HAT) and HDAC. The HAT adds an acetyl group, whereas HDAC removes acetyl groups from the ε-amino group of lysine residues (11, 61). In humans, there are 18 characterized HDAC, which are distributed in four classes based on function and DNA sequence similarity (20). Despite their name, HDAC target not only histone but also many non-histone proteins both in the nucleus and the cytoplasm and regulate a variety of functions, including transcription, cytoskeletal polymerization, and signaling pathways (28, 30, 51, 61).
Inhibition of HDAC activity exhibits immunosuppressive and anti-inflammatory properties by reducing cytokine production in both humans and mice (6, 22, 30, 59). HDAC inhibition also reduces disease severity in several animal models of inflammatory and autoimmune diseases (6, 43, 48). In endothelial cells, HDAC inhibitors have been shown to suppress thrombin-induced EBD (49). We now show that pharmacological and genetic inhibition of HDAC protects HLMVEC from LPS-mediated EBD.
We have demonstrated that LPS activates Hsp90 chaperone function by inducing Y-300 phosphorylation and that it promotes EBD in cultured HLMVEC (9, 26, 36). On the other hand, inhibition of Hsp90 prevents Y-300 phosphorylation, attenuates chaperone function, and protects against LPS-induced EBD in cultured HLMVEC. Furthermore, Hsp90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in a murine model of LPS-induced ALI (15). Hsp90 chaperone function is also regulated by reversible acetylation at residue K294. Acetylation suppresses whereas deacetylation promotes Hsp90 chaperone function (50). HDAC inhibitors abrogate deacetylation of Hsp90, resulting in the accumulation of acetylated Hsp90. Because we have previously shown that Hsp90 promotes EBD and ALI/ARDS (4, 16, 40), in this project we determined the role of HDAC in LPS-mediated EBD in cultured HLMVEC. We hypothesized and demonstrated that LPS-induced HDAC activity functions upstream of Hsp90, and inhibition of HDAC would suppress Hsp90 chaperone function. We also found that inhibition of HDAC activity blocked LPS-induced Y-300 phosphorylation of Hsp90, suggesting that HDAC inhibition also attenuates LPS-mediated and Src-dependent phosphorylation and activation of Hsp90 (9). Thus we propose that prevention of EBD by HDAC inhibitors is achieved by disrupting Hsp90 chaperone function. This is supported by directly demonstrating that HDAC inhibition prevents LPS-induced Akt phosphorylation (Fig. 2D). Akt is a Hsp90 client protein, and its activation (phosphorylation) by LPS (58) or other agonists is Hsp90 dependent.
The Rho-ROCK pathway modulates cell-cell adhesion and vascular permeability by regulating myosin-mediated contractile forces (13, 25). LPS-induced RhoA activity requires activation of Src, an Hsp90 client protein. Induction of RhoA activity phosphorylates ROCK, which in turn phosphorylates MLC, leading to an increase in myosin-mediated contractile forces and EBD (1, 25). In HLMVEC, two ROCK inhibitors, Y-27632 and GSK-429286, block MLC phosphorylation and provide significant protection against the LPS-mediated decrease in TER (31). Also, inhibition of Src or Hsp90 completely abolished the LPS-induced RhoA activity and MLC phosphorylation in HLMVEC (31). Because HDAC inhibition suppresses Hsp90 function, we hypothesized and demonstrated that treatment with HDAC inhibitor should also attenuate RhoA activity and MLC phosphorylation. We also demonstrated that, among HDACs, HDAC3 and HDAC6 are major contributors to the LPS-induced EBD and ALI.
HDAC3, a class I HDAC, is primarily a nuclear protein that regulates transcription (60). Phosphorylation at serine-424 increases HDAC3 activity and translocates HDAC3 to the cytoplasm where it interacts with and deacetylates Hsp90 and promotes its chaperone function (62). Cytoplasmic HDAC3 is a substrate of, and regulated by, Src and has been shown to localize at the plasma membrane (39). Consistent with these observations, our data demonstrate that LPS selectively induces HDAC3 activity by inducing serine-424 phosphorylation and that selective inhibition of HDAC3 protects against the LPS-induced decrease in TER, indicating that HDAC3 activity plays a crucial role in LPS-mediated EBD.
