

Abstract
Although conversion of the cellular form of the prion protein (PrPC) into a misfolded isoform is the underlying cause of prion diseases, understanding PrPC physiological functions has remained challenging. PrPC depletion or overexpression alters the proliferation and differentiation properties of various types of stem and progenitor cells in vitro by unknown mechanisms. Such involvement remains uncertain in vivo in the absence of any drastic phenotype of mice lacking PrPC. Here, we report PrPC enrichment at the base of the primary cilium in stem and progenitor cells from the central nervous system and cardiovascular system of developing mouse embryos. PrPC depletion in a neuroepithelial cell line dramatically altered key cilium-dependent processes, such as Sonic hedgehog signalling and α-tubulin post-translational modifications. These processes were also affected over a limited time window in PrPC–ablated embryos. Thus, our study reveals PrPC as a potential actor in the developmental regulation of microtubule dynamics and ciliary functions.
Prions are proteinaceous infectious agents responsible for a broad range of fatal neurodegenerative diseases in animals and humans. They are primarily composed of macromolecular assemblies of PrPSc, a misfolded isoform of the host-encoded PrPC. Whether prion toxicity in the central nervous system (CNS) is linked to the generation of toxic PrPSc (sub)species, to PrPC gain of toxic functions or to the activation of generic toxic pathways1 remains a fiercely debated issue, substantially due to the elusive physiological functions of PrPC2. Unravelling the functions of PrPC may have a broader significance given its increasingly apparent role in mediating toxic signalling associated with more common neurodegenerative diseases such as Alzheimer’s disease3. Moreover, PrPC possesses highly conserved primary and tertiary structures among mammals, and the presence of genes homologous to Prnp (the gene encoding PrPC) in birds, reptiles, amphibians and fish lends support for evolutionarily conserved functions2.
PrPC is a ubiquitously expressed, glycosylphosphatidylinositol-anchored cell surface sialoglycoprotein that is present in specific membrane domains termed lipid rafts, which are critical to the biology of the cell4. PrPC is involved in a variety of cytoprotective cellular functions and signal transduction pathways5. In the last decade, studies of cell systems depleted in or overexpressing PrPC have linked PrPC to the self-renewal of haematopoietic6 and embryonic7,8 stem cells and to the proliferation and/or differentiation of embryonic stem cells9 and neural progenitors10. However, our knowledge of the underlying cellular mechanisms remains limited. Further complicating the issue is the absence of any drastic phenotype in adult mice upon embryonic11 or post-natal12 inactivation of Prnp, although transient alterations in cell differentiation were later identified in these animals10,13. The upregulation of Prnp expression can be detected as early as embryonic day (E) 6.5 in extra-embryonic regions and E8 in the embryo proper14,15,16, thus supporting a developmental role for PrPC. However, the embryonic expression pattern of PrPC is poorly documented; its presence in neural progenitors can be questioned, with some studies reporting initial expression during differentiation10,16.
To characterize how the PrPC expression pattern is specified relative to stem/progenitor cell fate in vivo, we examined the spatiotemporal distribution of PrPC in early developing wild-type mouse embryos. From E8.25 onwards, we found that PrPC was enriched at the base of the primary cilium in stem cells and progenitors of the CNS, heart and forming blood vessels. In the mouse developing neural tube and even more markedly in a neuroepithelial cell line displaying neuroectodermal progenitor hallmarks, the depletion of PrPC altered key cilium-dependent processes and pathways, including α-tubulin post-translational modifications (PTMs) and the Sonic hedgehog signalling pathway. Collectively, these data reveal a new link between PrPC and microtubule dynamics as well as primary cilium functions during development.
Results
PrPC is expressed in stem and progenitor cells from the developing central nervous system and cardiovascular system
We examined the temporal and spatial PrPC expression pattern during mouse embryonic development by immunofluorescence microscopy after labelling with the anti-PrP antibody Sha31, which exhibits high affinity for both full-length and truncated isoforms of PrP17. Prnp−/− embryos were used as negative controls to check for any non-specific staining (e.g., Fig. S1g,h). Representative images are shown in Figs 1 and S1.
Figure 1. PrPC expression pattern in the nervous and cardiovascular systems from early developing mouse embryos.

PrPC (red) and nuclear marker 4’,6-diamidino-2-phenylindole (DAPI, blue) staining of transverse sections from FVB/N mouse embryos at E9 in the developing nervous (a–c) and cardiovascular systems (d,e). Section plans are indicated (f). PrPC was observed at the level of the apical face of the neuroepithelium in the head region (presumptive telencephalon; a), in the neural tube (NT) in the trunk region (b), and particularly in the floor plate (FP, c). The arrowhead indicates PrPC-positive differentiating cells in the mantle zone (c). Low-exposure acquisition of two juxtaposed embryo sections from the proximal and caudal trunk regions showed strong PrPC expression in the omphalomesenteric artery (OA) and in the embryonic heart ventricle (HtV) (d). Higher magnification of the omphalomesenteric artery (OA) (e). The asterisks indicate artefact signals found in FVB/N and in Prnp−/− mice (supplemental Figure S1), likely due to nonspecific Fc binding to maternal blood, and the arrowheads highlight specific punctuate or patchy PrPC signals. Scale bar: 10 μm, except in (b,d) 50 μm. DA: dorsal aorta, Hg: hindgut, L: lumen, MDA: midline dorsal aorta, som: somite.
At E8.25, in the embryo proper, PrPC was readily detected in the developing forebrain (optic pit region) as puncta at the apical face of the neuroepithelium (Fig. S1a) and as patches in endothelial cells of the forming dorsal aorta (Fig. S1b). PrPC expression was stronger in the developing heart, and the signal appeared mostly as a patchy pattern (Fig. S1c). More caudally, PrPC expression was observed neither in the neural groove (Fig. S1b) nor in the neural tube (Fig. S1e). PrPC patches were also detected in extra-embryonic regions such as the splanchnic mesoderm of the yolk sac (Fig. S1f). This region contains blood islands where primitive haematopoiesis18 and vasculogenesis19 occur.
At E9, PrPC expression was greater in the CNS (Figs 1a–c and S1j) and cardiovascular system (Figs 1d–f and S1k–m). PrPC puncta were abundant in the entire presumptive brain at the ventricular surface of the neuroepithelium (Fig. 1a) and in the floor plate in the trunk region, an essential organizing centre for the developing CNS (Fig. 1b,c). A fainter signal was observed at the apical face in other regions of the neural tube (Fig. S1j). PrPC patches and puncta together with membrane staining were observed in the mantle zone, where the first differentiating cells migrate (Fig. 1c). In the cardiovascular system, the strongest PrPC signals were observed in the omphalomesenteric artery (Fig. 1d–e), a site of definitive haematopoiesis20, and in the developing heart (Figs 1d and S1k–l). The venous pole of the heart, which corresponds at this early developmental stage to conduits draining to the inlet of the heart tube21, displayed PrPC membrane staining and intense puncta (Fig. S1k–l). In the omphalomesenteric artery, patches were present on endothelial cells (Fig. 1e). Other vessels, such as the dorsal aorta, also expressed PrPC in the endothelium, albeit at lower levels (Fig. S1m and data not shown). PrPC also remained expressed in the extra-embryonic yolk sac at E9 (not shown).
There was no drastic change in the regional expression pattern of PrPC between E9 and E10.5. Nonetheless, more intense labelling was observed in the neural tube mantle zone as the number of differentiating cells increased (data not shown).
