

Abstract
Maintenance of mitochondria is achieved by several mechanisms, including the regulation of mitochondrial proteostasis. The matrix protease CLPXP, involved in protein quality control, has been implicated in ageing and disease. However, particularly due to the lack of knowledge of CLPXP’s substrate spectrum, only little is known about the pathways and mechanisms controlled by this protease. Here we report the first comprehensive identification of potential mitochondrial CLPXP in vivo interaction partners and substrates using a combination of tandem affinity purification and differential proteomics. This analysis reveals that CLPXP in the fungal ageing model Podospora anserina is mainly associated with metabolic pathways in mitochondria, e.g. components of the pyruvate dehydrogenase complex and the tricarboxylic acid cycle as well as subunits of electron transport chain complex I. These data suggest a possible function of mitochondrial CLPXP in the control and/or maintenance of energy metabolism. Since bioenergetic alterations are a common feature of neurodegenerative diseases, cancer, and ageing, our data comprise an important resource for specific studies addressing the role of CLPXP in these adverse processes.
Mitochondria are essential eukaryotic organelles involved in different metabolic processes like energy conversion or the synthesis of iron sulfur clusters1, in cellular signalling2, and the control of apoptosis3,4. Not surprisingly, owing to their central role in cellular physiology, dysfunction of mitochondria and changes in mitochondrial bioenergetics are a common feature of neurodegenerative diseases5, cancer6, and ageing7,8,9. A complex network of different quality control pathways is active in an effort to maintain mitochondrial function10,11 and adapt it to stress conditions12. In this network, mitochondrial proteases are increasingly recognized as important regulatory components and no longer viewed as mere degradation machineries for damaged proteins13. To fully appreciate the biological function of mitochondrial proteases, in-depth knowledge of their substrates and interaction partners is necessary. One of the least characterized players in this regard is the soluble matrix serine protease CLPXP.
Like its bacterial counterpart, mitochondrial CLPXP is composed of a 14-mer CLPP proteolytic chamber, formed by two heptameric CLPP rings, and one or two hexameric rings of the AAA+ chaperone CLPX which recognizes and unfolds substrate proteins14. The proteolytic component CLPP participates in the mitochondrial unfolded protein response (UPRmt), a mitochondria-to-nucleus stress signalling pathway. In this context CLPP is thought, mostly based on observations made in C. elegans, to initiate the UPRmt by degrading misfolded proteins that accumulate under stress conditions15. In addition, a recent study applied a proximity-dependent labeling technique in human cells to determine 48 potential CLPP-interacting proteins. Although this particular technique does not allow discrimination of near-neighbors or direct physical interaction partners of the ‘bait’ protein16, it is intriguing that most identified proteins were components of the ETC or enzymes involved in mitochondrial metabolism17. However, direct interaction partners and, most importantly, potential in vivo substrates of the mitochondrial CLPXP protease remain as yet undetermined and its biological role is thus only very superficially understood. In part, this lack of knowledge is due to the fact that the proteolytic component CLPP is absent in Saccharomyces cerevisiae14, the most widely used eukaryotic model organism to address such foundational research questions.
In the fungal ageing model Podospora anserina18, deletion of the gene PaClpP, encoding the proteolytic subunit of CLPXP, led to an unexpected increase of the mutant strain’s healthy lifespan19. Importantly, it was possible to revert this longevity phenotype by expression of human ClpP in the fungal deletion background, demonstrating functional conservation of human and fungal CLPP. These features, together with well-established methods for experimental manipulation and genetic, biochemical, and cell biology analysis, make P. anserina a promising model organism to investigate conserved biological roles of mitochondrial CLPXP proteases.
In the present study, we set out to characterize the potential in vivo substrates and interaction partners of a chimeric human CLPP fungal CLPX protease in P. anserina using an unbiased approach. This was achieved with an experimental strategy originally developed for the identification of bacterial CLP protease substrates20 which, to our knowledge, has to date not been applied in eukaryotes. Overall, we uncovered at least 19 potential CLPXP substrates as well as more than 40 potential CLPP interaction partners. The vast majority of these proteins belong to fundamental mitochondrial metabolic pathways. Prominent potential targets of CLPXP that were identified are components of the pyruvate dehydrogenase complex and the tricarboxylic acid cycle, subunits of electron transport chain complex I, and enzymes involved in amino acid and fatty acid metabolism. These data strongly suggest that mitochondrial CLPXP in P. anserina functions in the control and/or maintenance of mitochondrial energy metabolism, a role that might be conserved across eukaryotic species, including humans.
Results
Establishing a CLPP substrate-trapping assay in P. anserina
The CLPP substrate-trapping assay is a well-established experimental strategy to comprehensively identify CLP protease substrates in an unbiased manner. This combined biochemical and proteomics approach was originally developed in Escherichia coli, where it served to reveal more than 100 potential substrates of CLPXP20,21 and was subsequently applied to successfully characterize the substrate spectra of other bacterial CLP proteases22,23. Briefly, tagged and catalytically inactive CLPP is used to trap and enrich substrates in the proteolytic chamber in vivo, followed by CLPP purification and mass spectrometry-based identification of potential substrates. Essential requirements to implement the assay are a strain deleted for wild-type ClpP which harbors mutationally inactivated CLPP plus the retained ability of inactive CLPP to oligomerize. Given these conditions, the CLP protease’s chaperone component is still able to translocate substrates into the CLPP proteolytic chamber but, since CLPP is now catalytically inactive, substrates can no longer be degraded and are thus trapped by CLPP. To date, no attempts to adopt this assay in a eukaryotic system have been reported.
We set out to establish a CLPP substrate-trapping assay using the already available P. anserina ClpP deletion strain (ΔPaClpP)19,24 to identify potential in vivo substrates and interaction partners of mitochondrial CLPXP. In a first attempt, a P. anserina CLPP variant (PaCLPPS135A), inactivated by mutating its catalytic serine to alanine, was introduced into ΔPaClpP. Western blot analyses of mitochondrial protein extracts from the resulting strain (ΔPaClpP/PaClpPS135A) revealed that inactive PaCLPP was present in the fungal mitochondria but apparently failed to form the proteolytic chamber necessary for the trapping of substrates in vivo (Fig. 1a). Additionally, an increase in the size of monomeric PaCLPPS135A compared to wild-type PaCLPP was observed. The altered size of inactive PaCLPP indicates a possible autocatalytic processing, i.e. possibly the self-cleavage of a propeptide, of this protease, which is blocked upon its catalytic inactivation. Indeed, autocatalytic cleavage of a propeptide has early on been described for E. coli CLPP25. Taken together, these results indicated that PaCLPP was not suitable for use in the CLPP substrate-trapping assay.
Figure 1. Catalytically inactive human CLPP in P. anserina is suitable for use in a CLPP substrate-trapping assay.

(a) Western blot analyses using a P. anserina CLPP-specific antibody after separation of mitochondrial protein extracts from the indicated strains with SDS-PAGE (above) or BN-PAGE (below). PaPORIN was detected as a loading control. Western blot analysis after SDS-PAGE reveals that monomeric, catalytically inactive PaCLPP (PaCLPPS135A) has an increased size compared to the wild-type PaCLPP monomer (predicted size of mature protein: ~25 kDa). After BN-PAGE, two distinct PaCLPP oligomers, corresponding to the heptameric CLPP ring (at around ~170 kDa) and the full proteolytic chamber formed by two heptamers (at around ~340 kDa), are only visible in the wild-type sample. (b) Western blot analyses as in a, using a Homo sapiens CLPP-specific antibody and mitochondrial protein extracts from the indicated strains. Monomeric, catalytically inactive HsCLPP (HsCLPPS153A) has the same size as the wild-type HsCLPP monomer (predicted size of mature protein: ~24 kDa) and, like wild-type HsCLPP, is able to form heptameric rings (at around ~220 kDa) and the full 14-mer proteolytic chamber (at around ~440 kDa) in P. anserina mitochondria. c, Lifespan of wild type (28.2 ± 0.7; n = 15), ΔPaClpP (61.3 ± 4.3; n = 15; P = 3.4E-06), and ΔPaClpP/HsClpPS153A (70.5 ± 12.8; n = 6; P = 3.7E-05) isolates at 27 °C. Data given in parentheses are mean lifespan ± s.e.m. in days. P-values were determined in comparison to the wild-type sample by two-tailed Wilcoxon rank-sum test.
We have previously demonstrated that human CLPP can functionally complement for the absence of PaCLPP, as it was able to completely revert ΔPaClpP’s longevity phenotype when expressed in the fungal PaClpP deletion background (resulting in the strain ΔPaClpP/HsClpP_Oex)19. Thus, use of HsCLPP represented a possible alternative to execute the intended assay in P. anserina. After introducing inactive human CLPP (HsCLPPS153A) into ΔPaClpP, the protein was present in mitochondria from the resulting transgenic fungal strain (ΔPaClpP/HsClpPS153A) and appeared identical in size to wild-type HsCLPP in the mitochondria from ΔPaClpP/HsClpP_Oex (Fig. 1b). This indicates, congruent with earlier observations26, that human CLPP might in fact not undergo autocatalytic processing.
