

Abstract
Genome wide association studies (GWAS) have been a significant technological advance in our ability to evaluate the genetic architecture of complex diseases such as Primary Biliary Cirrhosis (PBC). To date, six large-scale studies have been performed which identified 27 non-HLA risk loci associated with PBC. The identified risk variants emphasize important disease concepts; namely, that disturbances in immunoregulatory pathways are important in the pathogenesis of PBC and that such perturbations are shared among a diverse number of autoimmune diseases – suggesting the risk architecture may confer a generalized propensity to autoimmunity not necessarily specific to PBC. Furthermore, the impact of non-HLA risk variants, particularly in genes involved with IL-12 signaling, and ethnic variation in conferring susceptibility to PBC have been highlighted. While GWAS have been a critical stepping-stone in understanding common genetic variation contributing to PBC, limitations pertaining to power, sample availability, and strong linkage disequilibrium across genes have left us with an incomplete understanding of the genetic underpinnings of disease pathogenesis. Future efforts to gain insight into this missing heritability, the genetic variation that contributes to important disease outcomes and the functional consequences of associated variants will be critical if practical clinical translation is to be realized.
Keywords: GWAS, PBC, autoimmunity, genetics
INTRODUCTION
Primary biliary cirrhosis (PBC) is a female predominant autoimmune liver disease affecting 1 in 1000 women over the age of 401. It is characterized by chronic cholestasis, specific anti-mitochondrial antibodies (AMAs), destruction of the intralobular bile ducts, and can progress to liver failure2. Ursodeoxycholic acid (UDCA) is the only approved therapy for PBC and is beneficial to many patients3; however, approximately one-third of those treated do not respond well to UDCA and tend towards progressive liver disease requiring transplantation4.
As with most autoimmune diseases, PBC is genetically complex. That is, individual alleles are not likely deterministic per se, but rather act by modifying risk through subtle effects on disease-specific biological processes in context of the internal milieu and environmental exposures. Frequent co-expression of PBC with other autoimmune disorders suggests the susceptibility backgrounds influencing disease expression are shared to some extent. However, the mechanisms underlying these observations remain obscure. Multiple lines of evidence emphasize the importance of genetic predisposition in the development of PBC, including increased PBC prevalence in first-degree relatives (FDRs) of affected patients5 and high concordance in monozygotic twins6. Furthermore, a large study from the UK reported a sibling relative sibling relative risk (λs) of 10.57 and our group found a high prevalence of AMA positivity in FDRs of affected probands8. Despite this prior knowledge, significant advancements in PBC genetics have been slow to come, hampered by the rarity and late onset of disease. Until recently, genetic research in PBC was limited to surveys of candidate genes selected by biologic plausibility or known association with other autoimmune diseases. These studies, while an important step, were universally plagued by poor gene coverage and insufficient power to detect modest effects.
Genome wide association studies (GWAS) have significantly advanced our ability to evaluate the genetic architecture of complex diseases, with particular applicability to genetic variants that are common in the population (i.e. those with minor allele frequency (MAF) of > 5%). Current iterations of GWAS platforms effectively interrogate millions of single nucleotide polymorphisms (SNPs) across the entire genome, and are extendable through imputation to existing sources of genetic diversity data such as the 1,000 genomes project9. While the GWAS approach has matured, important limitations pertaining to power, sample availability, linkage disequilibrium and population differences remain; leaving us with an incomplete accounting of genetic contributors to disease. Moreover, the majority of detected GWAS associations have modest effect sizes (odds ratios in the range of 1.1–1.7) and the biological effects behind most of the associated variants remain undefined10. Thus, it is important to recognize that while GWAS have been critically informative, functional studies will be needed to identify precise causal variants and define their biologic effects.
PRE-GWAS: FOCUS ON IMMUNOLOGY AND CANDIDATE GENES
Prior and emerging knowledge of pathogenic disease mechanisms in PBC is a necessary basis for informed interpretation of GWAS findings. In PBC, past efforts have widely focused on features of immunology, and a broad range of immune cell types spanning both the innate and adaptive arms have been implicated (recently reviewed)2. The immunodominant PBC autoantigen is the E2 subunit of the pyruvate dehydrogenase complex (PDC-E2), a mitochondrial enzyme that is the target of disease-specific AMAs11. T-cells (CD4+ and CD8+) auto-reactive to PDC-E2 are enriched in the liver of PBC patients12,13, and produce high levels of the cytokine interferon-γ, suggesting T-helper 1 (TH1) polarization and a role for cellular immunity in mediating liver damage in PBC14. Despite the ubiquitous nature of PDC-E2, only the small intralobular bile ducts are targeted in patients with PBC15,16. This specificity derives from the unique nature of apoptotic protein degradation in biliary epithelial cells, which leaves an immunogenic form of PDC-E2 intact within the apoptotic bleb, a process that is not restricted to PBC17,18. Current consensus is that environmental factors such as certain infections or chemical exposures, in concert with an autoimmune-permissive genetic background, are the main contributors to loss of tolerance and conversion to frank autoimmunity in PBC19.
Prior to GWAS, genetic studies in PBC primarily focused on candidate genes selected based on the existing immunological knowledge. The most successful of these efforts was the study of human leukocyte antigen (HLA) genes located in the gene-dense and highly polymorphic major histocompatibility complex (MHC) at chromosome 6p21. HLA genes encode molecules responsible for antigen presentation, and thus, are integral in distinguishing self from non-self and enforcing immune tolerance20. The most commonly detected alleles from candidate gene studies were within the HLA DRB1*08 family, specifically DRB1*0801 in European and North American Caucasian populations21–23 and the DRB1*0803 in the Japanese24, suggesting ethnic variation in disease susceptibility. As well, DRB1*11 and HLA DRB*13 were found to be protective in studies from Italy and the UK22,25.
A number of non-HLA candidate gene studies in PBC interrogated genes known or suspected to be involved with autoimmunity including TNF, CTLA4, PTPN22, PDCD1, IL1, IL2, IL10, toll like receptors and the vitamin D receptor26–34. Many of these studies were limited by inadequate power and poor gene coverage (often just a single SNP) and there was little correlation between studies. Importantly, none of the genes investigated in the candidate-gene era have demonstrated robust findings in GWAS of PBC.