Collectively, these data indicate that PrPC is expressed during early embryonic stages in neuroepithelial cells, in cells from a stem cell niche associated with definitive haematopoiesis and in progenitors of the heart and blood vessels.
PrPC localizes at the base of the primary cilium
Using confocal imaging, we next sought to define the subcellular localization of the PrPC patches and puncta. The primary cilium is a microtubule-based versatile organelle composed of an axoneme extending from the mother centriole-derived basal body and surrounded by a ciliary membrane. Sections of E9-E9.5 FVB/N embryos were co-labelled with anti-PrP and either anti-acetylated tubulin (TubAc, Figs 2a, S2a–c and S3) or anti-γ-tubulin (γ-tub, Figs 2b–c, S2d–f and S3) antibodies as ciliary axoneme and centriole markers22, respectively. In endothelial cells from the omphalomesenteric artery (Fig. 2a,c, Fig. S3), in capillaries of the yolk sac (Fig. S2a), in cells from the venous pole (Fig. S2b,d) and in cardiomyocytes (Fig. S2e), PrPC patches and puncta were identified as punctiform structures at the ciliary base. PrPC was not detected along the axoneme. Quantitative analyses of the z-series showed that 90 ± 9% and 91 ± 10% of TubAc-positive primary cilia had positive PrPC signals at their base in the omphalomesenteric artery and capillaries of the yolk sac. Conversely 85 ± 8% and 83 ± 12% of PrPC patches localized at the ciliary base (n = 7 and n = 8 z-series analysed from n = 3 embryos, respectively). TubAc staining was not restricted spatially to the cilium in neuroepithelial cells, but the PrPC and TubAc co-labelling was compatible with the presence of PrPC at the ciliary base (Fig. S2c), as also suggested by the close association or co-localization of PrPC with γ-tub (Fig. 2b, Fig. S3). Quantitatively, 82 ± 6% of γ-tub signals in the floor plate co-localized with PrPC signals. Conversely 98 ± 4% of PrPC puncta co-localized with γ-tub signals (n = 5 z-series analysed from n = 3 embryos).
Figure 2. PrPC localization at the base of the primary cilium in progenitor cells of the developing nervous system and in the omphalomesenteric artery.

Confocal microscopy imaging of transverse sections of FVB/N mouse embryos at E9-9.5 co-stained for PrP (red) and (green) acetylated tubulin (TubAc) (a), γ-tubulin (γTub) (b,c) or Rab11 (d–f). Nuclei are stained with DAPI (blue). Merged confocal images or individual channels are shown. Z-series (a,b) or single optical sections (c–f) are presented. Co-localization of PrPC with TubAc at the level of the omphalomesenteric artery (a). The arrowhead highlights PrPC-positive punctiform structures at z = 1. Along the z-axis, a primary cilium, as probed by TubAc, progressively appears (from z = 3), while the PrPC signal progressively disappears. White arrows point to the centrosomes of a mitotic cell and highlight the absence of detectable PrPC. Co-localization of PrPC with γTub (arrowheads) at the apical face of the neuroepithelium (forebrain; b) and in cells from the omphalomesenteric artery (c). Co-localization (arrowheads) or co-regionalization (plain arrowheads) between PrPC and Rab11 at the apical face of the neuroepithelium in the forebrain (d), at the floor plate of the neural tube in the trunk region (e), and at the omphalomesenteric artery (f). The dotted lines delimit the lumen. Scale bar: 5 μm.
In differentiated cells from the mantle region of the neural tube, PrPC still co-localized with γ-tub, despite exhibiting wider expression (Fig. S2f). Importantly, the PrPC punctiform structures never co-localized with TubAc (Fig. 2a) or γ-tub (Fig. S2d) positive centrioles from the mitotic spindles, further reinforcing the view that at this developmental stage, PrPC was specifically enriched at the base of the primary cilium in a wide range of stem cells (or cells associated with stem cell niches) and progenitors.
Considering the localization of PrPC at the ciliary base and the intense trafficking of Golgi and endocytic vesicles there to insure proper ciliogenesis23, we next examined whether PrPC would co-localize with the recycling endosome marker Rab11. Rab11 is enriched at the ciliary base and is required for primary ciliogenesis24. A large proportion of PrPC co-localized with Rab11 or was present in its immediate vicinity in neuroepithelial cells (Fig. 2d–e), in the omphalomesenteric artery (Fig. 2f), in the yolk sac (Fig. S2g) and in the developing heart (Fig. S2h–i). In marked contrast, the most prominent PrPC-positive structures in differentiating cells from the mantle zone of the neural tube did not associate with Rab11 (Fig. S2j). Altogether, these data suggest that PrPC is specifically enriched in the pericentriolar recycling compartment of the primary cilium from progenitors and cells associated with stem cell niches of the developing central nervous and cardiovascular systems.
PrPC depletion affects Sonic hedgehog signalling
To examine the potential impact of PrPC depletion on primary cilium biology in stem cells/progenitors, we first used a neuroepithelial cell line that displays neuroectodermal progenitor hallmarks and has been instrumental in identifying PrPC-dependent signalling cascades25,26. PrPC can be constitutively knocked down in 1C11 cells by an anti-Prnp shRNA, resulting in a >95% decrease in PrPC levels27. These cells are referred to as PrPnull-1C11 cells. TubAc labelling of 1C11 versus PrPnull-1C11 cells revealed that the proportion of ciliated cells markedly increased by 3-fold (Fig. 3a), suggesting altered primary cilium turnover22 in the absence of PrPC. Primary cilia are essential for the transduction of the Sonic hedgehog (Shh) morphogen signal, a key signal in the developmental regulation pathways of multicellular organisms28. Cilium abnormalities are usually coupled to Shh signalling pathway alterations22,29, affecting the expression of effectors of the Shh pathway such as the Gli transcription factors and the Patched1 (Ptc1) Shh receptor. Our qPCR analyses of mRNA isolated from 1C11 and PrPnull-1C11 cells revealed that Gli1 and Gli2 expression levels were downregulated by >5-fold and 95-fold, respectively, in the absence of PrPC, whereas Gli3 levels were reduced by 20% (Fig. 3b). By contrast, Ptc1 transcripts were upregulated by >3-fold in the absence of PrPC (Fig. 3b). The Shh-induced transcription factor and Gli2-regulated FoxA230 showed nearly total transcriptional abolition (Fig. 3b). The increase in Ptc1 transcripts in PrPnull-1C11 cells, which stands in apparent contradiction to Gli1 downregulation31, may be readily explained by the absence of FoxA2, which negatively regulates the transcription of Ptc132. Additionally, mRNA expression of the transcription factor FoxO6, a recently identified Gli target31, was significantly lowered by 50% in PrPnull-1C11 cells (Fig. 3b). Finally, PrPC depletion had no impact on the mRNA levels of Smoothened (Smo, Fig. 3b), a Shh pathway signal transducer, arguing against a global, unspecific effect of PrPC depletion on the Shh pathway.
Figure 3. Primary cilium biology in PrPC-depleted 1C11 cells.