Most importantly, HsCLPPS153A, like wild-type HsCLPP, was still able to form the full 14-mer proteolytic chamber inside mitochondria from the PaClpP deletion strain (Fig. 1b). However, most likely reflecting its catalytic inactivation, inactive HsCLPP was not able to revert the lifespan of ΔPaClpP (Fig. 1c). It should be noted that the human CLPP oligomers appeared larger than their theoretically expected size. This phenomenon was already noted in a previous study19 and is likely explained by the fact that the migration of proteins during BN-PAGE is significantly influenced by their native shape, their intrinsic charge and the amount of bound Coomassie dye27. Taken together, inactive human CLPP appeared to fulfill the necessary prerequisites to be used for a CLPP substrate-trapping assay in P. anserina.
We also attempted to replace the P. anserina CLPX chaperone with its human homologue, but human CLPX was not stable in the fungal mitochondria (Supplementary Fig. 1). Therefore, we used a strain which harbors the inactive human CLPP protease and the P. anserina CLPX chaperone to perform the CLPP substrate-trapping in P. anserina.
For human CLPP purification, we constructed expression vectors coding for a C-terminally 3xFLAG-6xHis-tagged wild-type (HsCLPPWT-TAG) or inactive HsCLPP variant (HsCLPPTRAP-TAG; Fig. 2a). Transformation of each vector into ΔPaClpP spheroblasts and selection of transformants with single genomic vector integrations yielded the strains ΔPaClpP/HsClpPWT-TAG and ΔPaClpP/HsClpPTRAP-TAG, respectively (Supplementary Fig. 2). Presence of recombinant affinity-tagged active or inactive HsCLPP in the fungal mitochondria was validated by western blot analyses (Supplementary Fig. 2). Lifespan of ΔPaClpP/HsClpPWT-TAG was again identical to that of the P. anserina wild-type strain, directly confirming that the affinity tag doesn’t interfere with HsCLPP’s function inside the fungal mitochondria (Fig. 2b). At this point it should be emphasized that P. anserina represents a unique system for analyses of eukaryotic CLP proteases in vivo, since the deletion of PaClpP leads to a very specific phenotype: a pronounced increase in lifespan19. This easily detectable phenotype allows direct assessment of the functionality of different CLPP variants and constructs in vivo. In contrast to HsCLPPWT-TAG, we found that HsCLPPTRAP-TAG, as expected due to its catalytic inactivation, was not able to revert the extended lifespan of ΔPaClpP.
Figure 2. CLPP substrate-trapping assay using isolated mitochondria.
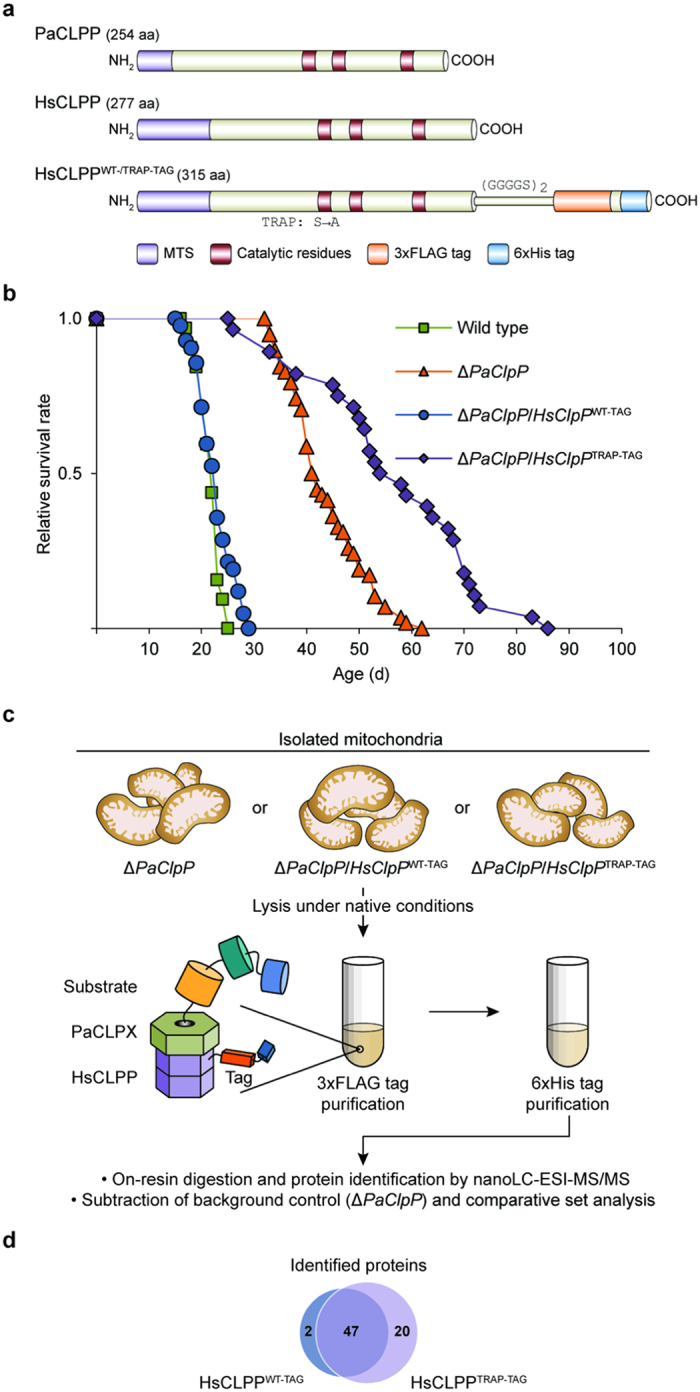
(a) Cartoon presentation of P. anserina and human CLPP homologues as well as recombinant HsCLPP variants. PaCLPP is 254 amino acids and HsCLPP 277 amino acids long. Both CLPP homologues display a conserved distribution of the canonical catalytic residues Ser, His, and Asp and contain a N-terminal mitochondrial targeting sequence (MTS). Recombinant human CLPP with a C-terminal (GGGGS)2 linker followed by a 3xFLAG-6xHis-tag has a length of 315 amino acids. Catalytic inactivation of recombinant HsCLPP was achieved by mutating its catalytic serine at position 153 of the full-length HsCLPP pre-protein to alanine. (b) Lifespan of wild type (21.8 ± 0.4; n = 32), ΔPaClpP (43.7 ± 1.0; n = 58; P = 8.3E-25), ΔPaClpP/HsClpPWT-TAG (22.7 ± 0.5; n = 42; P = 3.7E-01), and ΔPaClpP/HsClpPTRAP-TAG (56.9 ± 2.9; n = 28; P = 1.9E-17) isolates at 27 °C. Data given in parentheses are mean lifespan ± s.e.m. in days. P-values were determined in comparison to the wild-type sample by two-tailed Wilcoxon rank-sum test. (c) Overview of CLPP substrate-trapping assay and proteomics work flow. (d) Venn diagram displaying overlap of identified proteins co-purifying with 3xFLAG-6xHis-tagged active (HsCLPPWT-TAG) or inactive human CLPP (HSCLPPTRAP-TAG).
For execution of the assay, mitochondria isolated from biological triplicates of the strains ΔPaClpP, ΔPaClpP/HsClpPWT-TAG, or ΔPaClpP/HsClpPTRAP-TAG were used for native tandem affinity purification, which included rigorous washing steps to prevent unspecific adsorption to the bed material. Purified proteins were on-resin digested and identified by nanoLC-ESI-MS/MS (Fig. 2c). Stringent filtering criteria ensured that only reproducibly co-purifying proteins, found in at least 2 out of 3 independent biological replicates of the respective sample, were used for subsequent analyses (see Methods section for details). Unspecifically purifying proteins also present in the ΔPaClpP background control sample were subtracted from the sets of proteins co-purifying with either HsCLPPWT-TAG or HsCLPPTRAP-TAG (Supplementary Table 1). The remaining proteins that specifically co-purified with both HsCLPP variants over the background control sample were classified as potential interaction partners of CLPP. All proteins that fulfilled the selective criterion of co-purifying either exclusively or highly enriched with the catalytically inactive variant HsCLPPTRAP-TAG, as to be expected for proteins trapped by inactive CLPP in vivo, were classified as potential CLPXP substrates.
