

Abstract
Apoptosis is generally believed to be a process that requires several hours, in contrast to non-programmed forms of cell death that can occur in minutes. Our findings challenge the time-consuming nature of apoptosis as we describe the discovery and characterization of a small molecule, named Raptinal, which initiates intrinsic pathway caspase-dependent apoptosis within minutes in multiple cell lines. Comparison to a mechanistically diverse panel of apoptotic stimuli reveals Raptinal-induced apoptosis proceeds with unparalleled speed. The rapid phenotype enabled identification of the critical roles of mitochondrial voltage-dependent anion channel function, mitochondrial membrane potential/coupled respiration, and mitochondrial complex I, III and IV function for apoptosis induction. Use of Raptinal in whole organisms demonstrates its utility to study apoptosis in vivo for a variety of applications. Overall, rapid inducers of apoptosis are powerful tools that will be used in a variety of settings to generate further insight into the apoptotic machinery.
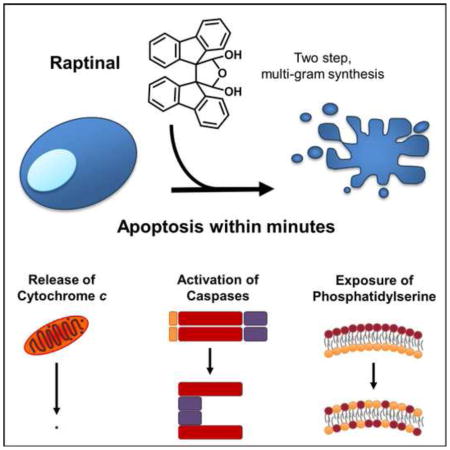
Graphical Abstract
INTRODUCTION
Apoptosis-inducing small molecules typically engage the intrinsic pathway, in which release of mitochondrial cytochrome c induces activation of caspase-9, followed by activation of caspase-3. The rate of apoptosis is dependent on the type and strength of the apoptotic stimuli and the cell type, and a minimum of several hours of activating stimulus is typically required for apoptosis induction through the intrinsic pathway (Goldstein et al., 2000). This lengthy induction period is likely due to rate limiting steps upstream of cytochrome c release such as transcription/translation (Dudgeon et al., 2009; Fridman and Lowe, 2003) or cell cycle dependent responses (Hamada et al., 2009). While the time to cytochrome c release varies, once initiated, cytochrome c release may be complete within 5–10 minutes (Goldstein et al., 2005; Luetjens et al., 2001) regardless of cell type. The kinetics of caspase activation following cytochrome c release may be dependent on the cell type, although in certain cells caspase activation is complete within 20 minutes after initiation (Luo et al., 2001; Rehm et al., 2002).
Agents capable of inducing intrinsic pathway-mediated apoptosis are widely employed in a range of biochemical experiments. Typically, the broad-spectrum kinase inhibitor staurosporine has been the small molecule of choice, as it requires a short time period for induction of apoptosis relative to other agents. Among numerous other experiments, staurosporine has been employed in studies that have identified fundamental regulators of the apoptotic pathway (including Bcl-2 (Yang et al., 1997), CAD/ICAD (Sakahira et al., 1998), AIF (Susin et al., 1999), and multiple others), in proteomics experiments examining the scope of cellular caspase protein substrates (Agard et al., 2012; Dix et al., 2008; Dix et al., 2012; Shimbo et al., 2012), and to help elucidate mechanisms of apoptotic death as induced by small molecules (Wolpaw et al., 2011). However, even staurosporine requires multiple hours for full cytochrome c release from the mitochondria (Bossy-Wetzel et al., 1998; Botham et al., 2014), with apoptotic cell death ensuing. In addition, the pan-kinase inhibition and ever-emerging biological effects elicited by staurosporine (Savitski et al., 2014) complicate interpretation of downstream readouts, as demonstrated by the fact that other proapoptotic agents (e.g. doxorubicin and bortezomib) induce a different pattern of caspase cleavage from staurosporine (Shimbo et al., 2012). A compound that rapidly induces mitochondrial cytochrome c release and apoptosis without prolonged engagement of upstream processes would be especially valuable in these and other cell biology experiments.
The overarching impact of apoptosis in diseases, such as cancer (Fulda, 2007), heart disease (Narula et al., 2006), and neurodegeneration (Ferrer, 2006), highlights the necessity of having resources to study and find regulators involved in programmed cell death. Given the importance of apoptosis, small molecules with unusual mechanistic properties will facilitate discovery of additional apoptotic regulators. Accordingly, we report the identification of a small molecule activator of apoptosis that induces initiation of cytochrome c release from the mitochondria within minutes, and displays potency across multiple cell types and animal models.
RESULTS
Raptinal Rapidly Induces Apoptosis
We discovered Raptinal (Figure 1A) while screening an in-house library of small molecules for cytotoxicity against HL-60 human leukemia cells; as described extensively below Raptinal was found to be an unusually rapid inducer of apoptosis in multiple cell lines. Aside from the skin-irritant properties of Raptinal (Curtin et al., 1965), no other biological activities regarding this small molecule have been previously reported. The compound was resynthesized for these studies and can be easily accessed in two steps on multi-gram scale (see Detailed Experimental Procedures). NMR spectroscopy experiments show that the hydrate as drawn in Figure 1A, rather than the dialdehyde form, is the dominant species in aqueous solution (Figure S1A and Detailed Experimental Procedures).
Figure 1. Raptinal Rapidly Induces Apoptosis.