FINDINGS FROM GWAS
To date, four GWAS and two Immunochip studies using well-characterized patient cohorts from North America, Europe or Japan have been performed for PBC. The strongest statistical associations have consistently been at the HLA locus, confirming its importance in the pathogenesis of disease. Dozens of new PBC-associated HLA variants have been identified by GWAS, particularly at the DRB1, DQA1 and DQB1 loci, but specific causal alleles have been challenging to define35. Analyses incorporating GWAS data have suggested that different HLA types may contribute to immunologic phenotypes, with SNPs at the HLA-DPB1 locus being strongly associated with disease in PBC patients who test positive for anti-sp100 antibodies but not in anti-sp100 negative individuals36. Similarly, a Japanese study reported that HLA-DRB1*0405 predisposed to anti-gp20 positivity and *0803 was associated with anti-centromere antibodies37. Notwithstanding these observations, a notable lesson from GWAS is that in PBC the HLA risk alleles are relatively uncommon among patients (often less than 15%) and the effect sizes, while statistically robust, are not striking relative to other autoimmune disease38. This suggests that though HLA is clearly an important contributor to PBC risk, the non-HLA loci are likely to play an equally critical role.
At present, 27 non-HLA genetic loci have demonstrated genome-wide significant associations with PBC (Table 1). The first GWAS, from Canada, identified SNPs at three loci, namely HLA, IL12A which encodes IL-12 p35, and IL12RB2 which encodes IL-12 receptor β239. Further fine-mapping efforts implicated a five allele haplotype at the 3′ flank of the IL12A gene as significantly associated with PBC, though the precise causal alleles remain unknown. Importantly, this study began to shed light on the potential importance of the IL-12 signaling axis in the pathophysiology of PBC. The second effort that used both Italian and Canadian subjects, confirmed associations from the initial GWAS, and identified three additional disease-associated loci mapping to regions containing IRF5-TNPO3, IKZF3, and SPIB, each of which plays a role in immunoregulation40. The largest PBC GWAS to date, which included more than 1800 cases and 5000 controls from the UK, identified 12 additional non-HLA loci reaching genome-wide significance and confirmed that all previously identified loci showed at least suggestive levels of association with PBC41. When data were combined with the Italian cohort in a meta-analysis, a further three novel loci were identified at a genome wide level of significance40. The most recent GWAS, which was performed in a Japanese population, identified two novel risk loci implicating the genes TNFSF15 and POU2AF1. Of interest, only five previously identified non-HLA loci (IL7R, IKZF3, CD80, STAT4 and NFKB1) from the North American and European GWAS were found to be associated with PBC at genome-wide or suggestive levels of significance42. Notably, despite the IL12A and IL12RB2 loci being among the strongest non-HLA associations in the Caucasian studies, they were not significantly associated with PBC in Japanese patients. This finding serves to highlight the importance of ethnic differences in the way common genetic variation impacts susceptibility to complex disease.
Table 1.
Non-HLA risk loci identified through GWAS as associated with PBC at genome wide level of significance
| Study | Platform | Cases (n) | Controls (n) | Locus | SNP | Associated Genes | OR (95% CI) | p-value |
|---|---|---|---|---|---|---|---|---|
| Hirschfield et al. | Illumina HumanHap370 | 1031 | 2713 | 1p31 | rs3790567 | IL12RB2 | 1.51 (1.33–1.70) | 2.76 × 10–11 |
| 3q25 | rs6441286 | IL12A | 1.54 (1.38–1.72) | 2.42 × 10–14 | ||||
| Liu et al. | Illumina 610K | 945 | 4651 | 7q32 | rs10488631 | IRF5-TNPO3 | 1.57 (1.38–1.77) | 8.66 × 10–13 |
| 17q12 | rs9303277 | IKZF3, ORMDL3 | 1.38 | 1.69 × 10–9 | ||||
| 19q13 | rs3745516 | SPIB | 1.46 | 7.97 × 10–11 | ||||
| Mells et al. | Illumina 660W-Quad | 1840 | 5163 | 1q31 | rs12134279 | DENND1B | 1.34 (1.25 – 1.45) | 2.06×10–14 |
| 2q32 | rs10931468 | STAT4, STAT1 | 1.50 (1.37 – 1.64) | 2.35×10–19 | ||||
| 3q13 | rs2293370 | CD80 | 1.35 (1.23 – 1.47) | 2.53×10–11 | ||||
| 3p24 | rs1372072 | PLCL2 | 1.20 (1.12–1.27) | 2.28×10–8 | ||||
| 4q24 | rs7665090 | NFKB1 | 1.26 (1.18 – 1.34) | 4.06×10–12 | ||||
| 5p13 | rs860413 | IL7R | 1.30 (1.21 – 1.40) | 1.02×10–11 | ||||
| 7p14 | rs6974491 | ELMO1 | 1.25 (1.16 – 1.36) | 4.44×10–8 | ||||
| 11q13 | rs538147 | RPS6KA4 | 1.23 (1.15–1.31) | 2.06×10–10 | ||||
| 11q23 | rs6421571 | CXCR5, DDX6 | 1.37 (1.25 – 1.50) | 2.69×10–12 | ||||
| 12p13 | rs1800693 | TNFRSF1A | 1.22 (1.14 – 1.30) | 1.80×10–9 | ||||
| 14q24 | rs911263 | RAD51B | 1.29 (1.20 – 1.39) | 1.76×10–11 | ||||
| 14q32 | rs8017161 | TNFAIP2 | 1.22 (1.16–1.27) | 2.61 × 10–13 | ||||
| 16p13 | rs12924729 | CLEC16A, SOCS1 | 1.29 (1.20 – 1.38) | 2.95×10–12 | ||||
| 16q24 | rs11117432 | IRF8 | 1.31 (1.21 – 1.43) | 4.66×10–11 | ||||
| 22q13 | rs968451 | TAB1, SYNGR1 | 1.27 (1.18 – 1.38) | 1.08×10–9 | ||||
| Nakamura et al. | Affymetrix Axiom | 1274 | 1091 | 9q32 | rs4979462 | TNFSF15 | 1.56 (1.39–1.76) | 2.84 × 10–14 |
| 11q23 | rs4938534 | POU2AF1 | 1.39 (1.24–1.56) | 2.38 × 10–8 | ||||
| Juran et al. | Immunochip | 2426 | 5731 | 13q14 | rs3862738 | TNFSF11 | 1.33 (1.20 – 1.47) | 2.18×10–8 |
| Liu et al. | Immunochip | 2861 | 8514 | 12q24 | rs11065979 | SH2B3 | 1.27 (1.19–1.34) | 1.18×10–14 |
| 17q21 | rs17564829 | MAPT | 1.25 (1.16–1.35) | 2.15×10–9 | ||||
| 19p12 | rs34536443 | TYK2 | 1.29 (1.21–1.38) | 1.29×10–13 | ||||
| Hirschfield et al.* | Illumina HumanHap370 | 1351 | 4700 | 1p36 | rs3748816 | MMEL1 | 1.33 (1.20–1.47) | 3.15 × 10–8 |
replication study of suggestive SNPs from previous GWAS in larger cohort
Two additional studies were performed using the Immunochip platform, which was designed as a tool to facilitate fine mapping of 186 known autoimmune loci43. The larger of the two studies, from the UKPBC consortium, added three new loci, implicating the genes SH2B3, MAPT, and TYK2, to the list of GWAS-level PBC associated variants44. In our study, which utilized American, Canadian and Italian cohorts, a novel association implicating the TNFSF11 gene was identified and many previously known associations were again confirmed45. Taken together, this collective body of evidence has implicated multiple genes in the pathogenesis of PBC, many of which have also demonstrated association with other autoimmune diseases35. Key among these are genes influencing IL-12 signaling.