Mitochondrial CLPXP is predominantly associated with metabolic pathways
Overall, we identified 47 proteins specifically co-purifying with both HsCLPP variants over the background control sample (termed HsCLPPWT-/TRAP-TAG-shared; Fig. 2d and Table 1), which were therefore classified as potential interaction partners of CLPP in P. anserina. Notably, 12 of these 47 shared proteins were enriched >1.5-fold in the ΔPaClpP/HsClpPTRAP-TAG sample (termed HsCLPPTRAP-TAG-enriched; Table 1). Only two proteins, homologues of the human methylcrotonoyl-CoA carboxylase beta chain (MCCC2; Swiss-Prot ID: Q9HCC0) and of a fungal cytochrome c peroxidase (ccp-1; Swiss-Prot ID: Q7SDV9), were preferentially co-purified with HsCLPPWT-TAG (Fig. 2d). Because their abundance was very low and they were also present in one biological replicate of the ΔPaClpP/HsClpPTRAP-TAG sample, these two proteins were excluded from subsequent analyses.
Table 1. Proteins co-purifying with HsCLPPWT-TAG and HsCLPPTRAP-TAG.
| P. anserina ID* | Avg. Number of Unique Peptides HsCLPPWT-TAG | Avg. Number of Unique Peptides HsCLPPTRAP-TAG | Swiss-Prot ID | Gene | Protein |
|---|---|---|---|---|---|
| Chaperones and Protein Import | |||||
| Pa_6_2570 | 27.0 | 28.3 | P38646 | HSPA9 | Stress-70 protein‡ |
| Pa_6_5750 | 16.7 | 13.7 | P10809 | HSPD1 | 60 kDa heat shock protein‡ |
| Pa_2_9700 | 14.3 | 15.3 | O94826 | TOMM70A | Mitochondrial import receptor subunit TOM70 |
| Pa_2_10580 | 9.0 | 9.3 | O96008 | TOMM40 | Mitochondrial import receptor subunit TOM40 |
| Pa_6_1920 | 3.0 | 2.7 | Q15388 | TOMM20 | Mitochondrial import receptor subunit TOM20 |
| Pa_2_12760 | 1.7 | 1.3 | P35232 | PHB | Prohibitin |
| Metabolism | |||||
| Pa_1_22300 | 12.0 | 15.3 | P23378 | GLDC | Glycine cleavage system P protein |
| Pa_3_10790 | 11.3 | 13.0 | O95571 | ETHE1 | Persulfide dioxygenase ETHE1 |
| Pa_5_5970 | 10.0 | 9.0 | Q99798 | ACO2 | Aconitate hydratase‡,§ |
| Pa_3_11290 | 8.7 | 9.3 | P00505 | GOT2 | Aspartate aminotransferase |
| Pa_6_1590 | 8.0 | 7.0 | P24752 | ACAT1 | Acetyl-CoA acetyltransferase |
| Pa_3_6780 | 7.3 | 14.0 | O75390 | CS | Citrate synthase |
| Pa_6_2730 | 7.3 | 6.3 | P50213 | IDH3A | Isocitrate dehydrogenase [NAD] subunit alpha |
| Pa_3_2310 | 6.7 | 12.0 | P10515 | DLAT | Pyruvate dehydrogenase E2 component‡ |
| Pa_5_11920 | 5.7 | 5.0 | Q13825 | AUH | Methylglutaconyl-CoA hydratase |
| Pa_2_1050 | 5.7 | 4.3 | P78827† | ilv-2 | Ketol-acid reductoisomerase |
| Pa_6_10000 | 5.0 | 3.7 | C7C436† | mcsA | 2-methylcitrate synthase |
| Pa_1_13140 | 4.7 | 6.3 | P31327 | CPS1 | Carbamoyl-phosphate synthetase I‡ (Pa_1_13140 matches to large chain/C-terminal region) |
| Pa_3_7700 | 4.3 | 7.7 | Q9P2R7 | SUCLA2 | Succinyl-CoA ligase subunit beta‡ |
| Pa_1_3450 | 4.0 | 6.0 | Q92506 | HSD17B8 | Estradiol 17-beta-dehydrogenase 8 |
| Pa_1_17280 | 3.3 | 5.0 | Q12428† | PDH1 | Probable 2-methylcitrate dehydratase |
| Pa_1_14630 | 3.0 | 6.3 | Q8N159 | NAGS | N-acetylglutamate synthase |
| Pa_4_7010 | 3.0 | 3.7 | Q16698 | DECR1 | 2.4-dienoyl-CoA reductase |
| Pa_3_10910 | 3.0 | 3.3 | P15937† | acu-8 | Acetyl-CoA hydrolase |
| Pa_4_8600 | 3.0 | 3.0 | O15382 | BCAT2 | Branched-chain-amino-acid aminotransferase |
| Pa_2_6200 | 2.3 | 5.0 | P23434 | GCSH | Glycine cleavage system H protein |
| Pa_6_8420 | 2.3 | 4.0 | P45954 | ACADSB | Short/branched chain specific acyl-CoA dehydrogenase |
| Pa_2_430 | 2.3 | 3.3 | Q9Y697 | NFS1 | Cysteine desulfurase‡ |
| Pa_3_2600 | 2.0 | 5.3 | P40926 | MDH2 | Malate dehydrogenase |
| Pa_1_15690 | 2.0 | 3.7 | Q10341† | cys2 | Probable serine-O-acetyltransferase cys2 |
| Pa_2_4980 | 2.0 | 3.3 | Q16836 | HADH | Hydroxyacyl-coenzyme A dehydrogenase |
| Pa_3_1420 | 2.0 | 2.0 | Q9UHQ9 | CYB5R1 | NADH-cytochrome b5 reductase 1 |
| Pa_3_9430 | 1.7 | 3.0 | P04181 | OAT | Ornithine aminotransferase‡ |
| Pa_4_3040 | 1.7 | 1.7 | P48735 | IDH2 | Isocitrate dehydrogenase [NADP] |
| Pa_1_7660 | 1.7 | 1.3 | Q02252 | ALDH6A1 | Methylmalonate-semialdehyde dehydrogenase |
| Pa_7_10210 | 1.3 | 3.0 | P30084 | ECHS1 | Enoyl-CoA hydratase |
| Pa_1_1980 | 1.3 | 1.7 | P51649 | ALDH5A1 | Succinate-semialdehyde dehydrogenase‡ |
| Pa_4_660 | 1.3 | 1.3 | P34897 | SHMT2 | Serine hydroxymethyltransferase‡ |
| Electron Transport Chain and Respiration | |||||
| Pa_1_14370 | 2.7 | 2.3 | O75947 | ATP5H | ATP synthase subunit d |
| Pa_4_7160 | 2.0 | 4.7 | O75489 | NDUFS3 | NADH dehydrogenase iron-sulfur protein 3 |
| Pa_1_8620 | 2.0 | 1.7 | O75306 | NDUFS2 | NADH dehydrogenase iron-sulfur protein 2§ |
| Pa_5_7500 | 1.3 | 1.7 | O14561 | NDUFAB1 | Acyl carrier protein |
| Pa_6_240 | 1.3 | 1.3 | P47985 | UQCRFS1 | Cytochrome b-c1 complex subunit Rieske§ |
| Other Pathways | |||||
| Pa_2_12010 | 8.7 | 10.0 | P49411 | TUFM | Mitochondrial elongation factor Tu‡ |
| Pa_4_1130 | 2.3 | 2.3 | nhd | — | — |
| Pa_5_8240 | 2.0 | 2.7 | P30044 | PRDX5 | Peroxiredoxin-5 |
| Pa_6_8740 | 2.0 | 2.3 | P10599 | TXN | Thioredoxin |
*P. anserina IDs correspond to the ‘P. anserina Genome Project’ database release version 6.32 (downloaded from http://podospora.igmors.u-psud.fr/).
†If no human homologue was determinable, if possible a homologue from a fungal species was selected for reference.
§Fe-S containing/binding protein.
Listed are all proteins that were specifically co-purified with both catalytically active (HsCLPPWT-TAG) and inactive human CLPP (HsCLPPTRAP-TAG) over the background control sample and therefore classified as potential CLPP interaction partners. For each protein the P. anserina ID, average number of unique peptides identified by MS analyses across all biological replicates of the respective sample as well as Swiss-Prot ID, and gene and protein name of the human homologue (Supplementary Table 1) are listed. Categories (e.g. ‘Metabolism’) were assigned based on annotations from the Swiss-Prot database and the general literature. nhd, no homologue determinable.
Proteins enriched >1.5-fold in the ΔPaClpP/HsClpPTRAP-TAG sample over the ΔPaClpP/HsClpPWT-TAG sample (HsCLPPTRAP-TAG-enriched) are in bold.