Recently, the HDAC6-specific inhibitor tubacin was shown to suppress thrombin-induced EBD in human pulmonary arterial cells (49). HDAC6 is a unique member of class II because it contains two homologous fully functional catalytic domains. The COOH-terminal catalytic domain of HDAC6 possesses α-tubulin deacetylase activity and regulates microtubule dynamics (12, 27). HDAC6 is primarily cytoplasmic and interacts with and deacetylates Hsp90 (5, 35). The HAT p300 acetylates HDAC6, promotes nuclear translocation, and regulates gene expression (38). Our data demonstrate that LPS does not induce HDAC6 activity (data not shown); however, selective pharmacological or genetic inhibition of HDAC6 protects against the LPS-induced decrease in TER. This suggests that basal HDAC6 activity is critical in inducing LPS-mediated EBD and that inhibition of HDAC6 provides a possible strategy to protect against LPS-mediated EBD.
Both HDAC3 and HDAC6 are involved in gene expression, protein function, kinase signaling, and cytoskeletal changes in endothelial cells (2, 32–34, 41, 49, 52, 63). In the cytoplasm, both HDAC3 and HDAC6 interact with the molecular chaperone Hsp90 and regulate its chaperone activity (47). Therefore, we speculated that combined inhibition of HDAC3 and -6 might provide better protection against LPS-mediated EBD. Our data demonstrate that combined pharmacological or genetic inhibition of HDAC3 and -6 provides significant protection against LPS-mediated EBD in HLMVEC and against LPS-induced ALI in mice.
Binding of LPS to the endothelial cells elicits a broad cellular response, including induction of kinase signaling pathways, production of proinflammatory cytokines, cytoskeletal remodeling, and loss of cell-cell adhesion. Together these effects cause an increase in transendothelial permeability and lead to EBD and ALI. Hsp90 regulates the stability and activity of many proteins in these pathways. Our present data uncover a novel mechanism of LPS-induced endothelial hyperpermeability (Fig. 9). We have recently demonstrated that src and Hsp90 interact in HLMVEC, resulting in mutually augmented activities, of kinase and chaperone functions, respectively. We now demonstrate that HDAC, specifically HDAC3 and HDAC6, are also involved. At minimum, the threesome of src, HDAC, and Hsp90 promotes enhancement of Hsp90 chaperone function triggered by LPS and possibly other inflammatory molecules and are thus critically important in the development of EBD and ALI. Consequently, the anti-inflammatory and endothelial barrier-protective effect of HDAC and Hsp90 inhibitors could be exploited to treat vascular diseases with minimal side effects.
Fig. 9.
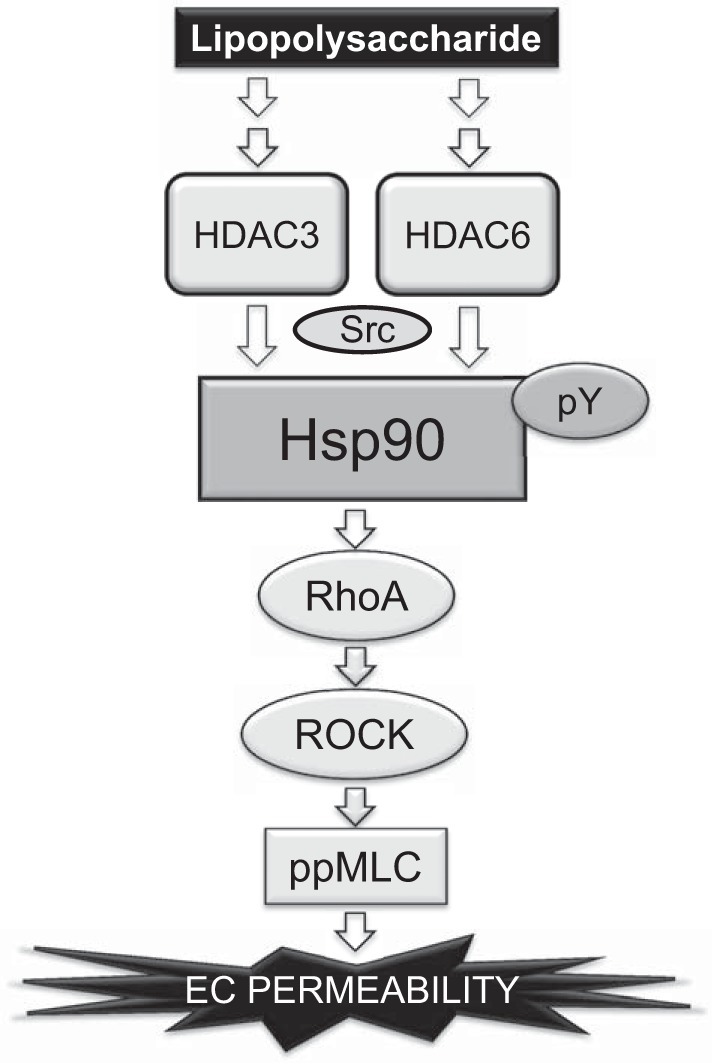
Proposed scheme for the role of HDAC3 and -6 in LPS-mediated EBD.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-101902.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.D.J. and J.D.C. conception and design of research; A.D.J., N.B., C.B., C.D., M.C.-S., and J.D. performed experiments; A.D.J., N.B., C.B., C.D., and J.D.C. analyzed data; A.D.J., N.B., C.B., C.D., G.T., and J.D.C. interpreted results of experiments; A.D.J., N.B., and J.D.C. prepared figures; A.D.J. drafted manuscript; A.D.J., N.B., C.B., C.D., G.T., M.C.-S., J.D., and J.D.C. approved final version of manuscript; J.D.C. edited and revised manuscript.