A) Structure of Raptinal. B) Scanning electron micrographs of U-937 cells show pronounced apoptotic blebbing after 1 hour of treatment with 10 μM Raptinal (right) versus vehicle control treated cells (left). C) AV/PI graphs of U-937 cells treated with 10 μM Raptinal for 2 hours show transition of cells through the early apoptotic AV+/PI− quadrant that is prevented by the pan-caspase inhibitor Q-VD-OPh. D) Immunoblots of U-937 cells treated with various toxins for 1 hour show more prominent activation of procaspase-3 (PC-3) to caspase-3 (C-3) and cleavage of PARP-1 (cPARP-1) by Raptinal (10 μM) versus 25 other toxins (all tested at 10 μM). E) Cell viability of U-937 cells assessed by AV/PI after 2 hour treatment with 10 μM Raptinal and 25 other small molecules (all tested at 10 μM). Data represent the mean ± SD from 3 independent experiments. F) Time course analysis of U-937 cell viability upon treatment with 10 μM of various anticancer agents and biological tool molecules. Cell viability was assessed by AV/PI analysis. G) Time course analysis of adherent cell viability upon treatment with Raptinal, 1541B, and Staurosporine (all tested at 10 μM). Cell viability was assessed by AV/PI analysis. See also Figure S1.
Apoptosis describes a form of programmed cell death characterized by blebbing of the cell membrane leading to the dissociation of apoptotic bodies from the cell body (Balasubramanian et al., 2007; Galluzzi et al., 2012). Consistent with apoptotic morphological changes, U-937 cells treated with 10 μM Raptinal exhibited modest (at 30 min) and extensive (at 60 min) blebbing, as observed by scanning electron and light microscopy (Figures 1B and S1B). Another hallmark of apoptosis is externalization of phosphatidylserine on the cell membrane prior to loss of cell membrane integrity, which can be monitored by annexin V/propidium iodide (AV/PI) staining (Galluzzi et al., 2012; Susin et al., 2000). Treatment with 10 μM Raptinal resulted in ~80% loss in U-937 cell viability after just 2 hours of exposure with cells progressing through the AV+/PI− apoptotic quadrant (Figure 1C). Consistent with caspase-dependent apoptosis, the pan-caspase inhibitor, Q-VD-OPh, quantitatively blocked the loss in cell viability and cellular blebbing (Figures 1C and S1B).
Time course analysis of apoptosis by AV/PI staining with various concentrations of Raptinal revealed the rate of apoptosis was concentration dependent and could be tailored for various applications (Figure S1C). Additionally, Raptinal was found to induce death against various cancer and non-cancerous cell lines with 24 hour IC50 values between 0.7–3.4 μM (Table 1, Figure S1D), indicating activity across a wide variety of cell lines.
Table 1. Raptinal Toxicity in Various Cell Lines.
The IC50 values of Raptinal against normal and cancer cell lines assessed after 24 hour incubation. Data represent the mean ± SD from 3 independent experiments.
| Cell Line | Cell Type | Average IC50 (μM) |
|---|---|---|
|
| ||
| HFF-1 | Human Foreskin Fibroblast | 3.3 ± 0.2 |
| MCF10A | Human Breast Tissue | 3.0 ± 0.2 |
| WT-MEF | Mouse Embryonic Fibroblasts | 2.4 ± 0.7 |
| 3T3 | Mouse Embryonic Fibroblasts | 1.0 ± 0.2 |
| MCF-7 | Human Breast Cancer | 3.4 ± 0.1 |
| BT-549 | Human Breast Cancer | 1.3 ± 0.4 |
| MDA-MB-436 | Human Breast Cancer | 1.1 ± 0.1 |
| 4T1 | Mouse Breast Cancer | 1.0 ± 0.2 |
| SK-MEL-5 | Human Melanoma | 0.7 ± 0.1 |
| B16-F10 | Mouse Melanoma | 1.6 ± 0.2 |
| 143B | Human Osteosarcoma | 1.2 ± 0.5 |
| HOS | Human Osteosarcoma | 1.0 ± 0.1 |
| Hs888Lu | Human Osteosarcoma | 1.7 ± 0.1 |
| SKW 6.4 | Human Lymphoma | 1.1 ± 0.1 |
| U-937 | Human Lymphoma | 0.7 ± 0.3 |
| Jurkat | Human Leukemia | 2.7 ± 0.9 |
| HL-60 | Human Leukemia | 2.1 ± 1.4 |
| HL-60 VCR | Human Leukemia Vincristine Resistant | 1.6 ± 0.5 |
| H460 | Human Lung Cancer | 1.1 ± 0.1 |
| H1993 | Human Lung Cancer | 1.2 ± 0.1 |
| HeLa | Human Cervical Cancer | 0.6 ± 0.4 |
| MIA PaCa-2 | Human Pancreatic Cancer | 1.9 ± 0.8 |
To assess if induction of apoptosis by Raptinal was unusually rapid, 25 other cell death inducing small molecules that encompass a diverse array of mechanisms and biological targets were assessed. Included were inhibitors of kinases (staurosporine and rapamycin), inhibitors of tubulin dynamics (paclitaxel, vincristine, colchicine), topoisomerase inhibitors (camptothecin, etoposide, doxorubicin), DNA alkylators (mitomycin C, cisplatin, MNNG), endoplasmic reticulum/proteasome inhibitors (geldanamycin, thapsigargin, tunicamycin), ROS inducing agents (DNQ (Bair et al., 2010), elesclomol, rotenone, antimycin A) and agents that directly target components of the apoptotic pathway such as Bcl-2 inhibitors (gossypol (Oliver et al., 2005) and HA14-1 (Chen et al., 2002)), XIAP inhibitor (SM-164 (Lu et al., 2008)), apoptosome promoter (AA2 (Nguyen and Wells, 2003)) and direct procaspase-3 activators (PAC-1 (Putt et al., 2006) and 1541/1541B (Wolan et al., 2009)). A concentration of 10 μM was used for all compounds, as most of these molecules are potent inducers of cell death with IC50 values typically ranging from the nM to low μM range. Immunoblots after a one hour treatment of U-937 cells showed complete activation of procaspase-3 to caspase-3 by Raptinal, partial activation by 1541, and no activation of procaspase-3 by the other agents (Figure 1D). Significantly more abundant cleavage of PARP-1 (from 116 to 89 kDa form), a substrate of active caspase-3, was observed in Raptinal treated cells versus the other toxins (Figure 1D). In SKW 6.4 cells, Raptinal was the only agent capable of inducing procaspase-3 activation after one hour of exposure (Figure S1E).