Most strikingly, 18 proteins were exclusively associated with HsCLPPTRAP-TAG (i.e. they were not co-purified with active HsCLPPWT-TAG) and two additional proteins were highly enriched (enrichment factor >3) in the ΔPaClpP/HsClpPTRAP-TAG sample over the ΔPaClpP/HsClpPWT-TAG sample. Hence, 20 proteins were identified that co-purified either exclusively or highly enriched with the catalytically inactive variant HsCLPPTRAP-TAG (termed HsCLPPTRAP-TAG-specific, Fig. 2d and Table 2). Notably, one of the HsCLPPTRAP-TAG-specific proteins was PaCLPX, thus confirming that inactive, affinity-tagged human CLPP, as a necessary prerequisite for the trapping of substrates, retains the ability to interact with the chaperone PaCLPX. A similar association of CLPX only with inactive but not with wild-type CLPP after stringent affinity purification, which most likely reflects stabilization of the CLPP-CLPX interaction in the presence of trapped substrates28, was previously observed in a prokaryotic CLPP substrate-trapping assay22. The remaining 19 HsCLPPTRAP-TAG-specific proteins were classified as potential CLPXP substrates in P. anserina (Table 2).
Table 2. Proteins identified as potential CLPXP substrates.
| P. anserina ID* | Avg. Number of Unique Peptides† | Swiss-Prot ID | Gene | Protein |
|---|---|---|---|---|
| Chaperones | ||||
| Pa_6_5510 | 7.3 | Q8NBU5 | ATAD1 | ATPase family AAA domain-containing protein 1 |
| Pa_6_5590 | 3.7 | O76031 | CLPX | ATP-dependent Clp protease ATP-binding subunit clpX-like§ |
| Metabolism | ||||
| Pa_6_5560 | 15.7 | Q02218 | OGDH | 2-oxoglutarate dehydrogenase E1 component |
| Pa_6_1640 | 7.7 | P31327 | CPS1 | Carbamoyl-phosphate synthetase I§ (Pa_6_1640 matches to small chain/N-terminal region) |
| Pa_7_9520 | 7.3 | O00330 | PDHX | Pyruvate dehydrogenase protein X component |
| Pa_5_5370 | 5.0 (x3.8) | P36957 | DLST | 2-oxoglutarate dehydrogenase E2 component |
| Pa_7_10050 | 4.0 | P08559 | PDHA1 | Pyruvate dehydrogenase E1 component subunit alpha |
| Pa_1_13750 | 2.7 | P48728 | AMT | Aminomethyltransferase |
| Pa_5_5810 | 2.3 | P09622 | DLD | Dihydrolipoyl dehydrogenase§ |
| Pa_1_15800 | 2.0 | P11177 | PDHB | Pyruvate dehydrogenase E1 component subunit beta |
| Pa_1_20100 | 1,3 | P26440 | IVD | Isovaleryl-CoA dehydrogenase |
| Pa_3_9520 | 1.3 | P35914 | HMGCL | Hydroxymethylglutaryl-CoA lyase |
| Electron Transport Chain | ||||
| Pa_3_4870 | 21.0 | P28331 | NDUFS1 | NADH dehydrogenase 75 kDa subunit‖ |
| Pa_4_7950 | 3.7 | P49821 | NDUFV1 | NADH dehydrogenase flavoprotein 1‖ |
| Pa_5_9670 | 3.3 | Q5T2R2 | PDSS1 | Decaprenyl-diphosphate synthase subunit 1 |
| Other Pathways | ||||
| Pa_3_11170 | 10.7 (x4.0) | O59778‡ | bio2 | Biotin synthase‖ |
| Pa_1_18430 | 2.7 | P22626 | HNRNPA2B1 | Heterogeneous nuclear ribonucleoproteins A2/B1 |
| Pa_2_10680 | 2.0 | Q86SX6 | GLRX5 | Glutaredoxin-related protein 5‖ |
| Pa_1_6330 | 1.7 | Q96RP9 | GFM1 | Mitochondrial elongation factor G§ |
| Pa_5_2590 | 1.7 | G2TRP3‡ | ymr31 | Mitochondrial 37S ribosomal protein YMR-31 |
*P. anserina IDs correspond to the ‘P. anserina Genome Project’ database release version 6.32 (downloaded from http://podospora.igmors.u-psud.fr/).
†If the protein was also co-purified with HsCLPPWT-TAG above threshold, its enrichment factor in the ΔPaClpP/HsClpPTRAP-TAG sample over the ΔPaClpP/HsClpPWT-TAG sample is provided in parentheses.
‡If no human homologue was determinable, if possible a homologue from a fungal species was selected for reference.
‖Fe-S containing/binding protein.
Listed are all proteins that were either exclusively or highly enriched co-purified with the catalytically inactive variant HsCLPPTRAP-TAG and therefore classified as potential CLPXP substrates. For each protein, the P. anserina ID, average number of unique peptides identified by MS analyses across all biological replicates as well as Swiss-Prot ID, and gene and protein name of the human homologue (Supplementary Table 2) are listed. Categories (e.g. ‘Metabolism’) were assigned based on annotations from the Swiss-Prot database and the general literature.
For the vast majority of identified proteins, human homologues, which were used for data annotation and analyses, were readily determinable (Supplementary Tables 2 and 3). Gene ontology (GO) analysis unveiled marked enrichment of essential mitochondrial metabolic pathways, e.g. the tricarboxylic acid (TCA) cycle and amino acid metabolism, among the genes encoding HsCLPPWT-/TRAP-TAG-shared and HsCLPPTRAP-TAG-specific proteins (Fig. 3a,b and Supplementary Tables 4 and 5).
Figure 3. Potential mitochondrial CLPP interaction partners and CLPXP substrates.

(a) Selected GO terms enriched among human homologues of genes coding for proteins co-purifying with both HsCLPP variants (HsCLPPWT-/TRAP-TAG-shared) that were classified as potential CLPP interaction partneres. Count refers to the number of genes associated with the respective GO term. Only GO terms with Bonferroni-corrected P-values <5.0E-02 were considered statistically significant. For full GO enrichment analysis data see Supplementary Table 4. (b) Selected GO terms enriched among human homologues of genes coding for potential CLPXP substrates. For full GO term enrichment analysis data see Supplementary Table 5. c, Legend for colour-coded circles indicating different characteristics of the identified proteins. (d) Overview of proteins identified as HsCLPPWT-/TRAP-TAG-shared (Table 1), HsCLPPTRAP-TAG-enriched (Table 1) or potential CLPXP substrates (Table 2). Proteins are denoted by their corresponding gene names which are listed in Tables 1 and 2. Colour-coded circles below each protein indicate its respective characteristics. For example, the TCA cycle enzyme dihydrolipoyl dehydrogenase (DLD; Swiss-Prot ID: P09622; Table 2) was identified as a potential CLPXP substrate in our analysis (magenta circle), its bacterial homologue is a known E. coli CLPXP substrate (purple circle), and it is involved in multiple cellular processes (blue circle). Proteins with no associated circles, e.g. the mitochondrial peroxiredoxin-5 (PRDX5; Swiss-Prot ID: P30044; Table 1), were identified as potential CLPP interaction partners (i.e. they were specifically co-purified with both HsCLPP variants) but do not possess any of the additional characteristics.
Proteins were assigned to their biological process, functional class, and/or associated protein complex (overview in Fig. 3c,d). Several of the proteins identified in our assay were chaperones or belonged to various mitochondrial processes, e.g. control of transcription, translation, and redox homeostasis. Unexpectedly, three components (TOMM20, TOMM40, and TOMM70A) of the translocase of the outer membrane (TOM) complex co-purified with both HsCLPP variants. This possibly indicates that a fraction of purified HsCLPPWT-TAG and HsCLPPTRAP-TAG might in fact be transport intermediates. Altogether 19 proteins were involved in amino acid metabolism, e.g. components of the glycine cleavage system or enzymes involved in the breakdown of branched-chain amino acids. Eight of these were classified as potential CLPXP substrates or were proteins markedly enriched in the ΔPaClpP/HsClpPTRAP-TAG sample. Likewise, four of the nine proteins related to beta-oxidation or other pathways of fatty acid metabolism were determined as HsCLPPTRAP-TAG-specific or HsCLPPTRAP-TAG-enriched. Strikingly, nearly all components of the pyruvate dehydrogenase complex (PDC), which is considered a mitochondrial gatekeeper linking glycolysis with mitochondrial energy metabolism, were identified as potential substrates of CLPXP in our analysis. This strongly suggests that the PDC in P. anserina is one of the major proteolytic targets of mitochondrial CLPXP. The TCA cycle appears to be another prominent target, with many of its enzymes identified as potential CLPXP substrates or potential CLPP-interacting proteins. The TCA cycle lies downstream of amino acid/fatty acid catabolism and the PDC, supplying the reducing equivalents to drive oxidative phosphorylation at the electron transport chain (ETC). Appropriately enough, core subunits of ETC complex I constitute yet another prevalent group containing several of the identified potential CLPXP substrates. Based on the overall results of the CLPP substrate-trapping assay we suggest that CLPXP in P. anserina, mainly through proteolytic control of PDC, TCA cycle, and ETC components, has a dedicated role in the control and/or maintenance of mitochondrial energy metabolism.