REFERENCES
- 1.Adamson RH, Curry FE, Adamson G, Liu B, Jiang Y, Aktories K, Barth H, Daigeler A, Golenhofen N, Ness W, Drenckhahn D. Rho and rho kinase modulation of barrier properties: cultured endothelial cells and intact microvessels of rats and mice. J Physiol 539: 295–308, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Advani A, Huang Q, Thai K, Advani SL, White KE, Kelly DJ, Yuen DA, Connelly KA, Marsden PA, Gilbert RE. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am J Pathol 178: 2205–2214, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 101: 3765–3777, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Antonov A, Snead C, Gorshkov B, Antonova GN, Verin AD, Catravas JD. Heat shock protein 90 inhibitors protect and restore pulmonary endothelial barrier function. Am J Respir Cell Mol Biol 39: 551–559, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aoyagi S, Archer TK. Modulating molecular chaperone Hsp90 functions through reversible acetylation. Trends Cell Biol 15: 565–567, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Balakin KV, Ivanenkov YA, Kiselyov AS, Tkachenko SE. Histone deacetylase inhibitors in cancer therapy: latest developments, trends and medicinal chemistry perspective. Anticancer Agents Med Chem 7: 576–592, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 280: 26729–26734, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Bannerman DD, Goldblum SE. Direct effects of endotoxin on the endothelium: barrier function and injury. Lab Invest 79: 1181–1199, 1999. [PubMed] [Google Scholar]
- 9.Barabutis N, Handa V, Dimitropoulou C, Rafikov R, Snead C, Kumar S, Joshi A, Thangjam G, Fulton D, Black SM, Patel V, Catravas JD. LPS induces pp60c-src-mediated tyrosine phosphorylation of Hsp90 in lung vascular endothelial cells and mouse lung. Am J Physiol Lung Cell Mol Physiol 304: L883–L893, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barginear MF, Van Poznak C, Rosen N, Modi S, Hudis CA, Budman DR. The heat shock protein 90 chaperone complex: an evolving therapeutic target. Curr Cancer Drug Targets 8: 522–532, 2008. [DOI] [PubMed] [Google Scholar]
- 11.Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J 25: 552–563, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Boyault C, Sadoul K, Pabion M, Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 26: 5468–5476, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Carbajal JM, Schaeffer RC Jr. RhoA inactivation enhances endothelial barrier function. Am J Physiol Cell Physiol 277: C955–C964, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Catravas JD, Snead C, Dimitropoulou C, Chang AS, Lucas R, Verin AD, Black SM. Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vasc Pharmacol 52: 175–181, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatterjee A, Dimitropoulou C, Drakopanayiotakis F, Antonova G, Snead C, Cannon J, Venema RC, Catravas JD. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. Am J Respir Crit Care Med 176: 667–675, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chatterjee A, Snead C, Yetik-Anacak G, Antonova G, Zeng J, Catravas JD. Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am J Physiol Lung Cell Mol Physiol 294: L755–L763, 2008. [DOI] [PubMed] [Google Scholar]
- 17.Choi SY, Ryu Y, Kee HJ, Cho SN, Kim GR, Cho JY, Kim HS, Kim IK, Jeong MH. Tubastatin A suppresses renal fibrosis via regulation of epigenetic histone modification and Smad3-dependent fibrotic genes. Vasc Pharmacol 72: 130–140, 2015. [DOI] [PubMed] [Google Scholar]
- 18.Chong DL, Sriskandan S. Pro-inflammatory mechanisms in sepsis. Contrib Microbiol 17: 86–107, 2011. [DOI] [PubMed] [Google Scholar]
- 19.Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest 86: 9–22, 2006. [DOI] [PubMed] [Google Scholar]
- 20.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370: 737–749, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Den RB, Lu B. Heat shock protein 90 inhibition: rationale and clinical potential. Ther Adv Med Oncol 4: 211–218, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Marcotullio L, Canettieri G, Infante P, Greco A, Gulino A. Protected from the inside: endogenous histone deacetylase inhibitors and the road to cancer. Biochim Biophys Acta 1815: 241–252, 2011. [DOI] [PubMed] [Google Scholar]