1C11 and PrPC-depleted cells (PrPnull-1C11) were compared with regard to the percentage of ciliary cells (a, n = 30 fields of view (~700 cells)). qPCR results showing the expression of Shh-related transcription factors, the Shh receptor Patched 1 (Ptc1) and Smoothened signal transducer transmembrane protein (Smo) are presented ((b), (n = 3)). The mitotic index ((c), n = 19 fields of view analysed per cell type, analysis on 3000–6000 cells), cell proliferation ((d), n = 3 MTT), proportion of cells in the different phases of the cell cycle ((e), n = 3), protein and transcriptional levels of Cyclin D1 ((f,g) and an immunoblot using Erk to normalize the protein levels ((f) n = 3) are shown. (*p < 0.05, Student test (a) or Mann-Whitney test (b–g)). The gels shown in panel (f) have been cropped for clarity and conciseness purposes, and have been run under the same experimental conditions; the original blots are shown in supplemental Figure S8a.
As Shh signalling controls the cycling of neural precursor cells33, we next investigated whether this process was impacted. No significant difference in the mitotic index was observed between 1C11 and PrPnull-1C11 cells (Fig. 3c). However, the mitotic index may not correlate with the proliferation rate as the duration of the M phase could vary between the two cell populations. We thus refined our cell proliferation studies by using MTT cell proliferation assay and cell cycle analyses34. PrPnull-1C11 cell proliferation was significantly decreased by >50%, as quantified by the MTT assay (Fig. 3d), and the proportion of cells in the G0/G1 and S phases was slightly but significantly increased (Fig. 3e). In line with the lack of difference in the mitotic index between 1C11 cells and PrPnull-1C11 cells, the proportion of PrPnull-1C11 cells in G2/M phases was not affected (Fig. 3e). In addition, qPCR and immunoblot analyses showed decreased transcriptional and protein levels of Cyclin D1 (Fig. 3f,g), a regulator of the entry into S phase35. Collectively, these data suggest that PrPC depletion affects 1C11 cell cycle regulation.
We next performed similar analyses in the embryo neuroepithelium, where primary cilia functions have been well documented29. Although no gross ultrastructural abnormalities (Fig. S4) or quantitative differences in the number of ciliary basal bodies were observed in the floor plate region between FVB/N and Prnp−/− embryos at E9 or E9.5 (Fig. 4A; Table S1), PrPC ablation exerted a recurrent impact on the expression of a number of Shh-related genes in neural tube-enriched preparations from E9 to E10.5 (Fig. 4b). Thus, Gli1 and Gli3 mRNA levels were slightly but significantly decreased by 20% and 30% at E10.5 and E9.5, respectively. In line with the in vitro data, Ptc1 was upregulated by 20%, and FoxO6 mRNA levels were significantly reduced by 30–40% throughout the time window of analysis. Finally, FoxJ1 expression, another forkhead family transcription factor upregulated by Shh signalling, was also decreased by up to 40–50% at E9 and E9.5.
Figure 4. Dorsoventral patterning and Shh-related transcripts in the neural tube of PrPC-ablated embryos.
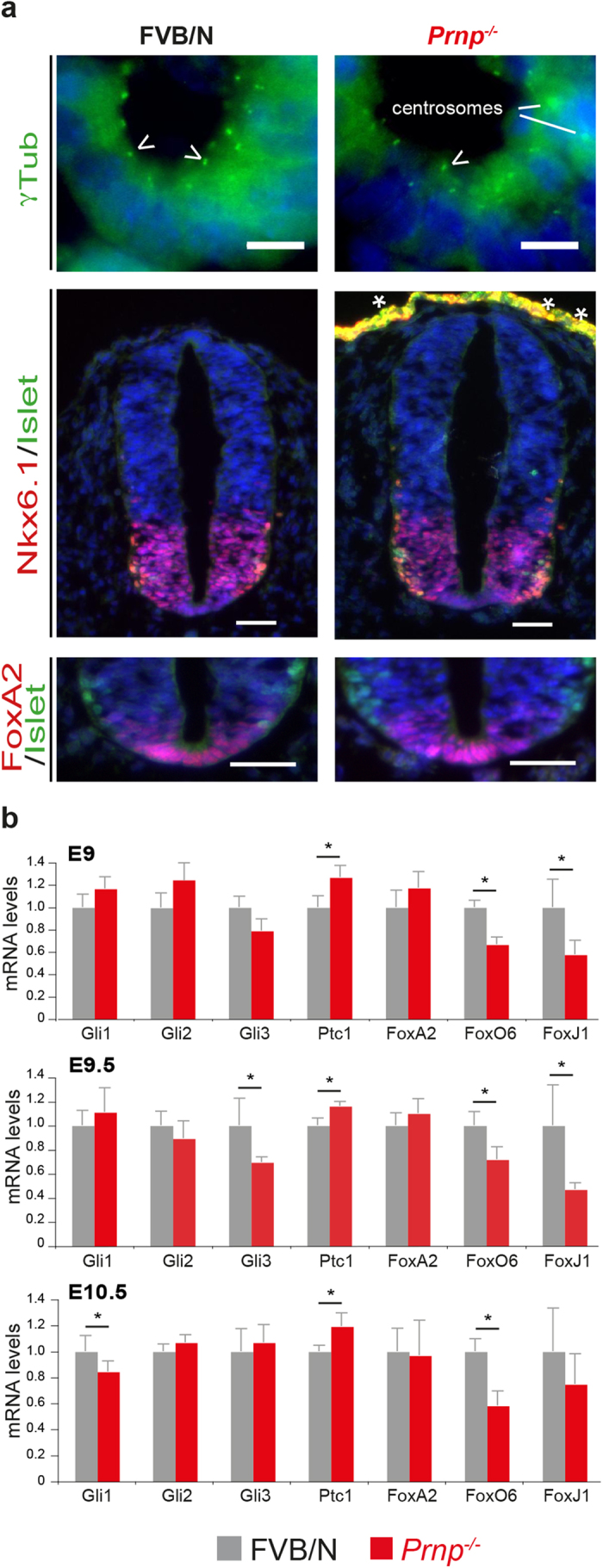
Immunofluorescence analysis of transverse sections from FVB/N and Prnp−/− E9 mouse embryos at the level of the mid-trunk region (a, representative images of n = 3 embryos analysed). Nuclei are stained with DAPI (blue). Arrowheads point to basal bodies. The asterisks indicate artefact signals. Scale bar: 50 μm, except for γTub staining, 10 μm. qPCR results showing the expression of Shh-related transcription factors and the Shh receptor Patched 1 (Ptc1) in FVB/N and Prnp−/− neural tube-enriched preparations from E9 to E10.5 are shown (b, n ≥ 5; *p < 0.05).
Primary cilia and Shh signalling are critical for proper patterning of both the neural tube along the dorsoventral axis28 and the heart36. These were negligibly affected in Prnp−/− E9 embryos, as shown by the conserved expression patterns of FoxA2 and Nkx6.1, another Shh pathway-dependent transcription factor (Fig. 4a and data not shown). Consistently, PrPC ablation had no significant effects on FoxA2 mRNA levels over the E9–E10.5 period (Fig. 4b). The number of Islet-positive cells (an early Shh-dependent marker of differentiation) in the neural tube was also stable between the two genotypes at E9 (Fig. 4a, Table S1).
Regarding cell cycling, the mitotic indexes in the neural tubes of FVB/N and Prnp−/− E9 embryos were similar (Table S1), and the cyclin D1 mRNA levels were conserved in FVB/N and Prnp−/− neural tube-enriched preparations from E9 to E10.5 (Fig. S5).
Collectively, these data indicate that PrPC depletion was detrimental to signalling functions related to primary cilium biology: in the 1C11 neuroepithelial cell line, the Shh signal transduction pathway was nearly switched off; in the developing mouse neural tube, the modest but consistent modulation of the transcript levels of effectors/regulators of the Shh pathway was accompanied by a robust decrease in the Shh targets FoxO6 and FoxJ1.