In line with this interpretation of our data, bacterial CLP proteases also degrade numerous metabolic enzymes20,21,23,29. In fact, bacterial homologues of 15 proteins we identified, e.g. of the PDC E2 component (DLAT; Swiss-Prot ID: P10515; Table 1) or the TCA cycle enzyme dihydrolipoyl dehydrogenase (DLD; Swiss-Prot ID: P09622; Table 2), are known substrates or interactors of E. coli CLPXP20,21. Strikingly, the murine homologue of one of these proteins, the carbamoyl-phosphate synthetase I (CPS1; Swiss-Prot ID: P31327; Tables 1 and 2), also preferentially interacted with a mutated version of human CLPX that lacks ATPase activity, indicating it as a putative physiological substrate of the human chaperone component30. As this protein is therefore identified in, including our own, three independent studies as a potential CLPXP-substrate in different organisms, it might in fact represent a substrate of this protease that is highly conserved during evolution. Furthermore, 48 potential human CLPP-interacting proteins were recently identified, most of them components of the ETC or enzymes involved in mitochondrial metabolism17. Overall, these observations strongly support a possible biological role of mitochondrial CLPXP connected to the energy metabolism in different eukaryotic organisms, which is already present in its bacterial ancestor.
The role of mitochondrial CLPXP in human disease
Strikingly, a number of previous studies implicate mitochondrial CLPXP in different human pathologies. We therefore further analysed our results to determine whether they might be of relevance in this regard and could shed light on certain observations concerning CLPP as reported in the literature.
Among the CLPP-associated pathologies is Friedreich’s Ataxia (FRDA), a neurodegenerative disease caused by failed assembly of Fe-S clusters due to defects in the mitochondrial iron chaperone frataxin31. In a FRDA mouse model, the proteolytic component CLPP is upregulated at mid-stage of the disease. This upregulation is concomitant with a loss of mitochondrial Fe-S proteins, indicating they are targets of CLPP in FRDA32. Indeed, several proteins we identified in our study contain or bind to Fe-S clusters, e.g. aconitase (ACO2; Swiss-Prot ID: Q99798; Table 1), biotin synthase (bio2; Swiss-Prot ID: O59778; Table 2), and complex I components such as the NADH-ubiquinone oxidoreductase 75 kDa subunit (NDUFS1; Swiss-Prot ID: P49821; Table 2). In addition, three of the proteins found as CLPXP interactors or substrates, the cysteine desulfurase NSF1 (Swiss-Prot ID: Q9Y697; Table 1), the chaperone HSPA9 (Swiss-Prot ID: P38646; Table 1), and the glutaredoxin-related protein 5 (GLRX5; Swiss-Prot ID: Q86SX6; Table 2), are known to be essential for Fe-S cluster biogenesis in eukaryotic cells including those of mammals33. Thus, our findings support the idea that CLPXP has a functional role in FRDA and might possibly be involved in regulating Fe-S cluster assembly and Fe-S cluster proteins.
In human Perrault syndrome (PS), characterized by sensorineural hearing loss and ovarian failure, ClpP mutations are assumed as causative for the disease34. ClpP knockout mice recapitulate the PS-associated phenotype, have severe growth retardation, and show bioenergetic and respiratory deficits in several tissues35. The finding that CLPXP is predominantly associated with metabolic pathways is well in concordance with these observations and suggests that PS could partially be driven by a dysregulation of mitochondrial energy metabolism following functional mutations in ClpP.
Finally, CLPP was found altered in different tumor types36,37,38, suggesting a link between mitochondrial CLPXP and cancer38. For instance, a recent study reported ClpP as overexpressed in 45% of 511 primary acute myeloid leukemia (AML) patient samples17. ClpP-knockdown in AML cell lines with high ClpP expression reduced their viability, coinciding with impaired oxidative phosphorylation17. These results, certainly in agreement with the main conclusion of our study, strongly imply a role of mitochondrial CLPXP in the control and/or maintenance of mitochondrial energy metabolism as well. It thus is reasonable to speculate that CLPXP, as a regulator of mitochondrial metabolism, is pathogenically altered in at least a subset of cancers and may in fact be involved in mediating and maintaining tumorigenic metabolic reprogramming.
Discussion
The CLPXP protease is, with a few exceptions, nearly ubiquitously present in mitochondria of eukaryotes where it participates in protein quality control13,14. Importantly, as exemplified above, the proteolytic component CLPP was found altered in different human diseases, making CLPXP an important target for future research. The precise biological role of CLPXP and thus the underlying molecular mechanisms of CLPP-associated pathologies, however, are barely understood. So far, the only well-elaborated pathway known to involve CLPP is the mitochondrial unfolded protein response in C. elegans, where CLPP degrades misfolded proteins during upstream UPRmt signalling15. Unspecific degradation of damaged proteins as well as processing of protein precursors were long thought to be the main functions of mitochondrial proteases. However, it is rapidly becoming clear that most proteases also perform highly regulated proteolytic activities, thereby influencing mitochondrial function, integrity, and homeostasis13.
Overall, our as yet unprecedented initial identification of mitochondrial CLPXP’s potential in vivo interaction partners and substrates contributes to a more detailed view of this as yet ill-defined protease. Notwithstanding the necessity of the independent validation of selected potential interaction partners and substrates in future studies, our results clearly argue for a function of P. anserina CLPXP in specifically targeting most of the central mitochondrial metabolic pathways (Fig. 4). This proposed novel role is in good agreement with correlative evidence in the literature and can be expected to be at least partially conserved during eukaryotic evolution.
Figure 4. The different roles of mitochondrial CLPXP.
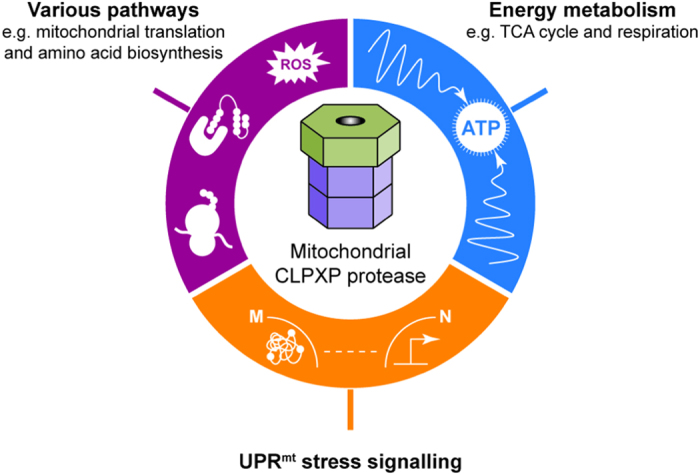
The proteolytic component CLPP degrades misfolded mitochondrial proteins in C. elegans to initiate UPRmt stress signalling, which leads to the induction of nuclear-encoded mitochondrial protective genes15. Our study identifies specific proteins and pathways that are likely targets of CLPXP, suggesting a dedicated role of this protease in regulation and maintenance of mitochondrial energy metabolism. The observation that CLPP-knockdown in cancer cells impairs oxidative phosphorylation is in support of this emerging view17. Furthermore, several chaperones and components of other mitochondrial pathways, e.g. those controlling translation, redox homeostasis, and amino acid biosynthesis, were found as additional potential targets or interactors of CLPXP.
Seemingly in opposition to this assumption is the fact that deletion or mutation of ClpP in mice35 or humans34 is associated with negative effects, while the P. anserina ClpP deletion strain in contrast shows an increase in its healthy lifespan19. These different outcomes may be due to distinctions in lifestyle and physiology of these organisms, which pose different challenges in adapting to internal or external insult to cellular and organismal homeostasis. It can be speculated, that in all these organisms absence or partial ablation of the mitochondrial CLPXP protease dysregulates mitochondrial and thus also cellular energy metabolism. While more complex eukaryotic organisms would be unable to cope with this adverse effect, as they require a fine-tuned adaptation of their metabolism in different tissues, the impaired mitochondrial integrity and cellular energy homeostasis in P. anserina could, at least under controlled laboratory conditions, potentially be compensated by activation of other quality control pathways, e.g. autophagy and/or mitophagy. Autophagy in particular is known to be induced in response to disturbances of cellular bioenergetics39,40 and was demonstrated to be a longevity assurance mechanism in P. anserina, mediating lifespan-extension under nutrient starvation conditions41.
Since dysfunction of mitochondria is associated with degenerative processes across a wide range of organisms, we anticipate the insights described herein to serve as a valuable precedent for detailed studies in various eukaryotic systems addressing CLPXP’s role in quality control and regulated proteolysis of mitochondrial proteins and in particular its impact on ageing and the development of disease.