AV/PI analyses of U-937 cells after two hours of treatment revealed Raptinal induced 80% loss in cell viability, 1541 reduced viability by 50% (average values, p values < 0.05), while the other 24 compounds were unable to affect cell viability (Figure 1E). Time course AV/PI experiments revealed conventional cytotoxic compounds with similar mechanisms of action shared similar times to 50% cell death (Figure 1F and Table S1), although no obvious trend was observed for alternative mechanistic apoptotic agents. The topoisomerase inhibitors required 5–6 hours, whereas agents affecting tubulin polymerization required 14–18 hours (Figure 1F). Evaluation of Raptinal in a caspase activity assay in cell lysate (utilizing the fluorescent substrate Ac-DEVD-AFC) showed faster induction of caspase-3/-7 activity and higher peak levels when compared to other apoptosis inducing agents, including staurosporine (Figure S1F). Raptinal, 1541B and staurosporine were evaluated in four adherent cell lines (HOS, H1993, SK-MEL-5, MIA PaCa-2) for their ability to induce cell death as observed by AV/PI staining (Figure 1G). Raptinal was found to be substantially faster than the other agents (Figure 1G) and was the only agent capable of activating procaspase-3 and inducing subsequent PARP-1 cleavage within 2 hours of treatment (Figure S1G). To assess if Raptinal has a direct effect on activation of procaspase-3, recombinant procaspase-3 was treated with up to 100 μM Raptinal; in this experiment (Figure S1H) no increase in enzymatic activity was observed, in contrast to 1541B, suggesting that Raptinal acts upstream of procaspase-3. A small set of derivatives of Raptinal were synthesized and assessed to obtain a structure-activity relationship. Evaluation of the ability of these compounds to induce cell death at 2 or 24 hours suggests that the presence of the aldehydes is critical to activity. Unrelated dialdehyde containing molecules were either non-cytotoxic or were slow inducers of death (see Table S2).
The Raptinal Mechanism Involves Mitochondrial-Mediated Intrinsic Pathway
To characterize whether Raptinal-induced apoptosis occurs through the intrinsic or extrinsic pathway, the kinetics of cytochrome c release were determined by fractionating between cytosolic or mitochondrial proteins using selective permeabilization with digitonin combined with analysis by immunoblotting or flow cytometry. Upon treatment of cells with 10 μM Raptinal, cytochrome c release occurs as early as 10 minutes and is nearly complete by 20–30 minutes in both U-937 (Figure 2A) and SKW 6.4 (Figure S2A) lymphoma cells by immunoblot and flow cytometry (Figure S2B). Concomitant with cytochrome c release at 20 minutes is the activation of the initiator caspase-9 (Figures 2A and S2A), which proceeds to completion by 45 minutes. Further investigation of caspase activation revealed complete cleavage of not only procaspase-9, but also the initiator procaspase-8 and the executioner procaspase-3 by 45 minutes (Figures 2B and S2C). Co-incubation of cells with the pan-caspase inhibitor Q-VD-OPh and Raptinal resulted in a 19 kDa inhibited form of caspase-3 (which has the pro-domain intact) and completely inhibited the processing of caspase-8 (Figures 2B and S2B). This result suggests that the proteolytic cleavage of procaspase-8, a known substrate of caspase-3, occurs downstream of caspase-3, and is consistent with the intrinsic pathway of caspase-dependent apoptosis. To assess the ability of Raptinal to induce apoptosis through other cell death pathways we assessed cells by Western blot for the cleavage of procaspase-1 (pyroptosis/pyronecrosis) and phosphorylation of the RIP3 kinase target MLKL (necroptosis). We saw neither cleavage of procaspase-1 (Figure S2D) or phosphorylation of MLKL (Figure S2E). In contrast, phosphorylation of MLKL was observed during the induction of necroptosis by TNFα in the presence of cycloheximide and the pan-caspase inhibitor Q-VD-OPh (Figure S2E).
Figure 2. Raptinal Activates the Intrinsic Pathway and Requires Functional Mitochondria for Apoptosis Induction.

A) Time course immunoblots of mitochondrial and cytosolic fractions of U-937 cells treated with 10 μM Raptinal show cytochrome c release and subsequent caspase-9 activation occur after 20 minutes of treatment. B) Time course immunoblot analysis of caspase-9, -3, and -8 activation in U-937 cells treated with 10 μM Raptinal. C) Relative percent caspase-3/-7 activity of MIA PaCa-2 cells treated with 10 μM Raptinal for 1 hour upon siRNA knockdown of apoptosis genes. D) Jurkat C8−/− are equally susceptible, Jurkat FADD−/− are more susceptible, while Jurkat Bcl-2 overexpressing cells are less susceptible than wild type cells to 10 μM Raptinal as assessed by AV/PI assay after 2 hours. Data represent the mean ± SD from 3 independent experiments. ** Indicates p values < 0.02 E) Transmission electron micrographs of U-937 cells treated with vehicle or 10 μM Raptinal for 5, 30 and 60 minutes. The images show rapid changes in mitochondrial morphology (arrows) after 5 minutes of treatment with Raptinal. At 30 minutes, mitochondria are largely devoid of cristae and at 60 minutes, peripheral nuclear condensation is apparent. Scale bars represent 1 micron. F) Raptinal does not induce cytochrome c release from the mitochondrial pellet into the supernatant of isolated mitochondria treated with Raptinal in vitro under non-respiring conditions. The positive control, pro-apoptotic Bid protein is able to induce cytochrome c release. G) U-937 cells pre-treated with various potential cytoprotective agents and inhibitors of various cellular pathways were treated with 10 μM Raptinal for 2 hours and protection from the effects of Raptinal was assessed by AV/PI. Data represent the mean % protection ± SD from 3 independent experiments. * Indicates p values < 0.05. H) Protective mitochondrial agents retard cytochrome c release and caspase 9 activation in U-937 cells. See also Figure S2.