- 23.Ding RY, Zhao DM, Zhang ZD, Guo RX, Ma XC. Pretreatment of Rho kinase inhibitor inhibits systemic inflammation and prevents endotoxin-induced acute lung injury in mice. J Surg Res 171: e209–e214, 2011. [DOI] [PubMed] [Google Scholar]
- 24.Drysdale MJ, Brough PA, Massey A, Jensen MR, Schoepfer J. Targeting Hsp90 for the treatment of cancer. Curr Opin Drug Discov Devel 9: 483–495, 2006. [PubMed] [Google Scholar]
- 25.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91: 1487–1500, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Duval M, Le Boeuf F, Huot J, Gratton JP. Src-mediated phosphorylation of Hsp90 in response to vascular endothelial growth factor (VEGF) is required for VEGF receptor-2 signaling to endothelial NO synthase. Mol Biol Cell 18: 4659–4668, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao YS, Hubbert CC, Lu J, Lee YS, Lee JY, Yao TP. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol Cell Biol 27: 8637–8647, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene 26: 5420–5432, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SG, Shah B, Sharma S, Chang JC, Melnick AM, Hiebert S, Bhalla KN. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 5: 5637–5650, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halili MA, Andrews MR, Sweet MJ, Fairlie DP. Histone deacetylase inhibitors in inflammatory disease. Curr Top Med Chem 9: 309–319, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Joshi AD, Dimitropoulou C, Thangjam G, Snead C, Feldman S, Barabutis N, Fulton D, Hou Y, Kumar S, Patel V, Gorshkov B, Verin AD, Black SM, Catravas JD. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am J Respir Cell Mol Biol 50: 170–179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung SB, Kim CS, Naqvi A, Yamamori T, Mattagajasingh I, Hoffman TA, Cole MP, Kumar A, Dericco JS, Jeon BH, Irani K. Histone deacetylase 3 antagonizes aspirin-stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circ Res 107: 877–887, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaluza D, Kroll J, Gesierich S, Yao TP, Boon RA, Hergenreider E, Tjwa M, Rossig L, Seto E, Augustin HG, Zeiher AM, Dimmeler S, Urbich C. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. Embo J 30: 4142–4156, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim Y, Kim K, Park D, Lee E, Lee H, Lee YS, Choe J, Jeoung D. Histone deacetylase 3 mediates allergic skin inflammation by regulating expression of MCP1 protein. J Biol Chem 287: 25844–25859, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 18: 601–607, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Legagneux V, Dubois MF, Morange M, Bensaude O. Phosphorylation of the 90 kDa heat shock protein in heat shocked HeLa cell lysates. FEBS Lett 231: 417–420, 1988. [DOI] [PubMed] [Google Scholar]
- 37.Li Y, Wu Y, Wang Z, Zhang XH, Wu WK. Fasudil attenuates lipopolysaccharide-induced acute lung injury in mice through the Rho/Rho kinase pathway. Med Sci Monit 16: BR112–BR118, 2010. [PubMed] [Google Scholar]
- 38.Liu Y, Peng L, Seto E, Huang S, Qiu Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J Biol Chem 287: 29168–29174, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Longworth MS, Laimins LA. Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene 25: 4495–4500, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol 77: 1763–1772, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margariti A, Zampetaki A, Xiao Q, Zhou B, Karamariti E, Martin D, Yin X, Mayr M, Li H, Zhang Z, De Falco E, Hu Y, Cockerill G, Xu Q, Zeng L. Histone deacetylase 7 controls endothelial cell growth through modulation of beta-catenin. Circ Res 106: 1202–1211, 2010. [DOI] [PubMed] [Google Scholar]