Methods
P. anserina strains and cultivation
In the present study, the P. anserina wild-type strain ‘s’42, a P. anserina ClpP deletion strain (ΔPaClpP)24, a PaClpP deletion strain expressing human ClpP (ΔPaClpP/HsClpP_OEx)19, newly generated PaClpP deletion strains expressing constructs coding for inactive variants of PaCLPP (ΔPaClpP/PaClpPS135A) or HsCLPP (ΔPaClpP/HsClpPS153A), newly generated PaClpP deletion strains expressing constructs coding for C-terminally 3xFLAG-6xHis-tagged variants of active HsCLPP (ΔPaClpP/HsClpPWT-TAG) or inactive HsCLPP (ΔPaClpP/HsClpPTRAP-TAG) as well as a newly generated P. anserina ClpX deletion strain (ΔPaClpX) and a newly generated PaClpX deletion strain containing a human ClpX expression construct (ΔPaClpX/HsClpX) were used. All mutant strains are in the genetic background of the wild-type strain ‘s’. Strains were grown on standard cornmeal agar (BMM) at 27 °C under constant light42, unless otherwise stated.
Cloning procedures and generation of P. anserina mutants
To generate a PaClpP deletion strain expressing a construct coding for inactive PaCLPP, ΔPaClpP spheroblasts were transformed with the plasmid pPaClpPExS135A. This plasmid contains a hygromycin B resistance gene in the pKO743 vector backbone and the full length PaClpP gene under control of its wild-type promoter and terminator, with the codon for catalytic serine-135 (TCT) replaced with a codon for alanine (GCC). Transformants were selected for hygromycin B resistance and verified by Southern blot analysis. Strains with a single integration of pPaClpPExS135A were termed ΔPaClpP/PaClpPS135A. To construct pPaClpPExS135A, the PaClpP promoter (~1 kbp of the gene’s upstream region), gene, and terminator (~500 bp of the gene’s downstream region) were amplified from genomic wild-type DNA by PCR using the oligonucleotides ClpPEx5′HindIIIfor (5′-GTAAGCTTTGTGTATCGCCCC GTCCC-3′) and ClpPEx3′KpnIrev (5′-CCGGTACCTTTCATTGGTCCAGCAGC-3′), introducing HindIII and KpnI restriction sites (underlined). The amplicon was cloned into the pKO7 vector backbone (HindIII/KpnI digested) to obtain the plasmid pPaClpPEx2. Subsequently, a codon substitution (catalytic serine-135 to alanine-135) was introduced by PCR amplification of two fragments using the oligonucleotides ClpPEx5′HindIIIfor (see above) and StoA-PaClpP_rev (5′-GGCTGCGGCGCCACCAACGCAC-3′) or StoA-PaClpP_for (5′-ATGGCGGCTATTCTCCTG-3′) and ClpPEx3′KpnIrev (see above) with pPaClpPEx2 as a template. These fragments were cloned into the pKO7 vector backbone (HindIII/KpnI digested), resulting in the plasmid pPaClpPExS135A.
To obtain a constitutive expression vector coding for inactive HsCLPP (catalytic serine-153 to alanine-153), two fragments were amplified by PCR using the oligonucleotides BamHI-HsClpP_3for (5′-TAGGATCCATGTGGCCCGGAATATTG-3′) and S153A-HsClpP_6rev (5′-GGCGGCGGCCTGGCCCACGCA-3′) or S153A-HsClpP_7for (5′-ATGGGCTCCCTGCTTCTC-3′) and XbaI-HsClpP_4rev (5′-ATTCTAGATCAGGTGCTAGCTGGGAC-3′) with pHsClpPEx119 as a template. These fragments were cloned into the pExMtterhph24 vector backbone (BamHI/XbaI digested), resulting in the plasmid pHsClpPExS153A. After transformation of ΔPaClpP spheroblasts with pHsClpPExS153A, selection and verification of transformants was performed as described above and strains with a single integration were termed ΔPaClpP/HsClpPS153A.
A constitutive expression vector coding for active HsCLPP with a C-terminal (GGGGS)2 linker followed by a 3xFLAG-6xHis-tag (full amino acid sequence: GGGGSGGGGS DYKDHDGDYKDHDIDYKDDDDK HHHHHH) was constructed by PCR amplification of two fragments using the oligonucleotides HsClpP-NotI-for (5′-CAGCGGCCGCCGCAGCGG-3′) and HsClpP-L3F6H-rev (5′-CTTGTAGTCGCCGTCGTGGTCCTTGTAGTCGCTGCCGCCGCCGCCGCTGCCGCCGCCGCCGGTGCTAGCTGGGACAGG-3′) or HsClpP-L3F6H-for (5′-GACCACGACATCGACTACAAGGACGACGACGACAAGCACCACCACCACCACCACTAATCTAGAGTTATTCCCTCACTCAC-3′)and TPamT1-HindIII-rev (5′-CGCCAAGCTTACATTTGATCC-3′) with pHsClpPEx1 as template. Subsequent cloning of these fragments into the pHsClpPEx1 vector backbone (NotI/HindIII digested) yielded the plasmid pHsClpPWT-TAG. Using pHsClpPWT-TAG as a template, two fragments were amplified by PCR using the oligonucleotides BamHI-HsClpP_3for (see above) and S153A-HsClpP_6rev (see above) or S153A-HsClpP_7for (see above) and TPaMT1-rev (5′-ACCCTAACCGACTAACAGAC-3′). Cloning of these fragments into the pExMtterhph vector backbone (BamHI/XbaI digested) resulted in the plasmid pHsClpPTRAP-TAG, coding for inactive HsCLPP (catalytic serine-153 to alanine-153) with a C-terminal (GGGGS)2 linker followed by a 3xFLAG-6xHis-tag. After transformation of ΔPaClpP spheroblasts with pHsClpPWT-TAG or pHsClpPTRAP-TAG, selection and verification of transformants was performed as described above and strains with a single integration of the respective vector were termed ΔPaClpP/HsClpPWT-TAG or ΔPaClpP/HsClpPTRAP-TAG.
The generation of a PaClpX deletion strain was performed using a two-step protocol previously described in detail44, using the oligonucleotides KoClpX1 (5′-ATGGTACCTCATCCAGGTGCTCTAAC-3′), KoClpX2 (5′-GGAAGCTTGGTATGCTTGAGCTACTG-3′), KoClpX3 (5′-GGGGCACTAGTAAGGCGTTTTGGGTATGG-3′) and KoClpX4 (5′-AAGCGGCCGCCTCACTGGCAGAATTACG-3′) for PCR amplification of the required 5′- and 3′-flanking regions of the PaClpX gene to ultimately obtain a ΔPaClpX cosmid. This gene-specific replacement cosmid was used to transform P. anserina wild-type spheroplasts. Transformants were selected for phleomycin resistance and successful deletion of PaClpX was verified by Southern blot analysis. Strains lacking the PaClpX gene and instead harbouring the phleomycin resistance gene (ble) were termed ΔPaClpX. The human ClpX cDNA open reading frame was amplified by PCR using the oligonucleotides BamHI-HsClpX-for (5′-TATAATAAGGATCCATGCCCAGCTGCGGTG-3′) and XbaI-HsClpX-rev (5′-CGCGTCTAGATTAGCTGTTTGCAGCATC-3′), introducing BamHI and XbaI restriction sites (underlined). The amplicon was cloned into the pExMtterhph vector backbone (BamHI/XbaI digested). The resulting plasmid pHsClpXEx1 was subsequently transformed into P. anserina ΔPaClpX spheroplasts. Transformants were selected for hygromycin B resistance and verified by Southern blot analysis. Strains containing a single integration of pHsClpXEx1 were termed ΔPaClpP/HsClpX.
Transformation of P. anserina spheroblasts
Production and regeneration of P. anserina spheroblasts were performed as previously described42,45. After integrative transformation of spheroblasts45, transformants were selected by growth on regeneration agar containing either 75 μg ml−1 hygromycin b or 6 μg ml−1 phleomycin.
Lifespan determination
Experiments to determine the lifespan of P. anserina strains at 27 °C under constant light on M2 agar race tubes were performed as previously described42. The period of linear growth was recorded as lifespan in days.
Southern blot analysis
Isolation of P. anserina total DNA, DNA digestion, gel electrophoresis and Southern blotting were performed according to standard protocols. For Southern blot hybridization and detection, Digoxigenin-labelled hybridization probes (DIG DNA Labelling and Detection Kit, Roche Applied Science) were used according to the manufacturer’s protocol.