In order to further investigate intrinsic pathway-mediated apoptosis, a focused siRNA screen was performed for 55 proteins (single siRNA per target) known to be involved in apoptosis (see Table S3 for the list of targets). MIA PaCa-2 cells were used for these screens due to the ease of siRNA transfection. Transfected cells were treated with Raptinal (10 μM for 1 hour) and 19 hit genes were identified that, upon siRNA knockdown, substantially reduced (by >30%) Raptinal-induced caspase-3/7 activity (Figure 2C). Consistent with an intrinsic pathway mechanism, siRNAs for APAF1, DIABLO, CASP3, BOK, and CASP3 afforded the greatest (75–96%) reduction in caspase-3/7 activity. Three of the most important genes (APAF1, CASP3, CASP9) were followed up using three distinct siRNA constructs for each gene, and the results validated knockdown of these genes efficiently inhibits Raptinal-induced caspase-3/-7 activity (Figure S2F).
Providing further evidence for apoptosis through the intrinsic pathway, Jurkat cells were utilized that contain either a vector control (WT), caspase-8 deletion, fas-activated death domain (FADD) deletion, or Bcl-2 upregulation. Jurkat caspase-8−/− and FADD−/− cell lines, which are resistant to extrinsic pathway stimuli such as Fas-L and TNF-α (Juo et al., 1998), were equally as sensitive to Raptinal as wild type cells against rapid apoptosis induction (Figure 2D). In addition, ectopic expression of anti-apoptotic Bcl-2 conferred partial resistance to Raptinal in short assays (2 hours by AV/PI, p value = 0.02, Figure 2D). These results indicate an involvement of mitochondrial-mediated apoptosis that is independent of caspase-8 and FADD, effectively ruling out the extrinsic pathway. In addition, observations in U-937 cells of morphological changes during Raptinal treatment reveal substantial mitochondrial swelling after 5 minutes with the cristae being clearly visible, in contrast to the untreated cells, as assessed by transmission electron microscopy (Figure 2E). At 30 and 60 minutes, mitochondria are devoid of cristae and at 60 minutes cells exhibit stage II chromatin condensation (peripheral nuclear condensation) consistent with caspase-activated DNAse (CAD) activity (Figure 2E) (Susin et al., 2000). Production of reactive oxygen species (ROS) in cells treated with Raptinal for 20 minutes was observed upon incubation with the flow cytometry dye dihydroethidium (DHE) (Figure S2G), which is sensitive to oxidation by superoxide anion radicals. The insensitivity to caspase-8 deletion, morphological mitochondrial changes, and ROS production all indicate Raptinal acts through the mitochondrial-mediated intrinsic pathway.
Due to the rapid nature of apoptotic induction, Raptinal was investigated for a direct effect on mitochondria. Mitochondria were isolated by differential centrifugation, and cytochrome c release was monitored by Western blot. Raptinal did not directly induce cytochrome c release in isolated mitochondria in vitro under respiring and non-respiring conditions (Figures 2F and S2H), whereas Bid, a known pro-apoptotic protein (Gillick and Crompton, 2008), was capable of releasing cytochrome c under both conditions. These results indicate that Raptinal is not able to directly induce cytochrome c release. Furthermore, Raptinal-treated cells did not undergo mitochondrial-permeability-transition-pore (MPTP) formation (Figure S2I), suggesting a mitochondrial outer membrane permeabilization (MOMP) model of cytochrome c release.
Maintenance of Mitochondrial Function is Crucial for Apoptosis
In order to identify important modulators that are involved in rapid apoptosis, a panel of small molecules that inhibit various processes in the cell were investigated for their effect on Raptinal-induced cell death (Figure 2G). The panel included the pan-caspase inhibitor Q-VD-OPh as a positive control for protection. Several agents that target the mitochondria were investigated, including inhibitors of respiration, the electron transport chain, and components of the mitochondrial transition pore, such as the voltage-dependent anion channel and cyclophilin D. Also studied were the importance of glycolysis, reactive oxygen species, calcium dependent pathways, granzyme B, transcription, and translation. Cells were pretreated with the putative protective agents for 2 hours at concentrations optimized or previously reported (see Table S4) and subsequently incubated with Raptinal (10 μM) for an additional 2 hours, at which point cell viability was assessed by annexin V/PI in order to identify agents critical to rapid apoptosis induction. Several compounds that affect mitochondrial function were able to afford significant to quantitative protection from Raptinal (Figure 2G). DIDS, a covalent inhibitor of the voltage-dependent anion channel (VDAC) afforded significant protection from apoptosis (Keinan et al., 2010; Shoshan-Barmatz et al., 2010). A mitochondrial respiration uncoupler (FCCP), electron transport chain inhibitors of complex I (rotenone), complex III (antimycin A), complex IV (sodium azide and potassium cyanide) and the ATP synthase inhibitor (oligomycin A) all provided significant to quantitative protection. Interestingly, complex II inhibition by TTFA or atpenin A5 did not confer significant protection in contrast to complex I, III, and IV inhibition. Other pathways, such as ROS, calcium signaling, transcription, necroptosis, ferroptosis and translation, proved to be less important, since only modest-to-no protection was observed with compounds modulating these processes (Figure 2G).
To further study the role of mitochondrial voltage dependent anion function and respiration in protection from cell death, cytochrome c release at 2 hours was evaluated in the presence of mitochondrial inhibitors. Immunoblots of mitochondrial and cytosolic fractions revealed that the small molecules that afforded protection in Figure 2G were able to delay Raptinal-induced cytochrome c release and subsequent caspase-9 activation (Figure 2H). These results are consistent with previous suggestions that proton transport may be required for Bax-mediated mitochondrial outer membrane permeabilization (Matsuyama et al., 1998). Utilizing a variety of small molecule modulators in combination with rapid induction of the intrinsic pathway by Raptinal has helped to further validate the importance of voltage dependent anion channel, coupled mitochondrial respiration, electron transport chain function and ATP synthase activity in apoptosis induction.