IL-12 GENETICS IN PBC
The list of genes identified through GWAS has emphasized the importance of immunoregulation in the pathogenesis of PBC (Table 2); and several potentially important pathways including antigen presentation, T and myeloid cell differentiation, and B cell function have been implicated as contributing to disease46. T lymphocyte differentiation and TH1 responses in particular, have been associated with several autoimmune diseases and may be involved in the development of auto-reactive TH1 cells associated with PBC47. The IL-12 cytokine family, which includes IL-12, IL-23, IL-27 and IL-35, is a diverse group of heterodimeric molecules sharing protein chains and conferring both positive and negative immunoregulation48 (Figure 1). IL-12 is a major cytokine involved in the development of TH1 responses49, and as mentioned, variants at the IL12A and IL12RB2 loci have been among the strongest and most reproducible associations with PBC in GWAS efforts48. Functional IL-12 is comprised of two subunits, IL-12 p35 (encoded by the IL12A gene) and IL-12 p40 (encoded by the IL12B gene) which interact with the cell–surface IL-12 receptor (composed of the IL-12 receptor β1 and β2 chains) on CD4+ T cells to induce Jak-STAT signaling leading to activation of a TH1 response50. Notably, genetic loci containing components of Jak-STAT signaling downstream of IL-12 (TYK2 and STAT4) have also demonstrated significant associations with PBC. Engagement of intact IL-12 with its receptor modulates the immune response by evoking interferon-γ (IFN-γ) production, which enforces expression of IL-12 receptor β2 and inhibits induction of proinflammatory T-helper 17 (TH17) cells by IL-23, which also uses the IL-12 p40 subunit and IL-12 receptor β1 chain19. Of interest, the components of IL-12 that demonstrate association with PBC, IL-12 p35 (IL12A) and IL-12 receptor β2 (IL12RB2), are also components of IL-35, an IL-12 family member with negative-regulatory activity which blocks TH1 and TH17 development and supports proliferation of naturally-occurring and inducible subsets of regulatory T cells51. Overall, the complexity of the IL-12 family, combined with lack of knowledge regarding the functional consequences of the observed genetic associations, obscures the precise mechanisms of IL-12 mediated pathogenesis in PBC.
Table 2.
Genes associated with PBC and other autoimmune diseases and their corresponding function.
| PBC Associated Genes | General Function | Diseases with Shared Risk Loci |
|---|---|---|
| IL12RB2 | T cell differentiation | Behcet’s |
| IL12A | T cell differentiation | Celiac |
| IRF5-TNPO3 | immune system activation | UC, RA, SLE, SSc |
| IKZF3, ORMDL3 | B cell proliferation and differentiation | UC, CD, RA, T1DM |
| SPIB | lymphoid specific enhancer, B cell signalling | n/a |
| DENND1B | clathrin mediated endocytosis | CD |
| STAT4, STAT1 | IL-12 signalling, T helper cell differentiation | Celiac, RA, SLE, SSc, Sjogren’s, Behcet’s, IBD |
| CD80 | T cell costimulatory signal | MS, celiac, vitiligo, SLE |
| PLCL2 | signal transduction | RA |
| NFKB1 | transcription regulation, involved in immune activation | UC |
| IL7R | lymphocyte development | MS, UC |
| ELMO1 | phagocytosis and cell migration | Celiac, RA |
| RPS6KA4 | regulator of inflammatory genes | IBD |
| CXCR5, DDX6 | B cell migration and/or differentiation | Celiac, vitiligo, RA, IBD |
| TNFRSF1A | regulatory of inflammation | MS |
| RAD51B | DNA repair | RA |
| TNFAIP2 | mediator of inflammation | n/a |
| CLEC16A, SOCS1 | negative regulation of cytokine signalling via JAK/STAT | MS, UC, T1DM |
| IRF8 | negative regulation of type I IFN stimulated genes | MS, IBD, RA, SSc |
| TAB1, SYNGR1 | mediates intracellular signaling | RA |
| TNFSF15 | mediates activation of NFKB and promotes apoptosis | UC, CD |
| POU2AF1 | essential for B cell response to antigens | n/a |
| TNFSF11 | T cell regulation and bone resorption | CD |
| SH2B3 | negative regulator of cytokine signalling | Celiac, RA, T1DM, Vitiligo, hypothyroidism |
| MAPT | maintenance of neuronal polarity | n/a |
| TYK2 | mediates intracellular signaling | IBD, RA, SLE, psoriasis, T1DM |
| MMEL1 | metalloproteinase involved in embryonic development | MS |
Figure 1.
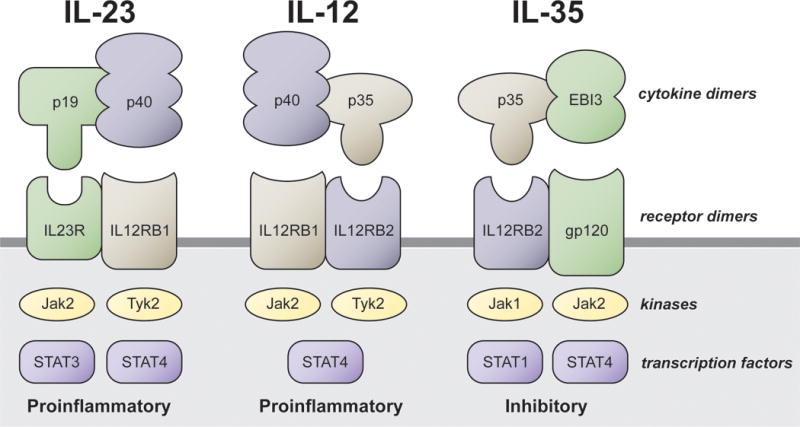
Chain-sharing between IL-12 and other members of the IL-12 cytokine family. Protein units comprising the dimeric cytokines and receptors, as well as the key downstream kinases and transcription factors, are shown for the proinflammatory IL-12 family members IL-23 and IL-12 and the inhibitory IL-12 family member IL-35. IL-12, the key cytokine in TH1 immune polarization, shares the IL-12 p40 and IL-12 receptor β1 subunits with TH17 polarizing IL-23 as well as the IL-12 p35 and IL-12 receptor β2 subunits with inhibitory IL-35. GWAS in PBC has identified associations with the genes encoding IL-12 p35 (IL12A) and IL-12 receptor β2 (IL12RB2), as well as with TYK2 and STAT4.