The HsClpP-specific hybridization probe was amplified by PCR using the oligonucleotides HsClpP_1for (5′-ATGTGGCCCGGAATATTG-3′) and HsClpP_2rev (5′-GCGAGTAGATGTCATAGG-3′) and corresponds to nucleotides 124–355 of the HsClpP cDNA sequence (GenBank: BC002956.1). The HsClpX-specific hybridization probe was amplified by PCR using the oligonucleotides HsClpX_1for (5′-CTGCAAAGAGCTCCTCTTAG-3′) and HsClpX_2rev (5′-ACGGGTGGATGATACAAAGG-3′) and corresponds to nucleotides 208–432 of the HsClpX cDNA sequence (GenBank: BC130373.1). The PaClpX-specific hybridization probe was amplified by PCR using the oligonucleotides PaClpX_1for (5′-TTTGAGCGCACAGGTTAC-3′) and PaClpX_2rev (5′-CGTCGAGGACAATGATTC-3′) and corresponds to a 530 bp fragment of the PaClpX genomic sequence (GenBank: FO904941.1; Region from 993541 to 995395). As a hybridization probe specific for the phleomycin resistance gene (ble), the 1293 bp BamHI-fragment of plasmid pKO344 was used.
Western blot analysis
Mitochondrial protein extracts from P. anserina strains were isolated according to a previously developed procedure42 and further purified by discontinuous sucrose gradient (20-36-50%) ultracentrifugation.
Separation of proteins by SDS-PAGE and subsequent transfer of proteins to PVDF membranes (Immobilon-FL, Merck Millipore) were performed following standard protocols. Blocking and antibody incubation of blotted PVDF membranes were performed according to the Odyssey ‘Western Blot Analysis’ handbook (LI-COR).
Primary antibodies were raised against a PaCLPP (UniProt: B2B591) specific synthetic peptide ([Ac]-CGTMLSADAKEGKH-[OH]; NEP) corresponding to amino acids 242–254 (antibody dilution: 1:400) and the PaPORIN (UniProt: B2B736) full-length protein (NEP; antibody dilution: 1:5,000). The human CLPP antibody (Abcam, product code: ab56455) was raised against an HsCLPP (Swiss-Prot: Q16740) recombinant fragment corresponding to amino acids 178–278 (antibody concentration: 1 μg ml−1). The human CLPX antibody (Abcam, product code: ab122644) was raised against an HsCLPX (Swiss-Prot: O76031) recombinant fragment corresponding to amino acids 39–132 (antibody dilution: 1:250). Recombinant active or inactive human CLPP with a C-terminal 3xFLAG-6xHis-tag was detected using a 3xFLAG- (Sigma-Aldrich; product code: F1804; antibody concentration: 1 μg ml−1) or a 6xHis-tag antibody (Abcam, product code: ab9136; antibody dilution: 1:1,000).
In all analyses, secondary antibodies conjugated with the infrared dyes IRDye 800CW or IRDye 680CW (LI-COR) were used (antibody dilution: 1:15,000–20,000). The Odyssey Infrared Imaging System (LI-COR) was used for detection of western blots.
Blue native PAGE
BN-PAGE was performed according to the protocol developed by Schägger and colleagues46. Sample preparation and western blotting of BN-PAGE gels was performed as previously described19.
Tandem affinity purification
Mitochondrial protein extracts from three independent biological replicates of the P. anserina strains ΔPaClpP, ΔPaClpP/HsClpPWT-TAG or ΔPaClpP/HsClpPTRAP-TAG were obtained as described above. For each individual purification experiment, 1 mg of freshly isolated mitochondria were pelleted by centrifugation at 20,000 g for 10 min at 4 °C in a total volume of 0.5 ml ice-cold mitochondria isotonic buffer (20 mM HEPES-KOH, pH 7.4, 0.6 M sorbitol). After discarding the supernatant, the mitochondrial pellets were flash-frozen in liquid nitrogen and stored at −80 °C until further use.
Mitochondria were lysed at 5 mg ml−1 in a total volume of 200 μl FLAG lysis buffer (20 mM HEPES-KOH, pH 7.4, 300 mM KCl, 1 mM EDTA, 10% w/v glycerol, 1x protease inhibitor cocktail set IV from Calbiochem) with 1% w/v digitonin for 30 min on ice. Lysates were cleared by centrifugation at 20,000 g for 10 min at 4 °C, adjusted to 1 ml with lysis buffer and incubated with 30 μl packed gel volume of anti-FLAG M2 magnetic beads (Sigma-Aldrich) for 2 hours at 4 °C with rotation. After binding, beads were washed with 3 × 200 μl FLAG wash buffer (20 mM HEPES-KOH, pH 7.4, 300 mM KCl, 10% w/v glycerol, 0.01% w/v digitonin) and elution was performed with 2× 150 μl FLAG elution buffer (20 mM HEPES-KOH, pH 7.4, 300 mM KCl, 10% w/v glycerol) containing 3xFLAG peptide (Sigma-Aldrich) at 150 ng µl-1. Pooled eluates were supplemented with imidazole (final concentration: 20 mM) and TWEEN 20 (final concentration: 0.01%) and adjusted to a final volume of 700 μl with FLAG elution buffer without 3xFLAG peptide.
The second round of purification was performed by incubating the adjusted first round eluates with 50 μl ‘Dynabeads for His-tag Isolation and Pulldown’ (Life Technologies/Thermo Fisher Scientific) for 30 min at 4 °C with rotation. After binding, beads were washed with 4 × 300 μl His wash buffer (20 mM HEPES-KOH, pH 7.4, 300 mM KCl, 20 mM imidazole, 10% glycerol, 0.01% TWEEN 20) and transferred to a new reaction tube during the last washing step for subsequent mass spectrometric analysis.
Peptide mass fingerprinting (PMF)
Dynabeads with bound affinity-purified proteins were washed three times using ABC buffer (50 mM NH4HCO3, pH 8.5, 300 mM KCl and 10% w/v glycerol). Proteins were subsequently denatured using 6 M urea for 2 min at 70 °C, reduced using 5 mM TCEP (Sigma-Aldrich) for 30 min at room temperature and alkylated using 20 mM iodoacetamide (Thermo Fisher Scientific) again for 30 min at room temperature. The samples were then incubated with the protease Lys-C (final concentration: 4 μg ml−1; Roche Pharmaceuticals) overnight at 37 °C. After 1:1 dilution and addition of CaCl2 to a final concentration of 1 mM, the samples were incubated with trypsin (final concentration: 10 μg ml−1; Serva) at 37 °C overnight. Subsequently, samples were centrifuged at 1,000 g for 5 min at room temperature, acidified using 1% formic acid and purified using C18-ZipTips (Merck Millipore) according to the manufacturer’s protocol. The elution fractions were dried using a SpeedVac (Thermo Fisher Scientfic) and stored at −20 °C until further use.
Proteolytic digests were dissolved in 5% acetonitrile with 0.1% formic acid and loaded on reverse phase columns (trapping column: particle size 3 μm, C18, L = 20 mm; analytical column: particle size <2 μm, C18, L = 50 cm; PepMap, Dionex/Thermo Fisher Scientific) using a nano-HPLC (Dionex – UltiMate 3000 RSLCnano, Thermo Fisher Scientific). Peptides were eluted in gradients of water (buffer A: water with 5% v/v acetonitrile and 0.1% formic acid) and acetonitrile (buffer B: 20% v/v water and 80% v/v acetonitrile and 0.1% formic acid). All LC-MS-grade solvents were purchased from Fluka. Typically, gradients were ramped from 5% to 70% buffer B in 200 minutes at flowrates of 300 nl min−1. Peptides eluting from the column were ionised online using a ‘nanoFlex ESI’-source and analysed either in a ‘Orbitrap Elite’ mass spectrometer or in a ‘Q Exactive Plus’ mass spectrometer (Thermo Fisher Scientific). Mass spectra were acquired over the mass range 250–1650 m z−1 and sequence information was acquired by computer-controlled, data-dependent automated switching to MS/MS mode using collision energies based on mass and charge state of the candidate ions. All samples were run in technical triplicates.
The data sets were processed using a standard proteomics script with the software ‘Proteome Discoverer 1.4’ (Thermo Fisher Scientific) and exported as mascot generic files. Proteins were identified by matching the derived mass lists against the ‘P. anserina Genome Project’ database release version 6.32 (downloaded from http://podospora.igmors.u-psud.fr/) on a local Mascot server (Matrix Science, UK). In general, a mass tolerance of 2 ppm for parent ions and 0.5 Da for fragment spectra, two missed cleavages, oxidation of Met, and fixed modification of carbamidomethyl cysteine were selected as matching parameters in the search program. For protein identification using spectral counting, a set of stringent selection criteria were applied to the dataset.