Raptinal Induces Apoptosis in a Whole Organism and Inhibits Tumor Growth In Vivo
The unique ability of Raptinal to induce rapid apoptosis may be desirable for investigation of programmed cell death both in cell culture and in vivo. Thus the ability of this compound to elicit rapid apoptosis in a transgenic zebrafish model with a secretory annexin V-YFP as a readout of phosphatidylserine externalization and apoptosis (van Ham et al., 2010) was assessed. Live transgenic zebrafish embryos (24 hours post fertilization) were treated with 10 μM Raptinal for 1.5 hours, and the appearance of the punctate annexin V-YFP signal following Raptinal-treatment was particularly evident in the tail section of the embryos (Figure 3A). Further analysis revealed 80 cells per fish were apoptotic following Raptinal treatment versus 26 cells per fish for DMSO-treated control (Figure 3B, p value = 0.0002).
Figure 3. Raptinal Exhibits Activity in vivo.

A) Zebrafish embryos expressing secretory annexin V-YFP exhibit pronounced punctate YFP signal indicating phosphatidylserine externalization following 1.5 hours of treatment with 10 μM Raptinal. B) Quantification of apoptotic cells in Raptinal- versus DMSO-treated zebrafish under the conditions in A. Data represent the mean ± SD (n = 5 and n = 7 embryos for Raptinal and DMSO, respectively; *** indicates p value < 0.001). Raptinal inhibits subcutaneous B16-F10 melanoma tumor growth in vivo as measured by tumor volume (** indicates p value < 0.005) in C and tumor mass after tumor excision in D (* indicates p value < 0.05). E–F) Raptinal inhibits subcutaneous 4T1 breast cancer tumor growth in vivo as measured by tumor volume (* indicates p value < 0.05) in E and tumor mass after tumor excision (* indicates p value < 0.05) in F. Arrows in C and E indicate intraperitoneal Raptinal administration at 20 mg/kg once a day. Tumor images in D and F are representative of tumor size at the conclusion of the studies. Data in C, D, E and F represent the mean ± SEM (n = 7 mice/group). See also Figure S3.
To characterize the in vivo pharmacokinetics and tolerability of Raptinal in rodents, C57BL/6 mice were administered intravenous Raptinal across a range of dosages as a one-time injection. When administered intravenously at a dosage of 37.5 mg/kg, the peak plasma concentration and elimination half-life of Raptinal were 54.4 ± 0.9 μg/mL (134.5 ± 2.2 μM) and 92.1 ± 5.8 minutes, respectively (Figure S3A). Single-dose intravenous Raptinal was well tolerated across a wide dose range (15–60 mg/kg) and did not cause hematologic toxicity as assessed 7 days post-administration (Figure S3B). To investigate the potential anticancer activities exerted by Raptinal, two syngeneic subcutaneous models of aggressive cancers were then used: B16-F10 melanoma and 4T1 breast cancer. Mice were administered 20 mg/kg of Raptinal via IP injection daily for 3–4 days. In the B16-F10 model, administration of Raptinal during the first three days was sufficient to retard tumor volume and tumor mass by 60% relative to controls (Figures 3C and 3D). Similar efficacy was observed for the 4T1 murine breast cancer tumor model with 50% growth inhibition after treatment with Raptinal (Figures 3E and 3F). These results indicate that Raptinal exerts anticancer activity in vivo, likely through the induction of apoptosis, and could be useful for studying rapid induction of apoptosis in in vivo settings. Overall, the activity of Raptinal in zebrafish embryos and aggressive cancer models in mice is promising and points to a conserved cross-species mechanism and the broad applicability of Raptinal’s potent and rapid apoptosis induction.
DISCUSSION
We report the discovery of the compound Raptinal, an unusually rapid inducer of caspase-dependent apoptosis in multiple cell lines and in vivo systems. Raptinal is able to induce apoptosis more rapidly than a diverse panel of 25 other small molecule anticancer agents and biological tool compounds tested at the same concentration (Figures 1D, 1E, 1F, S1E, S1F and S1G). Raptinal-induced apoptosis challenges the widely held belief that apoptosis takes a minimum time of several hours (Goldstein et al., 2000). A rapid apoptotic phenotype can have important implications in drug development and treatment regimens as supported by recent studies suggesting the rate and extent of apoptotic induction in cell culture can be used to identify effective regimens and drug combinations for the treatment of cancer patients in the clinic (Bosserman et al., 2012; Strickland et al., 2013).
Time course analyses reveal Raptinal induces cytochrome c release and caspase activation within minutes (Figures 2A, 2B, S2A and S2B). Raptinal induces apoptosis strongly in both suspension (Figures 1F, 2A, 2B, S2A, S2C) and adherent cell lines (Figures 1G, S1G). While we found the time to cell death to be slightly slower in some adherent cell lines (Figure 1G), Raptinal was still consistently the fastest apoptosis inducing agent in these cell lines. The induction of apoptosis by Raptinal occurs through the intrinsic pathway as supported by the sensitivity of cell lines devoid of functional extrinsic pathway signaling (Jurkat FADD−/− and Casp8−/−) and rapid morphological changes in mitochondria (Figures 2D and 2E), as well as the extensive protection observed by knockdown of apaf-1 and caspase-9 (Figure 2C and S2F).