PrPC depletion affects the levels of α-tubulin variants
The ciliary basal body is a microtubule organizing centre37 and the fine-tuning of cilium-dependent signalling pathways and regulation of microtubule dynamics are related processes38,39. Numerous subtypes of microtubules can be generated by tubulin PTMs, including acetylation, detyrosination, polyglutamylation and polyglycylation40. Tubulin PTMs involve a broad range of tightly regulated enzymes that are spatially and temporally key to microtubule assembly, stability and functions40 and thus emerge as key players in development. Detyrosinated α-tubulin (detyr-tub) is involved in proper neuronal organization41. The Δ2 variant, which is generated from detyr-tub by cytosolic carboxypeptidase cleavage42 (Fig. S6), is specifically enriched in neuronal cells and is linked to microtubule stability43. We thus assessed the impact of PrPC depletion on the levels of total α-tubulin (α-tub), TubAc, detyr-tub, and Δ2 in the embryo proper (E8.25 and E8.5), in neural tube-enriched preparations (E9 to E10.5 embryos) and in 1C11 cells (Fig. 5). The numbers of litters and of embryos per litter analysed are indicated in the supplementary experimental procedures. Although the levels of α-tub did not exhibit significant variations, the levels of the TubAc, detyr-tub and Δ2 variants varied markedly between PrPC-expressing and PrPC-depleted groups both in vivo (from E9 onwards) and in vitro. In vivo, despite noticeable individual variations, there was a transient and significant decrease in the mean levels of TubAc and Δ2 in Prnp−/− samples at E9 and E9.5, compared with wild-type counterparts (Fig. 5c,d). At E9, the values were lowered by 3- to 4-fold. The levels of Δ2 in Prnp−/− samples were still decreased at E10.5 (Fig. 5e). At E9 (Fig. 5c) and E10.5 (Fig. 5e), the levels of detyr-tub were also modestly but significantly decreased in Prnp−/− versus FVB/N neural tube-enriched embryos. In 1C11 cells, the mean levels of the three analysed α-tubulin variants were also affected by PrPC depletion (Fig. 5f). However, unlike the neural tube-enriched preparations, those levels were significantly increased by 2–3 fold.
Figure 5. Analysis of α-tubulin post-translational modifications in developing embryos and 1C11 cells lacking PrPC.

Immunoblot and densitometric quantification of total α-tubulin (α-tub), TubAc, detyr-tub and Δ2 variants in FVB/N embryos proper at E8.25 (a) and E8.5 (b), in neural tube-enriched preparations from embryos at E9 (c), E9.5 (d) and E10.5 (e) as well as in the 1C11 cell line (f) in the presence or absence of PrPC (*p < 0.05). Immunoblot of protein extracts prepared from individual embryos or cell lysate are shown. The gels shown have been cropped for clarity and conciseness purposes, and have been run under the same experimental conditions; the original blots are shown in supplemental Figures S9 to S12.
Collectively, these data indicate that PrPC depletion affects the steady-state levels of α-tubulin variants.
Blockade of primary cilium resorption does not reproduce a PrPnull-like phenotype
Upon PrPC depletion, the three α-tubulin variants were downregulated in neural tubes yet upregulated in 1C11 cells. The increased proportion of ciliated cells (Fig. 3a) in the PrPnull-1C11 population (which may suggest a defect in primary cilium disassembly22) may contribute to the increase in tubulin variants44, to the diminished proliferation rate and possibly to alterations of the Shh pathway due to the coordinated/intertwined nature of these processes28,45. To investigate this possibility, we artificially maintained 1C11 primary cilia in a PrP-expressing context by treatment with tubacin. Tubacin is a selective inhibitor of histone deacetylase 6 (HDAC6)46,47, the predominant enzyme that deacetylates tubulin, a process necessary to deciliation45. As negative control, 1C11 cells were cultured in the presence of niltubacin, an inactive congener of tubacin. As previously reported47, tubacin treatment increased TubAc levels by >30-fold compared to vehicle or niltubacin treatment (Fig. 6a). The detyr-tub and Δ2 variant levels were significantly decreased by ~2-fold and remained constant, respectively (Fig. 6a), an outcome contrasting with observations in PrPnull-1C11 cells (Fig. 5f). We next used qPCR to compare the levels of Gli1, Gli2 and Cyclin D1 transcripts in 1C11 cells cultured in the presence of tubacin or niltubacin. Gli2 and Cyclin D1 expression was reduced by 2-fold, whereas Gli1 expression doubled in tubacin-treated cells (Fig. 6b). Thus, although tubacin affects Cyclin D1 expression similarly to PrPC depletion, this treatment does not recapitulate the entire set of alterations of the Shh signalling pathway observed in PrPnull-1C11 cells. These results suggest that PrPC depletion, rather than impaired cilia resorption, is primarily driving the changes in tubulin PTM and Shh signalling observed in PrPnull-1C11 cells. PrPC depletion thus truly leads to different outcomes in cultured 1C11 cells and neural tubes as a whole with regard to α-tubulin PTM regulation.
Figure 6. Hallmarks of primary cilium biology upon treatment of 1C11 cells with tubacin.
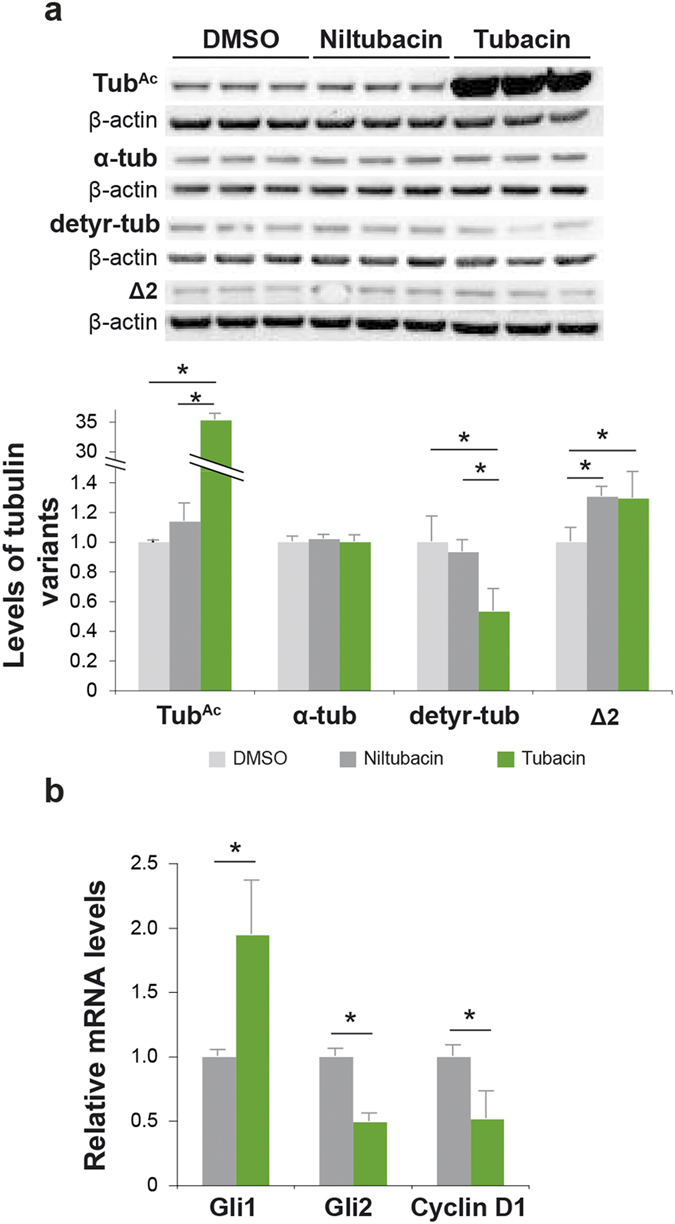
1C11 cells treated with tubacin, niltubacin and DMSO were compared with regards to protein levels of α-tub, TubAc, detyr-tub and Δ2 (a, n = 3). qPCR results showing the expression of Gli transcription factors and Cyclin D1 are shown (b, n = 3) (*p < 0.05). The gels shown have been cropped for clarity and conciseness purposes, and have been run under the same experimental conditions; the original blots are shown in supplemental Figure S8b.