Several other loci identified through GWAS including those containing IRF5, SOCS1, NFKB1, and TNFRSF1A suggest that pathways upstream of IL-12 production may be relevant to PBC52. For instance, interferon regulatory factor 5, the protein product of the IRF5 gene, interacts with NF-κB to cause expression of a number of TH1 cytokines, including IL-1253. In addition, IRF8 encodes a transcription factor that binds to the IL-12 promotors to modulate IL-12 and IFN-γ production52. In the Japanese GWAS, TNFSF15 was implicated, which interacts with death receptor 3 to promote TH1 and TH17 expansion and synergizes with IL-12 and IL-18 to promote IFN-γ production42. Thus, though the specific IL-12 loci were not significantly associated with PBC in this cohort, identification of TNFSF15 and STAT4 as PBC susceptibility genes may indicate that IL-12 signaling is also involved in the pathogenesis of PBC in the Japanese population.
SHARED LOCI IN AUTOIMMUNE DISEASE
Nearly one-third of patients with PBC are also affected by another autoimmune disease54. In keeping with this observation, the vast majority of SNPs implicated in PBC have also been found to be associated with other autoimmune diseases (Table 2). For example, SNPs within the IL12A gene have been associated with celiac disease55, and variants at the STAT4 locus (encoding a transcription factor critical to IL-12 signaling) have been associated with an increased risk of rheumatoid arthritis (RA), systemic lupus erythematosus (SLE)56, and Sjogren’s syndrome57, among others. NF-κB is a transcription factor that is activated in several autoimmune disorders including RA, multiple sclerosis (MS) and asthma, and regulates expression of many genes involved in the immune response58. Several loci containing genes involved in NF-κB activation have been associated with PBC including 4q24 which contains the NFKB1 gene itself, as well as 22q13 (TAB1), 12p13 (TNFSF1A), 3q13 (CD80) and 11q13 (RPS6KA4)41. The role this pathway plays in disease-specific manifestations is unclear, but its potential importance in PBC is suggested by evidence that NF-κB action modulates the balance of survival and apoptosis in activated stellate cells59 and NF-κB p50 knockout mice show aggressive hepatic inflammation and fibrosis60. Furthermore, in addition to their association with PBC, SNPs within a gene rich region on chromosome 17, particularly in the IKZF3 gene, have also been associated with T1DM61, Crohn’s disease (CD)62, and ulcerative colitis (UC)63,64. IKZF3 encodes IKAROS family zinc finger 3, a transcription factor that prevents apoptosis of IL-2 deprived B cells, regulates B cell activation, and has been associated with a lupus like syndrome in IKZF3 deficient mice35. Ultimately, the shared association of several SNPs among diverse autoimmune diseases suggests their pleiotropic effects and emphasizes that there are few disease-specific genetic associations in PBC, at least among common SNPs amenable to detection by GWAS. Whether organ-specificity in autoimmunity is the result of untested rare genetic variants, history of environmental exposures, epiphenomenon or some combination of factors remains to be determined and is the current challenge.
IMPACT OF GWAS ON PBC RESEARCH
Advancing our understanding of the genetics of PBC beyond what has been learned from GWAS, will involve complimentary approaches to investigation using animal models and next generation sequencing. Over the last several years, multiple spontaneous and inducible murine models of PBC have been developed that demonstrate characteristic serological, biochemical, and histological features of PBC and have provided valuable insight into the immunobiology of disease65–68. Knockout models of IL-12 subunits and associated downstream signals are particularly interesting in light of the GWAS-based evidence for the important role of this pathway in PBC. The dnTGFBRII mouse model results in transforming growth factor beta deficiency, which causes pleiotropic immunologic abnormalities including cholangitis, colitis, and early death69,70. Using this model, Yoshida et al. demonstrated that knockout of the IL-12 p40 subunit was associated with reduced levels of inflammatory cytokines, immune cell infiltration and bile duct damage71, whereas p35 knockout mice had liver inflammation and bile duct damage that was similar in severity to the control dnTGFBRII mice, though with delayed onset72. IL-12 p35−/− mice underwent a distinct cytokine profile shift from TH1 to TH17 and also demonstrated significant periportal fibrosis, suggesting the contribution of the TH17 family to progressive fibrosis in this model. The same group developed an induced model of PBC, using C57BL/6 mice, which developed high titer AMAs, portal inflammation, and autoimmune cholangitis when immunized with 2-octynoic acid (2-OA) coupled to bovine serum albumin73,74. Interestingly, in gene-deleted knockouts of this model, significantly less portal inflammation was observed in IL-12 p40−/− mice compared to IL-12 p35−/− or IL-12 p19−/−65, suggesting the potential role of an environmental trigger in development of disease amidst a permissive genetic background.
Despite advances in our understanding of the genetic contribution to disease in PBC, it is estimated that only about 15% of PBC heritability has been explained, partly because GWAS approaches are underpowered to detect rare genetic variation that could contribute to disease44. Though common variants likely play a significant role in PBC pathogenesis, it is plausible that highly-penetrant rare genetic variants with strong biological effect, non-SNP structural changes such as copy number variants or epigenetic modifications, and gene-gene or gene-environment interactions could account for the as yet undetermined “missing heritability” not explained by GWAS. Identifying rare genetic variants will require the use of next generation approaches including whole exome and whole genome sequencing; and for the time being, while costs and data handling capabilities pose significant limitations, targeted approaches using study designs with individuals at phenotypic extremes or family based studies with multiple affected relatives are perhaps a more practical and cost effective next step75. While initial application of these methods is likely to still leave a significant portion of heritability unexplained due to the small numbers of individuals used in such studies and the high likelihood for allelic heterogeneity within deleterious loci76, important lessons and concepts are still likely to emerge akin to those learned through initial GWAS efforts.