The delay in time between treatment with most apoptotic agents and initiation of apoptosis allows the influence of secondary events to confound interpretation of experimental results. For example, staurosporine is the current gold-standard compound for rapid apoptotic induction, and as such it has found wide use as a means to induce apoptosis in experiments designed to identify apoptotic regulators (Sakahira et al., 1998; Susin et al., 1999; Yang et al., 1997), in important proteomic profiling experiments (Agard et al., 2012; Dix et al., 2008; Dix et al., 2012; Shimbo et al., 2012), and as an ubiquitous control in cell death and caspase activation experiments. However, as a broad spectrum kinase inhibitor staurosporine initiates multiple events upstream of cytochrome c release, which causes staurosporine-induced apoptotic death to typically take ~6–12 hours (Figure 1F) and up to >24 h (Figure 1G), complicating the interpretation of downstream readouts. As shown herein Raptinal-induced apoptosis is markedly faster than staurosporine and induces caspase-3 activity considerably faster than staurosporine in six different cell lines in head-to-head experiments (Figures 1D, 1F, 1G, S1E, S1F, S1G). This rapid apoptotic induction limits the involvement of secondary inputs and permits the study of the intrinsic pathway in its latent condition. Consistent with this notion, inhibition of transcription or translation (by pre-treatment with actinomycin D and cycloheximide, respectively) did not prevent or delay Raptinal-induced apoptosis (Figure 2G). Therefore as a resource for cell biology, Raptinal will find use as a reagent to rapidly turn on intrinsic-pathway apoptosis while bypassing time-consuming upstream events. Used alone or in combination with other apoptotic modulators, Raptinal should also allow for precise evaluation of cellular caspase processing events. For example, data in Figure 2B suggests that Raptinal in combination with the appropriate caspase inhibitor would enable evaluating the consequences rapid caspase-9 activation without the confounding activation of caspase-8 following caspase-3 activation.
Using the rapid phenotype of Raptinal, we demonstrate the essential role of various mitochondrial functions in apoptosis induction. The link between mitochondrial processes and apoptosis has been a challenge to study due to the timing and toxicity issues related to genetic- or small molecule-based perturbations of mitochondrial functions that are otherwise essential in maintaining cell viability. Despite these challenges, studies have linked mitochondrial function to apoptosis. For example, alterations in mitochondrial respiration through genetic mutations have linked electron flux to cytochrome c release (Kwong et al., 2007; Matsuyama et al., 1998). In addition, inhibition of complex III by antimycin A and ATP-synthase activity by oligomycin A have been observed to protect against nitric oxide-induced cytochrome c release and apoptosis (Dairaku et al., 2004). Due to the rapid action of Raptinal, we were able to study this process with a variety of mitochondrial inhibitors and detail the requirement for complex III and ATP-synthase activity for apoptosis induction. We also demonstrate the additional requirement of coupled mitochondrial respiration/membrane potential, complex I and IV function and voltage-dependent anion channel function in engaging apoptosis. Inhibition of these various mitochondrial functions delays cytochrome c release and subsequent caspase activation (Figure 2H).
It is conceivable that by activating apoptosis with rapid modulators at various points along the pathway, either upstream of cytochrome c release with Raptinal, at the point of cytochrome c release with the organotin tributyltin (Nishikimi et al., 2001), or downstream by directly activating procaspase-3 with the combination of 1541B and PAC-1 (Botham et al., 2014), will permit elucidation of the myriad of apoptotic modulators yet to be identified. Raptinal is readily available (gram quantities can be produced in two synthetic steps), in contrast to the light-sensitive and expensive natural product staurosporine (ca $100 for purchase of 1 mg). Convenient access to gram quantities of Raptinal will facilitate experiments that are currently not feasible with staurosporine, such as examination of apoptotic phenotypes in whole organisms at various stages of development. The ability to rapidly induce apoptosis through the mitochondrial pathway using Raptinal will facilitate further study of known apoptotic regulators, and the identification of others; as such, Raptinal should find wide utility in both cell culture and in vivo studies as a superior alternative to staurosporine.
EXPERIMENTAL PROCEDURES
Annexin V/PI cell viability analysis
Suspension cells (U-937, SKW 6.4, or Jurkat cell lines) were plated into a 24 well plate (0.5 × 106 cells/mL). The drugs of interest were added from DMSO stocks (1% v/v final DMSO). The cells were incubated for the appropriate time after which they were centrifuged and resuspended in annexin V binding buffer containing 1 μg/mL propidium iodide (PI) and 100X dilution of FITC-annexin V (Southern Biotechnology). The samples were analyzed by cell flow cytometry on a Benton Dickinson LSRII flow cytometer. For protection assays, U-937 cells were pretreated with the prospective protective agents for 2 hours at concentrations listed in Detailed Experimental Procedures, after which the cells were then co-treated with 10 μM Raptinal for 2 hours prior to analysis.
Whole cell caspase activity assay
The caspase activity assay was conducted in 96-well plates using transfected cells (see Detailed Experimental Procedures for transfection protocol). Transfected cells were treated with 10 μM Raptinal for 1 hour, which corresponded to 40% of maximal caspase-3/-7 activity achievable by Raptinal. After treatment, the media was replaced with PBS (150 μL) and 50 μL of freshly made 4X caspase assay buffer (containing 20 μM Ac-DEVD-AFC) (Stennicke and Salvesen, 1997). After one hour incubation at room temperature in the dark, the fluorescence of cleaved AFC was read on a spectrofluorometer (Ex: 380nm, Em: 500nm). The results were expressed as the relative caspase-3/-7 activity and normalized by designating the vehicle (0.5% DMSO) as 0% activity and Raptinal treatment as 100% activity.
Apoptosis in zebrafish
Zebrafish experiments were performed as described (van Ham et al., 2010). TuAB zebrafish embryos were used for all experiments and were incubated at 28 °C in HEPES-buffered E3 zebrafish medium (pH 7.2) in the dark. Chemical treatments (10 μM Raptinal or vehicle, 1% v/v final DMSO) were performed in multi-well plates in buffered E3 medium. Images were taken using an AxioCam MRc camera and AxioVision 4.8 software (Zeiss). To quantify apoptosis, the number of secA5-YFP+ apoptotic cells was counted manually in embryos using a fluorescence dissection stereomicroscope (Discovery V8; Zeiss, Germany).
Syngeneic tumor models
All animal studies were performed with prior approval by the University of Illinois at Urbana-Champaign IACUC committee. B16-F10 cells in HBSS (100 μL of 1 × 107 cells/mL) or 4T1 murine breast cancer cells in HBSS (100 μL of 1 × 107 cells/mL) were injected subcutaneously into the right flank of shaved and sedated C57BL/6 and BALB/c female mice (6–8 weeks old) for B16-F10 and 4T1 models, respectively. Seven days (for B16-F10) and six days (for 4T1) after inoculation, the mice were randomized based upon tumor size with 7 mice per group. Vehicle or compound was administered intraperitoneally as a corn oil DMSO suspension (300 μL of 10% DMSO in trans fat-free corn oil, 20 mg/kg) once daily for 3 consecutive days for B16-F10 and 4 consecutive days for 4T1 models. Tumor measurements were performed every other day using a caliper and tumor volume was calculated using the equation (0.5 × lx w2).
