

Mutations in the TP53 tumour-suppressor gene are common in human tumours. Although these mutations invariably inactivate the normal activity of p53 (ref. 1), which is the transcription factor encoded by TP53, some mutations also endow p53 with ‘gain-of-function’ activities that promote cancer2. Whether diverse p53 mutants produce similar gain-of-function activities and how they do so remains a puzzle, but finding the answer might enable the design of strategies for treating many cancers. On page 206 of this issue, Zhu et al.3 provide a possible explanation: they show that gain-of-function mutant p53 proteins induce the production of enzymes that modify the histone proteins around which DNA is packaged as chromatin, thus altering gene expression.
Experimentally altering the expression of gain-of-function mutant p53 affects the expression of myriad genes, enhancing the invasiveness and proliferation of tumour cells in vitro4. Moreover, mice harbouring key gain-of-function mutations in TP53 develop tumours that differ from those lacking p53 (ref. 5). Lowering the level of gain-of-function p53 has antiproliferative effects in vitro and can reduce metastasis or trigger tumour regression in vivo2, 5, 6. A better understanding of these mutants is therefore desirable. Zhu et al. found that, in cultured human-cancer cell lines, gain-of-function mutant forms of p53 bind to different regions of DNA from the normal protein. In particular, the mutant proteins bind to the genes MLL1 and MLL2. Gain-of-function p53 seems to be recruited to these genes in part through binding to ETS2 — a transcription factor that is known7 to target gain-of-function p53 to different genes from those activated by normal p53.
MLL1 and MLL2 are members of the SET family of histone methyltransferase enzymes8. These act as parts of large complexes9 to modulate gene expression by attaching methyl groups to a lysine amino-acid residue (K4) of the histone H3 protein. Such H3K4 methylation allows increased transcription of the gene packaged around the histones. The authors found that gain-of-function p53 also activates expression of the gene MOZ, which encodes an enzyme that adds an acetyl group to K9 of H3, again allowing increased gene expression.
In agreement with the idea that gain-of-function p53 affects histone modification, reducing levels of the mutant p53 decreased H3K9 acetylation. However, p53 reduction had only a small effect on H3K4 methylation. Perhaps, as Zhu and colleagues suggest, this is because other members of the SET family have similar but p53-independent roles to MLL1 and MLL2. The authors demonstrated that gain-of-function p53 activates MLL1, MLL2 and MOZ, and showed that this activation is partly responsible for the ability of the mutant p53 to enhance cell proliferation in vitro (Fig. 1).
Figure 1. Gaining on p53.
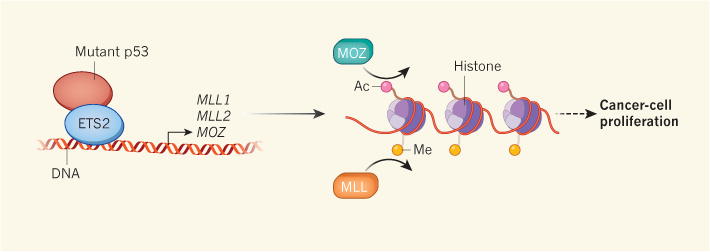
‘Gain-of-function’ mutations in the tumour-suppressor gene TP53 enable the transcription factor that it encodes to bind to abnormal targets, leading to cancer. Zhu et al.3 report that gain-of-function p53 binds to the transcription factor ETS2 and activates the genes MLL1, MLL2 and MOZ. MLL1 and MLL2 encode MLL enzymes that add methyl groups (Me) to the histone proteins around which genes are packaged as chromatin, and MOZ adds acetyl groups (Ac) to these histones. Both modifications increase local gene expression, leading to an increase in the proliferation of cancer cells through as-yet-unknown mechanisms.
Finally, mining human-cancer databases provided support for Zhu and colleagues’ data, indicating that expression of MLL1, MLL2 and MOZ is significantly upregulated in human tumours with select p53 gain-of-function mutants compared with tumours lacking p53 or those without mutant p53. This correlation is not obvious across breast cancers10, the tissue of origin for many cell lines studied in this work — although Zhu et al. clearly show that such a correlation exists in the cell lines that they used. It is likely that other variables affect MLL expression in some tumour types. As a result, it will be important to investigate the contextual factors that determine whether or when gain-of-function p53 can trigger changes in MLL and MOZ expression, and to analyse the mechanisms underlying these events.