CCP1 and PrPC co-localize in the embryo but the lack of PrPC has no overt impact on CCP1 distribution
Little is known about α-tubulin PTM regulation in vivo40,48. Acetylation primarily occurs on the Lys40 amino acid located on the microtubule lumenal surface, whereas tyrosination/detyrosination and subsequent removal of the penultimate glutamate residue to generate the Δ2 variant occur at the C-terminal tail of α-tubulin40,42,48 (Fig. S6). Although the enzymes responsible for the removal of the terminal tyrosine are unknown, cytosolic carboxypeptidases (CCPs), which are located in ciliary-based bodies (as PrPC) and other microtubule organizing centres49, are involved in the cleavage of detyr-tub to generate the Δ2 variant. This isotype was the most repressed isotype in Prnp−/− neural tube-enriched preparations (Fig. 5). In addition, recombinant PrP has been reported to directly interact with the C-terminal domains of tubulin50. Collectively, these data led us to hypothesize that CCP might co-localize with PrPC. We thus probed mouse sections of FVB/N embryos at E9 (Fig. 7a–c) and E9.5 (data not shown) for PrPC and CCP1, the only mouse CCP for which an antibody is available. In endothelial cells of the yolk sac (Fig. 7a) and the omphalomesenteric artery (Fig. 7b), CCP1 partially co-localized with PrPC punctiform structures, and the signal juxtaposed with γ-tub labelling (Fig. 7d and data not shown), strongly suggesting that CCP1 localizes with PrPC at the ciliary base.
Figure 7. Detection of cytosolic carboxypeptidase 1 in the mouse embryo.

Transverse sections from FVB/N (a–d,f,h) and Prnp−/− (e,g,i) E9 mouse embryos co-stained for CCP1 (green), DAPI (blue) and PrPC (a–c) or γTub (d–i) (red) are shown (representative images of n = 4 embryos analysed). The dotted lines and asterisks indicate the boundaries of the neural tube and artefact signals, respectively. PrPC and CCP1 co-localization (arrowheads) in the yolk sac (a), omphalomesenteric artery (b) and the mantle zone of the neural tube (c, inset), as observed by confocal microscopy. Note that no co-localization is observed at the apical face of the floor plate (c). γTub and CCP1 co-staining in the yolk sac at the base of the primary cilium (arrowheads; d,e). Expression patterns of γTub and CCP1 in the neural tube (f–i). Juxtaposition is rarely observed (arrowheads in h). Scale bar: 5 μm, except in c: 10 μm.
In the neural tube, CCP1 co-localized with PrPC in the mantle region but not at the ventricular face of the neuroepithelium (Fig. 7c). CCP1 and γ-tub labelling rarely overlapped (Fig. 7f,h), suggesting that in the neural tube, CCP1 localizes in zones of microtubule rearrangements distinct from the ciliary basal body. In the developing heart, no CCP1 labelling was observed (data not shown).
Our qPCR and immunoblot analyses of FVB/N versus Prnp−/− neural tube-enriched preparations from E9.0 to E10.5 embryos (Fig. S7a–b) did not reveal significant variations in CCP1 levels, except for a modest reduction in CCP1 mRNAs at E10.5 (Fig. S7a). Finally, the distribution of CCP1 in the yolk sac (Fig. 7e) and in the neural tube (Fig. 7g,i) was not affected by PrPC depletion at E9 and E9.5.
Taken together, these data suggest that although the lack of PrPC does not affect CCP1 distribution, PrPC and CCP1 co-localize at the ciliary base or in distinct zones of microtubule rearrangement, depending on the cell type and the differentiation state. These observations are consistent with a participatory role of PrPC in the fine-tuning of Δ2-tubulin variant levels.
Discussion
There is a fundamental and therapeutic interest in defining the physiological functions of the cellular form of the prion protein. In this work, we identify PrPC as a ciliary base component in different types of stem cells and progenitors in the developing central nervous and cardiovascular systems. Using neuroepithelial cells displaying neuroectodermal progenitor hallmarks and mouse embryos, we further show that PrPC depletion affects the fine-tuning of tubulin PTMs and cilium-related signalling.
Our data reveal that PrPC is detected in the mouse embryo in neuroepithelial cells, in a stem cell niche associated with definitive haematopoiesis and in progenitors of the heart and blood vessels. In these structures, the protein is detected as soon as E8.25, i.e. around the same time period as Prnp expression14,15,16. These observations are consistent with reports linking PrPC with the self-renewal of neural progenitors10 or haematopoietic stem cells6. We further document abundant PrPC expression in the mantle zone of the developing neural tube, which is consistent with its involvement in neuronal differentiation10,16. Notably, the present PrPC expression pattern is consistent with the biological pathways affected by Prnp invalidation, including cell adhesion, nervous system development, heart formation and angiogenesis14.
Although PrPC has been briefly mentioned in two distinct ciliomes from choroid plexus epithelial cells51 and the outer segment of photoreceptor cells52, our imaging of E9–E9.5 mouse embryos unambiguously links PrPC to the base of the primary cilium in the aforementioned stem cells and progenitors. PrPC was not detected along the ciliary axoneme, excluding a general distribution at the ciliary membrane. Importantly, PrPC does not localize to centrosomes in general but only at the ciliary base of a limited set of cells. In initial attempts to further characterize its subcellular distribution, PrPC was found to co-localize with the recycling endosome marker Rab11, suggesting enrichment in the pericentriolar recycling endocytic compartment. Although a major proportion of PrPC is detected as a lipid raft-clustered, cell surface protein in many differentiated cell types, this newly discovered intracellular topological localization is not unexpected. Indeed, a substantial fraction of PrPC is known to cycle constitutively between the plasma membrane and endocytic compartments2. Whether PrPC is localized in the ciliary pocket and/or in the membrane transition zone remains to be determined.
We propose that at the ciliary base, PrPC contributes to ciliary function. Proper ciliary function is critical for neural stem cells located at the apical face of the neuroepithelium to sense signals such as Shh29. The participation of PrPC in ciliary function is notably supported by the reduced levels of FoxJ1 in Prnp−/− neural tubes, a Shh-induced transcription factor reported to regulate ciliogenesis53. Reduced FoxJ1 expression may reflect defects in Shh signalling, a hypothesis supported by the decreased levels of FoxO6, another Shh target gene.