CLINICAL IMPACT OF GWAS ON PBC
A significant gap exists between the genetic information collected to date and its practical translation into the clinic – though efforts are undergoing to bridge this space. Recently, Tang et al. utilized 26 known PBC risk loci identified in GWAS to calculate a weighted genetic risk score (wGRS) in order to evaluate the ability of GWAS SNPs to predict disease in two independent cohorts77. The wGRS showed good ability to identify individuals at risk for developing PBC, with an area under the curve of 0.72 (95% CI 0.706–0.735). When the wGRS was divided into quartiles, individuals in the top quartile had a 9.31 times increase risk of PBC relative to those in the first quartile. Certainly, as clinical sequencing becomes commonplace and predictive values improve, one can imagine the value of such risk scores as part of the diagnostic armamentarium in patients and at risk populations.
To date, GWAS efforts have focused on the identification of genetic variants associated with PBC itself, but not specific sub-phenotypes of disease such as treatment response and liver-related complications. The ability to identify patients who are potentially at risk of an accelerated natural history or complication of their disease based on their genetic architecture would certainly be a valuable clinical tool for risk stratification and prognostication. While efforts are underway to identify variants associated with outcomes such as response to therapy and disease progression in PBC, given the number of patients and events required to achieve adequate statistical power, multicenter international collaborative efforts will undoubtedly be required to obtain meaningful results.
Perhaps one of the most obvious opportunities for clinical translation of findings from GWAS studies is the identification of novel therapeutic targets. There is clearly a clinical need in PBC given that UDCA is the only approved therapy, and for unknown reasons, a significant proportion of patients fail to respond to treatment4. As mentioned, the IL-12 pathway has been strongly implicated in the pathogenesis of PBC as well as other autoimmune diseases. The monoclonal antibody ustekinumab targets the IL-12 p40 subunit and thus, exerts its effect on both the IL-12/TH1 and IL-23/TH17 axes. While ustekinumab has demonstrated therapeutic benefit in patients with Crohn’s disease78 and psoriasis79, pilot studies in patients with PBC have been disappointing, with no patients achieving the predefined primary endpoint of biochemical response in a recently reported Phase II study (NCT01389973). Accumulating evidence suggests that co-inhibitory molecules are key in the prevention of autoimmune disease80. CTLA-4 is a co-inhibitor that suppresses T cell activation through binding to CD80 and CD86 on APCs with higher affinity than its co-stimulatory counterpart CD2881. CTLA-4 was a main focus of many candidate gene studies of PBC, but never demonstrated association at genome-wide significance; though interestingly, variants at the CD80 locus have been associated with PBC in GWAS efforts41. Abatacept, a fusion protein consisting of the extracellular domain for CTLA-4 and the constant region of IgG (CTLA-Ig)82, has been developed and improves outcomes in patients with rheumatoid arthritis83 and psoriasis84. Modulation and prevention of T cell priming via this pathway could have therapeutic implications in PBC and in fact pilot studies are underway to assess its efficacy in patients with poor response to UDCA (NCT02078882).
TNF-α is an activating factor for a number of intracellular pathways and plays an important role in liver homeostasis85. GWAS in PBC have identified several loci containing genes in the TNF-α signaling pathway including TNFRSF1A, DENND15, and TNFAIP241,42 and pathway-based analysis has indicated that TNF signaling may underlie the genetic predisposition of PBC46. Whether anti-TNF-α agents, which have clearly been effective in other autoimmune diseases such as CD, UC, and RA, will have a therapeutic role in selected subsets of PBC patients remains to be seen.
Though T lymphocytes seem to be the primary mediators of damage in PBC, the presence of disease specific antibodies in the vast majority of patients suggests a role for B cells as well. Several variants at loci associated with B cell function have been identified in GWAS. For example, POU2AF1 and IKZF3 encode transcription factors important in B cell maturation and the development of long term immunity86,87, SPI-B is a mediator of B cell signaling88, and IL-7R participates in pre-B cell expansion89. Selective depletion of B cells with the anti-CD20 antibody Rituximab was associated with biochemical improvement in a pilot study of PBC patients who were non-responders to UDCA90 (NCT00364819), though some safety concerns have been raised after mouse models demonstrated disease exacerbation following anti-CD20 therapy91. In sum, the biological pathways implicated in PBC through GWAS have provided a ripe landscape for exploration of alternative therapeutic options. However, a greater appreciation of the functional relevance of observed associations to each pathway will be crucial to successful application of targeted therapies in PBC.
CONCLUSIONS
Significant advancements have been made in our understanding of human genetics over the last decade and progress continues at a seemingly exponential pace. GWAS have been a critical stepping stone in the hunt for genetic variants contributing to PBC pathogenesis, particularly in identifying genes outside the HLA locus that might be involved in disease. However, it has become evident that the genes thus far implicated in PBC modulate non-specific immunologic pathways important in many different autoimmune diseases, rather than conferring organ specific risk. Given the number of shared risk loci among autoimmune diseases in general, it seems likely that a mosaic of genetic variations contribute to disease specificity, rather than true disease-specific associations. Certainly, the role of rare genetic variation and other structural and non-structural genetic changes, environmental influences, and interactions between the two in conferring disease specificity remain to be defined. Importantly, before meaningful clinical translation is realized, functional consequences of identified variants at the level of the end organ need to be understood. This will be especially important for therapeutics so that novel interventions can be disease specific rather than having systemic effects akin to the pleiotropic genes thus far identified.
Acknowledgments
This work was supported by a grant to Dr. Konstantinos N. Lazaridis from the National Institutes of Health (R01 DK80670).
Abbreviations
- AMA
anti-mitochondrial antibody
- APC
antigen presenting cell
- CD
Crohn’s Disease
- dnTGFBR2
dominant negative TGFβ receptor 2
- FDR
first degree relative
- GWAS
genome wide association study
- HLA
human leukocyte antigen
- IFN-γ
interferon gamma
- MHC
major histocompatibility complex
- MS
multiple sclerosis
- NF-κB
nuclear factor kappa beta
- PBC
primary biliary cirrhosis
- PDC
pyruvate dehydrogenase complex
- RA
rheumatoid arthritis
- SLE
systemic lupus erythematosis
- SNP
single nucleotide polymorphism
- T1DM
Type 1 diabetes mellitus
- TH1
T-helper 1
- TH17
T-helper 17
- TNF
tumor necrosis factor
- UC
ulcerative colitis
- UDCA
ursodeoxycholic acid
Contributor Information
Aliya F. Gulamhusein, Email: Gulamhusein.Aliya@mayo.edu, Division of Gastroenterology and Hepatology and the Mayo Clinic Center for Cell Signaling, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, Minnesota 55905.