Gain-of-function p53 was also recently shown11 to act with the SWI/SNF chromatin-remodelling complex to upregulate many genes that can themselves mediate the cancer-causing activities of gain-of-function p53. This finding, taken together with Zhu and colleagues’ demonstration that this p53 is linked to chromatin and, by extension, to the transcriptome (the complete gene-expression profile of the cell), could explain why so many genes are affected by the presence of gain-of-function p53. But precisely how p53 proteins with diverse mutations acquire similar capabilities remains to be discovered.
One possibility is that p53 mutants adopt a different structure from normal p53 that enables their interaction with ETS2. However, there is no obvious explanation for the evolution of such an interaction, and this model is at odds with the observation12 that some gain-of-function p53 proteins have similar structures to normal p53. Alternatively, the ability of normal p53 to bind to thousands of sites in the human genome might prevent it from associating with the factors with which gain-of-function p53 interacts. Or perhaps the expression of one or more target genes somehow actively prevents the normal protein from engaging in the interactions that are characteristic of the mutant p53.
The authors’ link between gain-of-function p53 and MLL1 and MLL2 is intriguing, given that members of the MLL family are frequently mutated in human cancers13. For example, chromosomal translocations involving MLL1 can drive leukaemia, and MLL2 mutations are common in several carcinomas. But MLL1 translocations eliminate the gene’s histone methyltransferase domain, and MLL2 mutations seem to be inactivating. The explanation for this apparent discrepancy with Zhu and colleagues’ findings is unclear, but probably reflects context-dependent differences in enzyme function.
Could targeting MLL1, MLL2 or MOZ be a strategy for treating tumours involving gain-of-function p53? Zhu et al. showed that two inhibitors of MLL-complex formation block proliferation in cells expressing mutant p53, but do not affect those lacking p53. Eliminating gain-of-function p53 or interfering with its mechanism of action can have anticancer effects in vitro and in mice6, 14, 15. Moreover, there is much enthusiasm for cancer treatments that affect chromatin modification, and compounds that target some chromatin-modifying activities have been approved for use in the clinic or are currently in clinical trials. But more work is required, because the specificity of the MLL inhibitors is not entirely established. Furthermore, MLL genes are active during embryonic development, and their inhibition can cause embryonic death, independent of TP53 mutations14. These observations, together with the previously mentioned fact that mutations disrupting MLL function are common in tumours, raise concerns that MLL inhibitors might be toxic, or might even promote tumours.
Nonetheless, given the frequency with which TP53 is mutated in cancer, continued efforts to modulate the effects of mutant p53 are clearly warranted. With the key targets of MLL and MOZ in hand, specific therapies might become possible. Thus, Zhu and colleagues’ study, and those of others, might point to treatments for tumours that harbour TP53 mutations.
Footnotes
The finding that genes encoding enzymes that modify histone proteins are among the targets of certain mutant forms of the p53 protein sheds light on how these mutations cause cancer beyond p53 inactivation.
Contributor Information
Carol Prives, Email: clp3@columbia.edu, Department of Biological Sciences, Columbia University, New York, New York 10027, USA.
Scott W. Lowe, Email: lowes@mskcc.org, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA.
References
- 1.Vogelstein B, Kinzler KW. Cell. 1992;70:523–526. doi: 10.1016/0092-8674(92)90421-8. [DOI] [PubMed] [Google Scholar]
- 2.Muller PAJ, Vousden KH. Nature Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 3.Zhu J, et al. Nature. 2015;525:206–211. doi: 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freed-Pastor WA, Prives C. Genes Dev. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia PB, Attardi LD. Semin Cell Dev Biol. 2014;27:74–85. doi: 10.1016/j.semcdb.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexandrova EM, et al. Nature. 2015;523:352–356. doi: 10.1038/nature14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Do PM, et al. Genes Dev. 2012;26:830–845. doi: 10.1101/gad.181685.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang P, Bergamin E, Couture JF. Biopolymers. 2013;99:136–145. doi: 10.1002/bip.22126. [DOI] [PubMed] [Google Scholar]
- 9.Shilatifard A. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.www.cBioPortal.org
- 11.Pfister NT, et al. Genes Dev. 2015;29:1298–1315. doi: 10.1101/gad.263202.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joerger AC, Ang HC, Fersht AR. Proc Natl Acad Sci USA. 2006;103:15056–15061. doi: 10.1073/pnas.0607286103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ford DJ, Dingwall AK. Cancer Genet. 2015;208:178–191. doi: 10.1016/j.cancergen.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Freed-Pastor WA, et al. Cell. 2012;148:244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weissmueller S, et al. Cell. 2014;157:382–394. doi: 10.1016/j.cell.2014.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]