The functional link between PrPC, cilia and Shh signalling was further corroborated by our data obtained for 1C11 neuroepithelial cells. In the 1C11-derived PrP null cells, PrPC has been knocked down by an RNAi approach, thus ruling out any contribution of Prnp-flanking genes to the resultant phenotype54. The PrPC knockdown drastically affected cilium turnover and cell cycle regulation, as well as Shh signalling. The dysregulation of these intertwined processes was not merely a by-product of primary cilium impaired resorption, as shown by tubacin treatment. The more limited effect of PrPC depletion on ciliary functions in vivo is not unexpected, as overt impairment of primary cilium functions is usually associated with pleiotropic developmental abnormalities22,29, which are not observed in adult mice with inactive Prnp11,12. Notably, the alterations found in the Shh regulatory circuitry in vivo appear to affect genes that are important for differentiation (e.g., FoxJ155) and subsequent synaptogenesis (e.g., FoxO656) rather than for dorsoventral patterning of the neural tube (e.g., FoxA228), which is consistent with mild but significant alterations. Indeed, FoxJ1 is important for floor plate cilia architecture, whereas its absence does not affect floor plate identity or ciliogenesis57. Furthermore, although viable and outwardly normal56, FoxO6 knockout mice exhibit impaired memory consolidation56, which recalls the phenotype of PrP knockout mice58.
A second important contribution of this study is the identification of a link between PrPC and the fine-tuning of microtubule PTMs. While it is not yet possible to conclude on the direct involvement of PrPC in microtubule PTM regulation, such a function would be truly consistent with PrPC topological location, the primary cilium basal body being a microtubule organizing centre37. Thus, the levels of TubAc, detyr-tub and Δ2 α-tubulin isoforms were markedly decreased in Prnp−/− neural tube-enriched preparations, at a stage in which PrPC begins to be broadly detected in the developing CNS. PrPC depletion in 1C11 cells yielded prominent, albeit opposite, changes in tubulin PTMs. Those changes were not driven by the concomitant defects in primary cilia disassembly. The limited time window for the PTM response to PrPC depletion in vivo would lend support for dynamic mechanisms of adaptation, which may not occur or may occur differently in 1C11 cells, as recently documented in Shh signalling59. Moreover, the components of the pathway or the PrPC interacting partners may qualitatively differ between the neural tube and 1C11 cells, resulting in opposite effects, as previously observed with respect to neurite outgrowth60,61. These mechanisms of regulation or adaptation may also vary according to the cellular differentiation state. In this respect, it is worth mentioning that data obtained using neural-tube enriched preparations cannot discriminate between neural progenitors and differentiating neurons, in contrast with 1C11 cells.
The tubulin variants studied here appear to be important in both cell contexts. They may, on the one hand, participate in ciliary maintenance in both progenitors and differentiating cells. On the other hand, their accumulation appears to accompany neuronal differentiation40. Their deregulation in PrPC-deficient progenitors may thus affect ciliary functions, as suggested in PrPnull-1C11 cells and also by the alterations in Shh downstream targets in vivo. Because certain tubulin PTM alterations persist at later stages (E10.5) in the neural tubes of Prnp−/− embryos, those alterations may also affect neuronal differentiation, in accordance with the contribution of PrPC to this process. In direct support of this hypothesis, we found drastically reduced expression of the neuronal differentiation marker NFL in the neural tubes of E10.5 Prnp−/− embryos (SML, unpublished observations). Refining the link between PrPC and tubulin PTM may thus yield further insight into how PrPC contributes to ciliary function and neuronal differentiation.
How PrPC might regulate α-tubulin PTM mechanistically in the developing mouse embryo remains an open question that is further obscured by the absence of a coherent picture of the generation and enzymatic regulations of these PTMs and by the dynamic nature of their interactions40,48. We conducted a series of preliminary experiments on the expression pattern of CCP1 (which had not been previously documented in the mouse embryo), a recently described cytosolic carboxypeptidase responsible for the generation of Δ2-tubulin, the mostly altered α-tubulin isoform in neural tube-enriched preparations from PrP-null mice. CCP1 was found to co-localize with PrPC in yolk sac vessels, the omphalomesenteric artery and the mantle zone of the neural tube. PrPC could thus participate in CCP1 activity through a signalling cascade/platform, as commonly observed with this protein3,4,5. Alternatively, as recombinant PrP can interact directly with the C-terminal tail of tubulin50, where Δ2 generation occurs40,48, PrPC may directly regulate CCP1 access to the C-terminal domains of tubulin. Such regulation (i.e., CCP1 enzymatic activity) would be consistent with the absence of any significant effect of PrPC ablation on the subcellular localization or the protein levels of CCP1. In vitro studies with purified components will help to determine whether these interactions do physically occur.
In summary, our study links PrPC to the primary cilium biology and to the developmental regulation of microtubule subtypes, two tightly coordinated processes controlling stem cell fate. This information will certainly help to elucidate how PrPC and differentiation intersect mechanistically. The emerging links between microtubule PTM regulation and neurodegeneration42 and evidence that a significant number of signalling pathways altered in Prnp−/− embryos are over-activated at late stage of prion disease pathogenesis14 warrant the study of possible alterations in microtubule dynamics during PrPC-dependent neurodegeneration in prion diseases and Alzheimer’s disease.
Methods
Mouse
Animal experiments were conducted in strict accordance with EU directive 2010/63 and were approved by the local ethics committee of the authors’ institution (Comethea; permit number 12/034). FVB/N Prnp−/− mice were kindly provided by S.B. Prusiner (UCSF, San Francisco). E8.25 to E10.5 FVB/N and FVB/N Prnp−/− embryos were dissected in ice-cold phosphate buffered saline (PBS) and immediately frozen in liquid nitrogen for RNA and protein analysis or fixed for electron microscopy and immunofluorescence analyses.
Cell culture, cell viability and cell cycle analyses
The 1C11 cells and their PrP−null counterparts were grown as described previously27. Cell cycle, MTT assays and immunofluorescence analyses were performed as described in the supplemental procedures.
Immunofluorescence microscopy
Mouse embryos were prepared and transverse sections were analysed by immunofluorescence as previously described14. Specific primary antibodies are described in Supplementary Table S2. Sections were imaged using an inverted microscope with epifluorescence illumination (Zeiss Axio Observer.Z1, France) and a cooled CDD camera (CoolSNAP HQ2; Photometrics, Roper Scientific, Lisses, France). Images were captured using AxioVision (Zeiss-France). Z-series were acquired on a Zeiss LSM 700 confocal microscope. Images were analysed with ImageJ and when necessary with the Bio-Formats plugin.
Real-Time PCR
Total RNA from embryos or 1C11 cells was isolated using Trizol (Invitrogen, Cergy Pontoise, France) according to the manufacturer’s instructions. The corresponding cDNA was synthesized with oligo(dT)17 primer using 200 units of Superscript III™ reverse transcriptase (Invitrogen) according to the manufacturer’s protocol. Real-time PCR was performed using Absolute QPCR SYBR Green ROX Mix (Thermo-Scientific) on an ABI PRISM 7900HT (Applied Biosystems, Illkirsch, France). The sequences of the RT-PCR primers are given in Supplementary Table S3.
Immunoblotting
Embryos proper, neural tube-enriched samples and 1C11 cells were homogenized in anti-protease (Sigma)-containing buffer (50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 300 mM NaCl). The total protein concentration was determined by a bicinchoninic acid assay (Thermo-Scientific). 5–10 μg protein samples were then run on 12% Bis-Tris polyacrylamide gels (Bio-Rad, Marnes-la-Coquette, France), electrotransferred and blotted onto nitrocellulose membranes with the antibodies of interest (Table S2). Immunoreactivity was visualized by enhanced chemiluminescence (Amersham Pharmacia Biosciences, GE Healthcare Europe, Velizy-Villacoublay, France). The protein levels were quantified with the GeneTools software, after acquisition of chemiluminescent signals with a GeneGnome digital imager (Syngene, Frederick, Maryland, United States). The blots were also incubated with anti-β-actin antibody (Table S2) to normalize signals to β-actin as a loading control for quantification.