Brian D. Juran, Email: Juran.Brian@mayo.edu, Division of Gastroenterology and Hepatology and the Mayo Clinic Center for Cell Signaling, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, Minnesota 55905.
Konstantinos N. Lazaridis, Email: Lazaridis.Konstantinos@mayo.edu, Division of Gastroenterology and Hepatology and the Mayo Clinic Center for Cell Signaling, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, Minnesota 55905. Phone: (507) 538-4877. Fax: (507) 284-0762.
References
- 1.Kim WR, Lindor KD, Locke GR, 3rd, et al. Epidemiology and natural history of primary biliary cirrhosis in a US community. Gastroenterology. 2000;119(6):1631–1636. doi: 10.1053/gast.2000.20197. [DOI] [PubMed] [Google Scholar]
- 2.Hirschfield GM, Gershwin ME. The immunobiology and pathophysiology of primary biliary cirrhosis. Annu Rev Pathol. 2013;8:303–330. doi: 10.1146/annurev-pathol-020712-164014. [DOI] [PubMed] [Google Scholar]
- 3.Lindor KD, Gershwin ME, Poupon R, et al. Primary biliary cirrhosis. Hepatology. 2009;50(1):291–308. doi: 10.1002/hep.22906. [DOI] [PubMed] [Google Scholar]
- 4.Corpechot C, Chazouilleres O, Poupon R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long-term outcome. J Hepatol. 2011;55(6):1361–1367. doi: 10.1016/j.jhep.2011.02.031. [DOI] [PubMed] [Google Scholar]
- 5.Bach N, Schaffner F. Familial primary biliary cirrhosis. J Hepatol. 1994;20(6):698–701. doi: 10.1016/s0168-8278(05)80137-0. [DOI] [PubMed] [Google Scholar]
- 6.Selmi C, Mayo MJ, Bach N, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127(2):485–492. doi: 10.1053/j.gastro.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Jones DE, Watt FE, Metcalf JV, Bassendine MF, James OF. Familial primary biliary cirrhosis reassessed: a geographically-based population study. J Hepatol. 1999;30(3):402–407. doi: 10.1016/s0168-8278(99)80097-x. [DOI] [PubMed] [Google Scholar]
- 8.Lazaridis KN, Juran BD, Boe GM, et al. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrhosis. Hepatology. 2007;46(3):785–792. doi: 10.1002/hep.21749. [DOI] [PubMed] [Google Scholar]
- 9.Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Juran BD, Lazaridis KN. Genomics in the post-GWAS era. Semin Liver Dis. 2011;31(2):215–222. doi: 10.1055/s-0031-1276641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung PS, Van de Water J, Coppel RL, Gershwin ME. Molecular characterization of the mitochondrial autoantigens in primary biliary cirrhosis. Immunol Res. 1991;10(3–4):518–527. doi: 10.1007/BF02919751. [DOI] [PubMed] [Google Scholar]
- 12.Kita H, Matsumura S, He XS, et al. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. 2002;109(9):1231–1240. doi: 10.1172/JCI14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimoda S, Nakamura M, Ishibashi H, Hayashida K, Niho Y. HLA DRB4 0101-restricted immunodominant T cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: evidence of molecular mimicry in human autoimmune diseases. J Exp Med. 1995;181(5):1835–1845. doi: 10.1084/jem.181.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harada K, Van de Water J, Leung PS, et al. In situ nucleic acid hybridization of cytokines in primary biliary cirrhosis: predominance of the Th1 subset. Hepatology. 1997;25(4):791–796. doi: 10.1002/hep.510250402. [DOI] [PubMed] [Google Scholar]
- 15.Yeaman SJ, Fussey SP, Danner DJ, et al. Primary biliary cirrhosis: identification of two major M2 mitochondrial autoantigens. Lancet. 1988;1(8594):1067–1070. doi: 10.1016/s0140-6736(88)91894-6. [DOI] [PubMed] [Google Scholar]
- 16.Fussey SP, Guest JR, James OF, Bassendine MF, Yeaman SJ. Identification and analysis of the major M2 autoantigens in primary biliary cirrhosis. Proc Natl Acad Sci U S A. 1988;85(22):8654–8658. doi: 10.1073/pnas.85.22.8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lleo A, Selmi C, Invernizzi P, et al. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology. 2009;49(3):871–879. doi: 10.1002/hep.22736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lleo A, Bowlus CL, Yang GX, et al. Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology. 2010;52(3):987–998. doi: 10.1002/hep.23783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Wang FS, Chang C, Gershwin ME. Breach of tolerance: primary biliary cirrhosis. Semin Liver Dis. 2014;34(3):297–317. doi: 10.1055/s-0034-1383729. [DOI] [PubMed] [Google Scholar]
- 20.Invernizzi P. Human leukocyte antigen in primary biliary cirrhosis: an old story now reviving. Hepatology. 2011;54(2):714–723. doi: 10.1002/hep.24414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Begovich AB, Klitz W, Moonsamy PV, et al. Genes within the HLA class II region confer both predisposition and resistance to primary biliary cirrhosis. Tissue Antigens. 1994;43(2):71–77. doi: 10.1111/j.1399-0039.1994.tb02303.x. [DOI] [PubMed] [Google Scholar]
- 22.Donaldson PT, Baragiotta A, Heneghan MA, et al. HLA class II alleles, genotypes, haplotypes, and amino acids in primary biliary cirrhosis: a large-scale study. Hepatology. 2006;44(3):667–674. doi: 10.1002/hep.21316. [DOI] [PubMed] [Google Scholar]
- 23.Mullarkey ME, Stevens AM, McDonnell WM, et al. Human leukocyte antigen class II alleles in Caucasian women with primary biliary cirrhosis. Tissue Antigens. 2005;65(2):199–205. doi: 10.1111/j.1399-0039.2005.00351.x. [DOI] [PubMed] [Google Scholar]
- 24.Onishi S, Sakamaki T, Maeda T, et al. DNA typing of HLA class II genes; DRB1*0803 increases the susceptibility of Japanese to primary biliary cirrhosis. J Hepatol. 1994;21(6):1053–1060. doi: 10.1016/s0168-8278(05)80617-8. [DOI] [PubMed] [Google Scholar]
- 25.Invernizzi P, Selmi C, Poli F, et al. Human leukocyte antigen polymorphisms in Italian primary biliary cirrhosis: a multicenter study of 664 patients and 1992 healthy controls. Hepatology. 2008;48(6):1906–1912. doi: 10.1002/hep.22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsushita M, Tanaka A, Kikuchi K, et al. Association of single nucleotide polymorphisms of the interleukin-10 promoter gene and susceptibility to primary biliary cirrhosis: immunogenetic differences in Italian and Japanese patients. Autoimmunity. 2002;35(8):531–536. doi: 10.1080/0891693021000056703. [DOI] [PubMed] [Google Scholar]
- 27.Gordon MA, Oppenheim E, Camp NJ, et al. Primary biliary cirrhosis shows association with genetic polymorphism of tumour necrosis factor alpha promoter region. J Hepatol. 1999;31(2):242–247. doi: 10.1016/s0168-8278(99)80220-7. [DOI] [PubMed] [Google Scholar]