- 42.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM, Acute Lung Injury in Animals Study. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mottet D, Castronovo V. Histone deacetylases: anti-angiogenic targets in cancer therapy. Curr Cancer Drug Targets 10: 898–913, 2010. [DOI] [PubMed] [Google Scholar]
- 44.Murch O, Collin M, Hinds CJ, Thiemermann C. Lipoproteins in inflammation and sepsis. I. Basic science. Intensive Care Med 33: 13–24, 2007. [DOI] [PubMed] [Google Scholar]
- 45.Oda M, Han JY, Nakamura M. Endothelial cell dysfunction in microvasculature: relevance to disease processes. Clin Hemorheol Microcirc 23: 199–211, 2000. [PubMed] [Google Scholar]
- 46.Opal SM. The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int J Med Microbiol 297: 365–377, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Park JH, Kim SH, Choi MC, Lee J, Oh DY, Im SA, Bang YJ, Kim TY. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem Biophys Res Commun 368: 318–322, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Ryan GB, Majno AG. Acute inflammation. A review. Am J Pathol 86: 183–276, 1977. [PMC free article] [PubMed] [Google Scholar]
- 49.Saito S, Lasky JA, Guo W, Nguyen H, Mai A, Danchuk S, Sullivan DE, Shan B. Pharmacological inhibition of HDAC6 attenuates endothelial barrier dysfunction induced by thrombin. Biochem Biophys Res Commun 408: 630–634, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scroggins BT, Robzyk K, Wang D, Marcu MG, Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, Rosen N, Neckers L. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell 25: 151–159, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Rev Anticancer Ther 10: 935–954, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stein S, Matter CM. Protective roles of SIRT1 in atherosclerosis. Cell Cycle 10: 640–647, 2011. [DOI] [PubMed] [Google Scholar]
- 53.Takami Y, Nakayama T. N-terminal region, C-terminal region, nuclear export signal, and deacetylation activity of histone deacetylase-3 are essential for the viability of the DT40 chicken B cell line. J Biol Chem 275: 16191–16201, 2000. [DOI] [PubMed] [Google Scholar]
- 54.Tobias PS, Gegner J, Han J, Kirkland T, Kravchenko V, Leturcq D, Lee JD, Moriarty A, Mathison JC, Pugin J. LPS binding protein and CD14 in the LPS dependent activation of cells. Prog Clin Biol Res 388: 31–39, 1994. [PubMed] [Google Scholar]
- 55.Travers J, Sharp S, Workman P. HSP90 inhibition: two-pronged exploitation of cancer dependencies. Drug Discov Today 17: 242–252, 2012. [DOI] [PubMed] [Google Scholar]
- 56.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks PA. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA 102: 673–678, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wells CE, Bhaskara S, Stengel KR, Zhao Y, Sirbu B, Chagot B, Cortez D, Khabele D, Chazin WJ, Cooper A, Jacques V, Rusche J, Eischen CM, McGirt LY, Hiebert SW. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PloS One 8: e68915, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu H, Song J, Gao X, Xu Z, Xu X, Xia Y, Dai Y. Paeoniflorin attenuates lipopolysaccharide-induced permeability of endothelial cells: involvements of F-actin expression and phosphorylations of PI3K/Akt and PKC. Inflammation 36: 216–225, 2013. [DOI] [PubMed] [Google Scholar]
- 59.Xu SS, Alam S, Margariti A. Epigenetics in vascular disease: therapeutic potential of new agents. Curr Vasc Pharmacol 12: 77–86, 2014. [DOI] [PubMed] [Google Scholar]
- 60.Yang WM, Yao YL, Sun JM, Davie JR, Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J Biol Chem 272: 28001–28007, 1997. [DOI] [PubMed] [Google Scholar]
- 61.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26: 5310–5318, 2007. [DOI] [PubMed] [Google Scholar]
- 62.Zhang X, Ozawa Y, Lee H, Wen YD, Tan TH, Wadzinski BE, Seto E. Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev 19: 827–839, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou B, Margariti A, Zeng L, Xu Q. Role of histone deacetylases in vascular cell homeostasis and arteriosclerosis. Cardiovasc Res 90: 413–420, 2011. [DOI] [PubMed] [Google Scholar]