Statistical analysis
Data are presented as the means of replicates (as indicated) ± SDs. Significant differences between groups were examined using the nonparametric Mann-Whitney and Wilcoxon tests (small sample tests; Analyse-it software) unless specified otherwise, and p-values <0.05 were considered significant.
Additional Information
How to cite this article: Halliez, S. et al. Prion protein localizes at the ciliary base during neural and cardiovascular development, and its depletion affects α-tubulin post-translational modifications. Sci. Rep. 5, 17146; doi: 10.1038/srep17146 (2015).
Supplementary Material
Acknowledgments
The authors thank the MIMA2 platform (INRA-Jouy) for access to confocal microscopy, M. Vilotte (INRA-Jouy) for help with animal dissection, M. Moudjou for help with immunoblotting (INRA-Jouy), J. Bouchet (Pasteur Institute, Paris, France) for the gift of the anti-Rab11 antibody, S. Simon (CEA Saclay, France) and P. Clayette (Bertin Pharma, France) for the kind gift of Sha31, T.M. Jessell, S. Brenner-Morton and O.D. Madsen who kindly contributed to the DSHB, and M. Wassef (IBENS, Paris) for critical reading of the manuscript. This work was partially supported by the ANR-09-BLAN-0015-01 and by the ARC (grant SFI2011205489). S.M-L was supported by a fellowship from Region Ile de France (DIM-Stem Pôle).
Footnotes
Author Contributions S.H., S.M.L., H.L., J.L.V., S.M.R. and V.B. conceived and designed the experiments. S.H., S.M.L., B.P., J.H.R., J.C., C.U., S.C., H.L., J.L.V., S.M.R. and V.B. performed the experiments. S.H., S.M.L., B.P., J.H.R., J.L.V., S.M.R. and V.B. analysed the data. S.H., S.M.L., J.L.V., S.M.R. and V.B. wrote the manuscript. All authors reviewed the manuscript.
References
- Halliday M., Radford H. & Mallucci G. R. Prions: generation and spread versus neurotoxicity. The Journal of biological chemistry 289, 19862–19868, doi: 10.1074/jbc.R114.568477 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden R. et al. Physiology of the prion protein. Physiological reviews 88, 673–728, doi: 10.1152/physrev.00007.2007 (2008). [DOI] [PubMed] [Google Scholar]
- Hirsch T. Z., Hernandez-Rapp J., Martin-Lanneree S., Launay J. M. & Mouillet-Richard S. PrP(C) signalling in neurons: from basics to clinical challenges. Biochimie 104, 2–11, doi: 10.1016/j.biochi.2014.06.009 (2014). [DOI] [PubMed] [Google Scholar]
- Lewis V. & Hooper N. M. The role of lipid rafts in prion protein biology. Frontiers in bioscience 16, 151–168 (2011). [DOI] [PubMed] [Google Scholar]
- Martins V. R. et al. Prion protein: orchestrating neurotrophic activities. Curr Issues Mol Biol 12, 63–86, doi: v12/63 (2010). [PubMed] [Google Scholar]
- Zhang C. C., Steele A. D., Lindquist S. & Lodish H. F. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proceedings of the National Academy of Sciences of the United States of America 103, 2184–2189, doi: 10.1073/pnas.0510577103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. J. & Baskakov I. V. The cellular form of the prion protein is involved in controlling cell cycle dynamics, self-renewal, and the fate of human embryonic stem cell differentiation. Journal of neurochemistry 124, 310–322, doi: 10.1111/j.1471-4159.2012.07913.x (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda A., Ramos-Ibeas P., Pericuesta E., Ramirez M. A. & Gutierrez-Adan A. The role of prion protein in stem cell regulation. Reproduction 146, R91–99, doi: 10.1530/REP-13-0100 (2013). [DOI] [PubMed] [Google Scholar]
- Peralta O. A., Huckle W. R. & Eyestone W. H. Expression and knockdown of cellular prion protein (PrPC) in differentiating mouse embryonic stem cells. Differentiation 81, 68–77, doi: 10.1016/j.diff.2010.09.181 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele A. D., Emsley J. G., Ozdinler P. H., Lindquist S. & Macklis J. D. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proceedings of the National Academy of Sciences of the United States of America 103, 3416–3421, doi: 10.1073/pnas.0511290103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueler H. et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577–582, doi: 10.1038/356577a0 (1992). [DOI] [PubMed] [Google Scholar]
- Mallucci G. et al. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302, 871–874, doi: 10.1126/science.1090187 (2003). [DOI] [PubMed] [Google Scholar]
- Bribian A. et al. Role of the cellular prion protein in oligodendrocyte precursor cell proliferation and differentiation in the developing and adult mouse CNS. PLoS One 7, e33872, doi: 10.1371/journal.pone.0033872 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalife M. et al. Transcriptomic analysis brings new insight into the biological role of the prion protein during mouse embryogenesis. PLoS One 6, e23253, doi: 10.1371/journal.pone.0023253 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson J. et al. The prion protein gene: a role in mouse embryogenesis? Development 115, 117–122 (1992). [DOI] [PubMed] [Google Scholar]
- Tremblay P., Bouzamondo-Bernstein E., Heinrich C., Prusiner S. B. & DeArmond S. J. Developmental expression of PrP in the post-implantation embryo. Brain Res 1139, 60–67, doi: 10.1016/j.brainres.2006.12.055 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feraudet C. et al. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. The Journal of biological chemistry 280, 11247–11258, doi: 10.1074/jbc.M407006200 (2005). [DOI] [PubMed] [Google Scholar]
- Dzierzak E. & Speck N. A. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol 9, 129–136, doi: 10.1038/ni1560 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson J. E. 3rd, Kelley R. W. & Patterson C. Mechanisms of endothelial differentiation in embryonic vasculogenesis. Arterioscler Thromb Vasc Biol 25, 2246–2254, doi: 10.1161/01.ATV.0000183609.55154.44 (2005). [DOI] [PubMed] [Google Scholar]
- Zovein A. C. et al. Vascular remodeling of the vitelline artery initiates extravascular emergence of hematopoietic clusters. Blood 116, 3435–3444, doi: 10.1182/blood-2010-04-279497 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R. H., Brown N. A. & Moorman A. F. Development and structures of the venous pole of the heart. Dev Dyn 235, 2–9, doi: 10.1002/dvdy.20578 (2006). [DOI] [PubMed] [Google Scholar]
- Ishikawa H. & Marshall W. F. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol 12, 222–234, doi: 10.1038/nrm3085 (2011). [DOI] [PubMed] [Google Scholar]
- Hsiao Y. C., Tuz K. & Ferland R. J. Trafficking in and to the primary cilium. Cilia 1, 4, doi: 10.1186/2046-2530-1-4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco I. et al. PI3K class II alpha controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev Cell 28, 647–658, doi: 10.1016/j.devcel.2014.01.022 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouillet-Richard S. et al. Signal transduction through prion protein. Science 289, 1925–1928, doi: 8827 (2000). [DOI] [PubMed] [Google Scholar]
- Mouillet-Richard S. et al. Regulation by neurotransmitter receptors of serotonergic or catecholaminergic neuronal cell differentiation. The Journal of biological chemistry 275, 9186–9192 (2000). [DOI] [PubMed] [Google Scholar]
- Loubet D. et al. Neuritogenesis: the prion protein controls beta1 integrin signaling activity. FASEB J 26, 678–690, doi: 10.1096/fj.11-185579 (2012). [DOI] [PubMed] [Google Scholar]