- 28.Graham AM, Dollinger MM, Howie SE, Harrison DJ. Identification of novel alleles at a polymorphic microsatellite repeat region in the human NRAMP1 gene promoter: analysis of allele frequencies in primary biliary cirrhosis. J Med Genet. 2000;37(2):150–152. doi: 10.1136/jmg.37.2.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker EJ, Hirschfield GM, Xu C, et al. CTLA4/ICOS gene variants and haplotypes are associated with rheumatoid arthritis and primary biliary cirrhosis in the Canadian population. Arthritis Rheum. 2009;60(4):931–937. doi: 10.1002/art.24412. [DOI] [PubMed] [Google Scholar]
- 30.Juran BD, Atkinson EJ, Larson JJ, et al. Carriage of a tumor necrosis factor polymorphism amplifies the cytotoxic T-lymphocyte antigen 4 attributed risk of primary biliary cirrhosis: evidence for a gene-gene interaction. Hepatology. 2010;52(1):223–229. doi: 10.1002/hep.23667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donaldson P, Agarwal K, Craggs A, et al. HLA and interleukin 1 gene polymorphisms in primary biliary cirrhosis: associations with disease progression and disease susceptibility. Gut. 2001;48(3):397–402. doi: 10.1136/gut.48.3.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogel A, Strassburg CP, Manns MP. Genetic association of vitamin D receptor polymorphisms with primary biliary cirrhosis and autoimmune hepatitis. Hepatology. 2002;35(1):126–131. doi: 10.1053/jhep.2002.30084. [DOI] [PubMed] [Google Scholar]
- 33.Juran BD, Atkinson EJ, Schlicht EM, et al. Interacting alleles of the coinhibitory immunoreceptor genes cytotoxic T-lymphocyte antigen 4 and programmed cell-death 1 influence risk and features of primary biliary cirrhosis. Hepatology. 2008;47(2):563–570. doi: 10.1002/hep.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Juran BD, Atkinson EJ, Schlicht EM, Fridley BL, Lazaridis KN. Primary biliary cirrhosis is associated with a genetic variant in the 3′ flanking region of the CTLA4 gene. Gastroenterology. 2008;135(4):1200–1206. doi: 10.1053/j.gastro.2008.06.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirschfield GM, Invernizzi P. Progress in the genetics of primary biliary cirrhosis. Semin Liver Dis. 2011;31(2):147–156. doi: 10.1055/s-0031-1276644. [DOI] [PubMed] [Google Scholar]
- 36.Hirschfield GM, Liu X, Han Y, et al. Variants at IRF5-TNPO3, 17q12–21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet. 2010;42(8):655–657. doi: 10.1038/ng.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura M, Yasunami M, Kondo H, et al. Analysis of HLA-DRB1 polymorphisms in Japanese patients with primary biliary cirrhosis (PBC): The HLA-DRB1polymorphism determines the relative risk of antinuclear antibodies for disease progression in PBC. Hepatol Res. 2010;40(5):494–504. doi: 10.1111/j.1872-034X.2010.00631.x. [DOI] [PubMed] [Google Scholar]
- 38.Fernando MM, Stevens CR, Walsh EC, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirschfield GM, Liu X, Xu C, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med. 2009;360(24):2544–2555. doi: 10.1056/NEJMoa0810440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Invernizzi P, Lu Y, et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet. 2010;42(8):658–660. doi: 10.1038/ng.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mells GF, Floyd JA, Morley KI, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011;43(4):329–332. doi: 10.1038/ng.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakamura M, Nishida N, Kawashima M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet. 2012;91(4):721–728. doi: 10.1016/j.ajhg.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis Res Ther. 2011;13(1):101. doi: 10.1186/ar3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu JZ, Almarri MA, Gaffney DJ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2012;44(10):1137–1141. doi: 10.1038/ng.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Juran BD, Hirschfield GM, Invernizzi P, et al. Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants. Hum Mol Genet. 2012;21(23):5209–5221. doi: 10.1093/hmg/dds359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kar SP, Seldin MF, Chen W, et al. Pathway-based analysis of primary biliary cirrhosis genome-wide association studies. Genes Immun. 2013;14(3):179–186. doi: 10.1038/gene.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carbone M, Lleo A, Sandford RN, Invernizzi P. Implications of genome-wide association studies in novel therapeutics in primary biliary cirrhosis. Eur J Immunol. 2014;44(4):945–954. doi: 10.1002/eji.201344270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Wanrooij RL, Zwiers A, Kraal G, Bouma G. Genetic variations in interleukin-12 related genes in immune-mediated diseases. J Autoimmun. 2012;39(4):359–368. doi: 10.1016/j.jaut.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 49.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3(2):133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 50.Lleo A, Gershwin ME, Mantovani A, Invernizzi P. Towards common denominators in primary biliary cirrhosis: the role of IL-12. J Hepatol. 2012;56(3):731–733. doi: 10.1016/j.jhep.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13(8):722–728. doi: 10.1038/ni.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirschfield GM, Chapman RW, Karlsen TH, et al. The genetics of complex cholestatic disorders. Gastroenterology. 2013;144(7):1357–1374. doi: 10.1053/j.gastro.2013.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krausgruber T, Blazek K, Smallie T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12(3):231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 54.Gershwin ME, Selmi C, Worman HJ, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. 2005;42(5):1194–1202. doi: 10.1002/hep.20907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hunt KA, Zhernakova A, Turner G, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40(4):395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Remmers EF, Plenge RM, Lee AT, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357(10):977–986. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, Zhang K, Chen H, et al. A genome-wide association study in Han Chinese identifies a susceptibility locus for primary Sjogren’s syndrome at 7q11. 23 Nat Genet. 2013;45(11):1361–1365. doi: 10.1038/ng.2779. [DOI] [PubMed] [Google Scholar]