Other methods
For compound synthesis procedures, compound characterization, high throughput toxicity screen assay, immunoblotting procedures, microscopy techniques, reactive-oxygen species assay, cytochrome c release assays, mitochondrial permeability transition pore (MPTP) assay, mitochondrial membrane potential assay, transfection protocol and pharmacokinetic/toxicity profiling, see Detailed Experimental Procedures.
Supplementary Material
Acknowledgments
We would like to thank the University of Illinois at Urbana-Champaign flow cytometry core facility, the W.M. Keck Center for comparative and functional genomics systems biology core, the Roy J. Carver Biotechnology Center for LC/MS analysis, and the electron microscopy facility at the college of veterinary medicine. R.C.B. is a National Science Foundation predoctoral fellow and a Robert C. and Carolyn J. Springborn graduate fellow. M.J.L and R.C.B are members of the NIH Chemistry-Biology Interface Training Grant (NRSA 1-T32-GM070421). This work was supported by the University of Illinois, the National Institutes of Health (R01-CA120439 to P.J.H.) and (NS079201 to R. T. P.) and the Charles and Ann Sanders MGH Scholar Award (to R.T.P.).
Footnotes
AUTHOR CONTRIBUTIONS
R.P. conceived the study, designed and conducted the experiments, analyzed the data, and wrote the manuscript; M.J.L., R.C.B., and K.C.P. designed and conducted the experiments, analyzed the data and wrote the manuscript; T.J.V.H., K.S.P., R.T.P. and T.M.F. designed and conducted the experiments and analyzed the data; L.T.N. and S.H.K conducted the experiments; R.T.P and T.M.F. reviewed the manuscript; P.J.H conceived the study, designed the experiments, analyzed the data, and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agard NJ, Mahrus S, Trinidad JC, Lynn A, Burlingame AL, Wells JA. Global kinetic analysis of proteolysis via quantitative targeted proteomics. Proc Natl Acad Sci U S A. 2012;109:1913–1918. doi: 10.1073/pnas.1117158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bair JS, Palchaudhuri R, Hergenrother PJ. Chemistry and biology of deoxynyboquinone, a potent inducer of cancer cell death. J Am Chem Soc. 2010;132:5469–5478. doi: 10.1021/ja100610m. [DOI] [PubMed] [Google Scholar]
- Balasubramanian K, Mirnikjoo B, Schroit AJ. Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J Biol Chem. 2007;282:18357–18364. doi: 10.1074/jbc.M700202200. [DOI] [PubMed] [Google Scholar]
- Bosserman L, Prendergast F, Herbst R, Fleisher M, Salom E, Strickland S, Raptis A, Hallquist A, Perree M, Rajurkar S, et al. The microculture-kinetic (MiCK) assay: the role of a drug-induced apoptosis assay in drug development and clinical care. Cancer Res. 2012;72:3901–3905. doi: 10.1158/0008-5472.CAN-12-0681. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. Embo J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botham RC, Fan TM, Im I, Borst LB, Dirikolu L, Hergenrother PJ. Dual small-molecule targeting of procaspase-3 dramatically enhances zymogen activation and anticancer activity. J Am Chem Soc. 2014;136:1312–1319. doi: 10.1021/ja4124303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Freeman A, Liu J, Dai Q, Lee RM. The apoptotic effect of HA14-1, a Bcl-2-interacting small molecular compound, requires Bax translocation and is enhanced by PK11195. Mol Cancer Ther. 2002;1:961–967. [PubMed] [Google Scholar]
- Curtin DY, Kampmeie Ja, Farmer ML. Nitrosation Reactions of Primary Vinylamines. 3-Amino-2-Phenylindenone. J Am Chem Soc. 1965;87:874. [Google Scholar]
- Dairaku N, Kato K, Honda K, Koike T, Iijima K, Imatani A, Sekine H, Ohara S, Matsui H, Shimosegawa T. Oligomycin and antimycin A prevent nitric oxide-induced apoptosis by blocking cytochrome C leakage. J Lab Clin Med. 2004;143:143–151. doi: 10.1016/j.lab.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Dix MM, Simon GM, Cravatt BF. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell. 2008;134:679–691. doi: 10.1016/j.cell.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix MM, Simon GM, Wang C, Okerberg E, Patricelli MP, Cravatt BF. Functional interplay between caspase cleavage and phosphorylation sculpts the apoptotic proteome. Cell. 2012;150:426–440. doi: 10.1016/j.cell.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudgeon C, Qiu W, Sun QH, Zhang L, Yu J. Transcriptional Regulation of Apoptosis. Essentials of Apoptosis. (2) 2009:239–260. [Google Scholar]
- Ferrer I. Apoptosis: future targets for neuroprotective strategies. Cerebrovasc Dis. 2006;21(Suppl 2):9–20. doi: 10.1159/000091699. [DOI] [PubMed] [Google Scholar]
- Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030–9040. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- Fulda S. Inhibitor of apoptosis proteins as targets for anticancer therapy. Expert Rev Anticancer Ther. 2007;7:1255–1264. doi: 10.1586/14737140.7.9.1255. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillick K, Crompton M. Evaluating cytochrome c diffusion in the intermembrane spaces of mitochondria during cytochrome c release. J Cell Sci. 2008;121:618–626. doi: 10.1242/jcs.021303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JC, Kluck RM, Green DR. A single cell analysis of apoptosis. Ordering the apoptotic phenotype. Ann N Y Acad Sci. 2000;926:132–141. doi: 10.1111/j.1749-6632.2000.tb05607.x. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Munoz-Pinedo C, Ricci JE, Adams SR, Kelekar A, Schuler M, Tsien RY, Green DR. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005;12:453–462. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- Hamada H, Tashima Y, Kisaka Y, Iwamoto K, Hanai T, Eguchi Y, Okamoto M. Sophisticated Framework between Cell Cycle Arrest and Apoptosis Induction Based on p53 Dynamics. PLoS ONE. 2009;4:e4795. doi: 10.1371/journal.pone.0004795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juo P, Kuo CJ, Yuan J, Blenis J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Current Biology. 1998;8:1001–1008. doi: 10.1016/s0960-9822(07)00420-4. [DOI] [PubMed] [Google Scholar]