- Dessaud E., McMahon A. P. & Briscoe J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development 135, 2489–2503, doi: 10.1242/dev.009324 (2008). [DOI] [PubMed] [Google Scholar]
- Guemez-Gamboa A., Coufal N. G. & Gleeson J. G. Primary cilia in the developing and mature brain. Neuron 82, 511–521, doi: 10.1016/j.neuron.2014.04.024 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavromatakis Y. E. et al. Foxa1 and Foxa2 positively and negatively regulate Shh signalling to specify ventral midbrain progenitor identity. Mechanisms of development 128, 90–103, doi: 10.1016/j.mod.2010.11.002 (2011). [DOI] [PubMed] [Google Scholar]
- Lee E. Y. et al. Hedgehog pathway-regulated gene networks in cerebellum development and tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America 107, 9736–9741, doi: 10.1073/pnas.1004602107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzakopian E. et al. Genome-wide characterization of Foxa2 targets reveals upregulation of floor plate genes and repression of ventrolateral genes in midbrain dopaminergic progenitors. Development 139, 2625–2634, doi: 10.1242/dev.081034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A. et al. Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat Cell Biol 13, 402–411, doi: 10.1038/ncb2218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Xie Y., Yang P., Chen P. & Zhang L. HCV core protein-induced down-regulation of microRNA-152 promoted aberrant proliferation by regulating Wnt1 in HepG2 cells. PLoS One 9, e81730, doi: 10.1371/journal.pone.0081730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey D. W. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol 15, 158–163, doi: S0955067403000085 (2003). [DOI] [PubMed] [Google Scholar]
- Clement C. A. et al. The primary cilium coordinates early cardiogenesis and hedgehog signaling in cardiomyocyte differentiation. J Cell Sci 122, 3070–3082, doi: 10.1242/jcs.049676 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- May-Simera H. L. & Kelley M. W. Cilia, Wnt signaling, and the cytoskeleton. Cilia 1, 7, doi: 10.1186/2046-2530-1-7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loktev A. V. et al. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell 15, 854–865, doi: 10.1016/j.devcel.2008.11.001 (2008). [DOI] [PubMed] [Google Scholar]
- Pathak N., Austin C. A. & Drummond I. A. Tubulin tyrosine ligase-like genes ttll3 and ttll6 maintain zebrafish cilia structure and motility. The Journal of biological chemistry 286, 11685–11695, doi: 10.1074/jbc.M110.209817 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C. & Bulinski J. C. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 12, 773–786, doi: 10.1038/nrm3227 (2011). [DOI] [PubMed] [Google Scholar]
- Erck C. et al. A vital role of tubulin-tyrosine-ligase for neuronal organization. Proceedings of the National Academy of Sciences of the United States of America 102, 7853–7858, doi: 10.1073/pnas.0409626102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogowski K. et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 143, 564–578, doi: 10.1016/j.cell.2010.10.014 (2010). [DOI] [PubMed] [Google Scholar]
- Paturle-Lafanechere L. et al. Accumulation of delta 2-tubulin, a major tubulin variant that cannot be tyrosinated, in neuronal tissues and in stable microtubule assemblies. J Cell Sci 107 (Pt 6), 1529–1543 (1994). [DOI] [PubMed] [Google Scholar]
- Berbari N. F. et al. Microtubule modifications and stability are altered by cilia perturbation and in cystic kidney disease. Cytoskeleton (Hoboken) 70, 24–31, doi: 10.1002/cm.21088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H., Inoko A. & Inagaki M. Cell cycle progression by the repression of primary cilia formation in proliferating cells. Cell Mol Life Sci 70, 3893–3905, doi: 10.1007/s00018-013-1302-8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty S. J., Koeller K. M., Wong J. C., Grozinger C. M. & Schreiber S. L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proceedings of the National Academy of Sciences of the United States of America 100, 4389–4394, doi: 10.1073/pnas.0430973100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva E. N., Jablonski S. A., Hartman T. R., Henske E. P. & Golemis E. A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129, 1351–1363, doi: 10.1016/j.cell.2007.04.035 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y. & Brady S. T. Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends Cell Biol 25, 125–136, doi: 10.1016/j.tcb.2014.10.004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez de la Vega Otazo M., Lorenzo J., Tort O., Aviles F. X. & Bautista J. M. Functional segregation and emerging role of cilia-related cytosolic carboxypeptidases (CCPs). FASEB J 27, 424–431, doi: 10.1096/fj.12-209080 (2013). [DOI] [PubMed] [Google Scholar]
- Nieznanski K., Podlubnaya Z. A. & Nieznanska H. Prion protein inhibits microtubule assembly by inducing tubulin oligomerization. Biochem Biophys Res Commun 349, 391–399, doi: 10.1016/j.bbrc.2006.08.051 (2006). [DOI] [PubMed] [Google Scholar]
- Narita K. et al. Proteomic analysis of multiple primary cilia reveals a novel mode of ciliary development in mammals. Biology open 1, 815–825, doi: 10.1242/bio.20121081 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. et al. The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics 6, 1299–1317, doi: 10.1074/mcp.M700054-MCP200 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choksi S. P., Lauter G., Swoboda P. & Roy S. Switching on cilia: transcriptional networks regulating ciliogenesis. Development 141, 1427–1441, doi: 10.1242/dev.074666 (2014). [DOI] [PubMed] [Google Scholar]
- Nuvolone M. et al. SIRPalpha polymorphisms, but not the prion protein, control phagocytosis of apoptotic cells. J Exp Med 210, 2539–2552, doi: 10.1084/jem.20131274 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquet B. V. et al. FoxJ1-dependent gene expression is required for differentiation of radial glia into ependymal cells and a subset of astrocytes in the postnatal brain. Development 136, 4021–4031, doi: 10.1242/dev.041129 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih D. A. et al. FoxO6 regulates memory consolidation and synaptic function. Genes Dev 26, 2780–2801, doi: 10.1101/gad.208926.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz C. et al. Foxj1 regulates floor plate cilia architecture and modifies the response of cells to sonic hedgehog signalling. Development 137, 4271–4282, doi: 10.1242/dev.051714 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coitinho A. S. et al. The interaction between prion protein and laminin modulates memory consolidation. Eur J Neurosci 24, 3255–3264, doi: 10.1111/j.1460-9568.2006.05156.x (2006). [DOI] [PubMed] [Google Scholar]
- Cohen M. et al. Ptch1 and Gli regulate Shh signalling dynamics via multiple mechanisms. Nat Commun 6, 6709, doi: 10.1038/ncomms7709 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M. H. et al. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J Neurosci 25, 11330–11339, doi: 10.1523/JNEUROSCI.2313-05.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanathan V. et al. Cellular form of prion protein inhibits Reelin-mediated shedding of Caspr from the neuronal cell surface to potentiate Caspr-mediated inhibition of neurite outgrowth. J Neurosci 30, 9292–9305, doi: 10.1523/JNEUROSCI.5657-09.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.