- 58.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2(10):725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 59.Elsharkawy AM, Oakley F, Lin F, et al. The NF-kappaB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J Hepatol. 2010;53(3):519–527. doi: 10.1016/j.jhep.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oakley F, Mann J, Nailard S, et al. Nuclear factor-kappaB1 (p50) limits the inflammatory and fibrogenic responses to chronic injury. Am J Pathol. 2005;166(3):695–708. doi: 10.1016/s0002-9440(10)62291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barrett JC, Clayton DG, Concannon P, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42(12):1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43(3):246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bianchi I, Carbone M, Lleo A, Invernizzi P. Genetics and epigenetics of primary biliary cirrhosis. Semin Liver Dis. 2014;34(3):255–264. doi: 10.1055/s-0034-1383725. [DOI] [PubMed] [Google Scholar]
- 65.Kawata K, Tsuda M, Yang GX, et al. Identification of potential cytokine pathways for therapeutic intervention in murine primary biliary cirrhosis. PLoS One. 2013;8(9):e74225. doi: 10.1371/journal.pone.0074225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawata K, Yang GX, Ando Y, et al. Clonality, activated antigen-specific CD8(+) T cells, and development of autoimmune cholangitis in dnTGFbetaRII mice. Hepatology. 2013;58(3):1094–1104. doi: 10.1002/hep.26418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ando Y, Yang GX, Kenny TP, et al. Overexpression of microRNA-21 is associated with elevated pro-inflammatory cytokines in dominant-negative TGF-beta receptor type II mouse. J Autoimmun. 2013;41:111–119. doi: 10.1016/j.jaut.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dhirapong A, Yang GX, Nadler S, et al. Therapeutic effect of cytotoxic T lymphocyte antigen 4/immunoglobulin on a murine model of primary biliary cirrhosis. Hepatology. 2013;57(2):708–715. doi: 10.1002/hep.26067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ebert EC, Panja A, Das KM, et al. Patients with inflammatory bowel disease may have a transforming growth factor-beta-, interleukin (IL)-2- or IL-10-deficient state induced by intrinsic neutralizing antibodies. Clin Exp Immunol. 2009;155(1):65–71. doi: 10.1111/j.1365-2249.2008.03802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kel JM, Girard-Madoux MJ, Reizis B, Clausen BE. TGF-beta is required to maintain the pool of immature Langerhans cells in the epidermis. J Immunol. 2010;185(6):3248–3255. doi: 10.4049/jimmunol.1000981. [DOI] [PubMed] [Google Scholar]
- 71.Yoshida K, Yang GX, Zhang W, et al. Deletion of interleukin-12p40 suppresses autoimmune cholangitis in dominant negative transforming growth factor beta receptor type II mice. Hepatology. 2009;50(5):1494–1500. doi: 10.1002/hep.23132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsuda M, Zhang W, Yang GX, et al. Deletion of interleukin (IL)-12p35 induces liver fibrosis in dominant-negative TGFbeta receptor type II mice. Hepatology. 2013;57(2):806–816. doi: 10.1002/hep.25829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wakabayashi K, Lian ZX, Leung PS, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology. 2008;48(2):531–540. doi: 10.1002/hep.22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wakabayashi K, Yoshida K, Leung PS, et al. Induction of autoimmune cholangitis in non-obese diabetic (NOD). 1101 mice following a chemical xenobiotic immunization. Clin Exp Immunol. 2009;155(3):577–586. doi: 10.1111/j.1365-2249.2008.03837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11(6):415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 76.Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69(1):124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang R, Chen H, Miao Q, et al. The cumulative effects of known susceptibility variants to predict primary biliary cirrhosis risk. Genes Immun. 2015 doi: 10.1038/gene.2015.2. [DOI] [PubMed] [Google Scholar]
- 78.Mannon PJ, Fuss IJ, Mayer L, et al. Anti-interleukin-12 antibody for active Crohn’s disease. N Engl J Med. 2004;351(20):2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 79.Tan JY, Li S, Yang K, et al. Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: a meta-analysis. J Dermatolog Treat. 2011;22(6):323–336. doi: 10.3109/09546634.2010.487890. [DOI] [PubMed] [Google Scholar]
- 80.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 81.Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143–155. doi: 10.1111/j.1600-065X.2008.00639.x. [DOI] [PubMed] [Google Scholar]
- 82.Najafian N, Sayegh MH. CTLA4-Ig: a novel immunosuppressive agent. Expert Opin Investig Drugs. 2000;9(9):2147–2157. doi: 10.1517/13543784.9.9.2147. [DOI] [PubMed] [Google Scholar]
- 83.Genovese MC, Becker JC, Schiff M, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med. 2005;353(11):1114–1123. doi: 10.1056/NEJMoa050524. [DOI] [PubMed] [Google Scholar]
- 84.Abrams JR, Lebwohl MG, Guzzo CA, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest. 1999;103(9):1243–1252. doi: 10.1172/JCI5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36(1):4–12. doi: 10.1007/s12016-008-8091-0. [DOI] [PubMed] [Google Scholar]
- 86.Strubin M, Newell JW, Matthias P. OBF-1, a novel B cell-specific coactivator that stimulates immunoglobulin promoter activity through association with octamer-binding proteins. Cell. 1995;80(3):497–506. doi: 10.1016/0092-8674(95)90500-6. [DOI] [PubMed] [Google Scholar]
- 87.Cortes M, Georgopoulos K. Aiolos is required for the generation of high affinity bone marrow plasma cells responsible for long-term immunity. J Exp Med. 2004;199(2):209–219. doi: 10.1084/jem.20031571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Garrett-Sinha LA, Su GH, Rao S, et al. PU.1 and Spi-B are required for normal B cell receptor-mediated signal transduction. Immunity. 1999;10(4):399–408. doi: 10.1016/s1074-7613(00)80040-0. [DOI] [PubMed] [Google Scholar]
- 89.Mackall CL, Fry TJ, Gress RE. Harnessing the biology of IL-7 for therapeutic application. Nat Rev Immunol. 2011;11(5):330–342. doi: 10.1038/nri2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Myers RP, Swain MG, Lee SS, Shaheen AA, Burak KW. B-cell depletion with rituximab in patients with primary biliary cirrhosis refractory to ursodeoxycholic acid. Am J Gastroenterol. 2013;108(6):933–941. doi: 10.1038/ajg.2013.51. [DOI] [PubMed] [Google Scholar]
- 91.Dhirapong A, Lleo A, Yang GX, et al. B cell depletion therapy exacerbates murine primary biliary cirrhosis. Hepatology. 2011;53(2):527–535. doi: 10.1002/hep.24044. [DOI] [PMC free article] [PubMed] [Google Scholar]