- Keinan N, Tyomkin D, Shoshan-Barmatz V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol Cell Biol. 2010;30:5698–5709. doi: 10.1128/MCB.00165-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Henning MS, Starkov AA, Manfredi G. The mitochondrial respiratory chain is a modulator of apoptosis. J Cell Biol. 2007;179:1163–1177. doi: 10.1083/jcb.200704059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Bai L, Sun H, Nikolovska-Coleska Z, McEachern D, Qiu S, Miller RS, Yi H, Shangary S, Sun Y, et al. SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res. 2008;68:9384–9393. doi: 10.1158/0008-5472.CAN-08-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luetjens CM, Kogel D, Reimertz C, Dussmann H, Renz A, Schulze-Osthoff K, Nieminen AL, Poppe M, Prehn JH. Multiple kinetics of mitochondrial cytochrome c release in drug-induced apoptosis. Mol Pharm. 2001;60:1008–1019. doi: 10.1124/mol.60.5.1008. [DOI] [PubMed] [Google Scholar]
- Luo KQ, Yu VC, Pu Y, Chang DC. Application of the fluorescence resonance energy transfer method for studying the dynamics of caspase-3 activation during UV-induced apoptosis in living HeLa cells. Biochem Biophys Res Commun. 2001;283:1054–1060. doi: 10.1006/bbrc.2001.4896. [DOI] [PubMed] [Google Scholar]
- Matsuyama S, Xu Q, Velours J, Reed JC. The Mitochondrial F0F1-ATPase proton pump is required for function of the proapoptotic protein Bax in yeast and mammalian cells. Mol Cell. 1998;1:327–336. doi: 10.1016/s1097-2765(00)80033-7. [DOI] [PubMed] [Google Scholar]
- Narula J, Haider N, Arbustini E, Chandrashekhar Y. Mechanisms of disease: apoptosis in heart failure--seeing hope in death. Nat Clin Pract Cardiovasc Med. 2006;3:681–688. doi: 10.1038/ncpcardio0710. [DOI] [PubMed] [Google Scholar]
- Nguyen JT, Wells JA. Direct activation of the apoptosis machinery as a mechanism to target cancer cells. Proc Natl Acad Sci U S A. 2003;100:7533–7538. doi: 10.1073/pnas.1031631100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikimi A, Kira Y, Kasahara E, Sato EF, Kanno T, Utsumi K, Inoue M. Tributyltin interacts with mitochondria and induces cytochrome c release. Biochem J. 2001;356:621–626. doi: 10.1042/0264-6021:3560621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CL, Miranda MB, Shangary S, Land S, Wang S, Johnson DE. (−)-Gossypol acts directly on the mitochondria to overcome Bcl-2- and Bcl-X(L)-mediated apoptosis resistance. Mol Cancer Ther. 2005;4:23–31. [PubMed] [Google Scholar]
- Putt KS, Chen GW, Pearson JM, Sandhorst JS, Hoagland MS, Kwon JT, Hwang SK, Jin H, Churchwell MI, Cho MH, et al. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat Chem Biol. 2006;2:543–550. doi: 10.1038/nchembio814. [DOI] [PubMed] [Google Scholar]
- Rehm M, Dussmann H, Janicke RU, Tavare JM, Kogel D, Prehn JHM. Single-cell fluorescence resonance energy transfer analysis demonstrates that caspase activation during apoptosis is a rapid process - Role of caspase-3. J Biol Chem. 2002;277:24506–24514. doi: 10.1074/jbc.M110789200. [DOI] [PubMed] [Google Scholar]
- Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S, et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346:1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- Shimbo K, Hsu GW, Nguyen H, Mahrus S, Trinidad JC, Burlingame AL, Wells JA. Quantitative profiling of caspase-cleaved substrates reveals different drug-induced and cell-type patterns in apoptosis. Proc Natl Acad Sci U S A. 2012;109:12432–12437. doi: 10.1073/pnas.1208616109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Keinan N, Abu-Hamad S, Tyomkin D, Aram L. Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochim Biophys Acta. 2010;1797:1281–1291. doi: 10.1016/j.bbabio.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Stennicke HR, Salvesen GS. Biochemical characteristics of caspases-3, -6, -7, and -8. J Biol Chem. 1997;272:25719–25723. doi: 10.1074/jbc.272.41.25719. [DOI] [PubMed] [Google Scholar]
- Strickland SA, Raptis A, Hallquist A, Rutledge J, Chernick M, Perree M, Talbott MS, Presant CA. Correlation of the microculture-kinetic drug-induced apoptosis assay with patient outcomes in initial treatment of adult acute myelocytic leukemia. Leuk Lymphoma. 2013;54:528–534. doi: 10.3109/10428194.2012.722217. [DOI] [PubMed] [Google Scholar]
- Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prevost MC, et al. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–579. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- van Ham TJ, Mapes J, Kokel D, Peterson RT. Live imaging of apoptotic cells in zebrafish. Faseb J. 2010;24:4336–4342. doi: 10.1096/fj.10-161018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolan DW, Zorn JA, Gray DC, Wells JA. Small-Molecule Activators of a Proenzyme. Science. 2009;326:853–858. doi: 10.1126/science.1177585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpaw AJ, Shimada K, Skouta R, Welsch ME, Akavia UD, Pe’er D, Shaik F, Bulinski JC, Stockwell BR. Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proc Natl Acad Sci U S A. 2011;108:E771–780. doi: 10.1073/pnas.1106149108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.