We set a manual Mascot score threshold of 40 and only considered proteins present in at least two of the three independent biological replicates of each sample. Proteins also found in the background control sample (ΔPaClpP) were subtracted from the sets of proteins co-purifying with either HsCLPP variant. Manual inspection of the data showed that a small subset of proteins found in all three samples was highly enriched in the ΔPaClpP/HsClpPWT-TAG and/or the ΔPaClpP/HsClpPTRAP-TAG sample over the background control sample (using spectral counting, enrichment factor >3) and these proteins were also retained for subsequent analyses. The resulting sets of proteins co-purifying with each of the two HsCLPP variants over the background control sample were compared and proteins classified as specifically co-purifying with HsCLPPWT-TAG, HsCLPPTRAP-TAG, or with both. Proteins that were highly enriched in the ΔPaClpP/HsClpPTRAP-TAG sample over the ΔPaClpP/HsClpPWT-TAG sample (using spectral counting, enrichment factor >3) were also classified as specifically co-purifying with HsCLPPTRAP-TAG. The resulting sets were further refined by eliminating all proteins not present with at least more than one unique peptide on average across all biological replicates of the respective sample. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD003264.
Additional Information
Data availability: Access to the proteome data associated with this manuscript is available upon request.
How to cite this article: Fischer, F. et al. Identification of potential mitochondrial CLPXP protease interactors and substrates suggests its central role in energy metabolism. Sci. Rep. 5, 18375; doi: 10.1038/srep18375 (2015).
Supplementary Material
Acknowledgments
We would like to thank Imke Wüllenweber for excellent technical assistance. This study was supported by resources of the state of Hesse to H.D.O. He is a member of the DFG-funded Cluster of Excellence-Macromolecular Complexes.
Footnotes
Author Contributions H.D.O. and F.F. designed this study. F.F. and J.L. performed the experiments and analysed the data. F.F. and H.D.O. wrote the manuscript. H.D.O supervised the work. All authors read and approved the final manuscript.
References
- Lill R. Function and biogenesis of iron-sulphur proteins. Nature 460, 831–838 (2009). [DOI] [PubMed] [Google Scholar]
- Chandel N. S. Evolution of mitochondria as signaling organelles. Cell Metab 22, 204–206 (2015). [DOI] [PubMed] [Google Scholar]
- Kroemer G. & Reed J. C. Mitochondrial control of cell death. Nat. Med. 6, 513–519 (2000). [DOI] [PubMed] [Google Scholar]
- Hamann A., Brust D. & Osiewacz H. D. Apoptosis pathways in fungal growth, development and ageing. Trends Microbiol. 16, 276–283 (2008). [DOI] [PubMed] [Google Scholar]
- Lin M. T. & Beal M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 (2006). [DOI] [PubMed] [Google Scholar]
- Gaude E. & Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab 2, 10, 10.1186/2049-3002-2-10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace D. C., Fan W. & Procaccio V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 5, 297–348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C., Blasco M. A., Partridge L., Serrano M. & Kroemer G. The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osiewacz H. D. & Bernhardt D. Mitochondrial quality control: impact on aging and life span - a mini-review. Gerontology 59, 413–420 (2013). [DOI] [PubMed] [Google Scholar]
- Fischer F., Hamann A. & Osiewacz H. D. Mitochondrial quality control: an integrated network of pathways. Trends Biochem. Sci. 37, 284–292 (2012). [DOI] [PubMed] [Google Scholar]
- Szklarczyk R., Nooteboom M. & Osiewacz H. D. Control of mitochondrial integrity in ageing and disease. Philos. Trans. R. Soc. Lond B Biol. Sci. 369, 20130439, 10.1098/rstb.2013.0439 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C. M., Fiorese C. J. & Lin Y. F. Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 23, 311–318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros P. M., Langer T. & Lopez-Otin C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 16, 345–359 (2015). [DOI] [PubMed] [Google Scholar]
- Yu A. Y. & Houry W. A. ClpP: a distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 581, 3749–3757 (2007). [DOI] [PubMed] [Google Scholar]
- Pellegrino M. W., Nargund A. M. & Haynes C. M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 1833, 410–416 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux K. J., Kim D. I., Raida M. & Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole A. et al. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 27, 864–876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osiewacz H. D. Mitochondrial quality control in aging and lifespan control of the fungal aging model Podospora anserina. Biochem. Soc. Trans. 39, 1488–1492 (2011). [DOI] [PubMed] [Google Scholar]
- Fischer F., Weil A., Hamann A. & Osiewacz H. D. Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nat. Commun. 4, 1397, 10.1038/ncomms2397 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J. M., Neher S. B., Kim Y. I., Sauer R. T. & Baker T. A. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 11, 671–683 (2003). [DOI] [PubMed] [Google Scholar]
- Neher S. B. et al. Proteomic profiling of ClpXP substrates after DNA damage reveals extensive instability within SOS regulon. Mol. Cell 22, 193–204 (2006). [DOI] [PubMed] [Google Scholar]
- Bhat N. H., Vass R. H., Stoddard P. R., Shin D. K. & Chien P. Identification of ClpP substrates in Caulobacter crescentus reveals a role for regulated proteolysis in bacterial development. Mol. Microbiol. 88, 1083–1092 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J. et al. Trapping and proteomic identification of cellular substrates of the ClpP protease in Staphylococcus aureus. J. Proteome. Res. 12, 547–558 (2013). [DOI] [PubMed] [Google Scholar]
- Luce K. & Osiewacz H. D. Increasing organismal healthspan by enhancing mitochondrial protein quality control. Nat. Cell Biol. 11, 852–858 (2009). [DOI] [PubMed] [Google Scholar]
- Maurizi M. R., Clark W. P., Kim S. H. & Gottesman S. Clp P represents a unique family of serine proteases. J. Biol. Chem. 265, 12546–12552 (1990). [PubMed] [Google Scholar]
- Corydon T. J. et al. A human homologue of Escherichia coli ClpP caseinolytic protease: recombinant expression, intracellular processing and subcellular localization. Biochem. J. 331 (Pt 1), 309–316 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause F. Detection and analysis of protein-protein interactions in organellar and prokaryotic proteomes by native gel electrophoresis: (Membrane) protein complexes and supercomplexes. Electrophoresis 27, 2759–2781 (2006). [DOI] [PubMed] [Google Scholar]
- Joshi S. A., Hersch G. L., Baker T. A. & Sauer R. T. Communication between ClpX and ClpP during substrate processing and degradation. Nat. Struct. Mol. Biol. 11, 404–411 (2004). [DOI] [PubMed] [Google Scholar]
- Gerth U. et al. Clp-dependent proteolysis down-regulates central metabolic pathways in glucose-starved Bacillus subtilis. J. Bacteriol. 190, 321–331 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowth B. R. et al. Substrate recognition and processing by a Walker B mutant of the human mitochondrial AAA+ protein CLPX. J. Struct. Biol. 179, 193–201 (2012). [DOI] [PubMed] [Google Scholar]
- Puccio H. & Koenig M. Friedreich ataxia: a paradigm for mitochondrial diseases. Curr. Opin. Genet. Dev. 12, 272–277 (2002). [DOI] [PubMed] [Google Scholar]
- Guillon B. et al. Frataxin deficiency causes upregulation of mitochondrial Lon and ClpP proteases and severe loss of mitochondrial Fe-S proteins. FEBS J. 276, 1036–1047 (2009). [DOI] [PubMed] [Google Scholar]
- Rouault T. A. Mammalian iron-sulphur proteins: novel insights into biogenesis and function. Nat. Rev. Mol. Cell Biol. 16, 45–55 (2015). [DOI] [PubMed] [Google Scholar]
- Jenkinson E. M. et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 92, 605–613 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gispert S. et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 22, 4871–4887 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roblick U. J. et al. Sequential proteome alterations during genesis and progression of colon cancer. Cell Mol. Life Sci. 61, 1246–1255 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishigaki R. et al. Proteomic identification of differentially-expressed genes in human gastric carcinomas. Proteomics. 5, 3205–3213 (2005). [DOI] [PubMed] [Google Scholar]
- Goard C. A. & Schimmer A. D. Mitochondrial matrix proteases as novel therapeutic targets in malignancy. Oncogene 33, 2690–2699 (2014). [DOI] [PubMed] [Google Scholar]
- Rabinowitz J. D. & White E. Autophagy and metabolism. Science 330, 1344–1348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kundu M., Viollet B. & Guan K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuppertz L., Hamann A., Pampaloni F., Stelzer E. & Osiewacz H. D. Identification of autophagy as a longevity-assurance mechanism in the aging model Podospora anserina. Autophagy. 10, 822–834 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osiewacz H. D., Hamann A. & Zintel S. Assessing organismal aging in the filamentous fungus Podospora anserina. Methods Mol. Biol. 965, 439–462 (2013). [DOI] [PubMed] [Google Scholar]
- Kunstmann B. & Osiewacz H. D. The S-adenosylmethionine dependent O-methyltransferase PaMTH1: a longevity assurance factor protecting Podospora anserina against oxidative stress. Aging (Albany NY) 1, 328–334 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann A., Krause K., Werner A. & Osiewacz H. D. A two-step protocol for efficient deletion of genes in the filamentous ascomycete Podospora anserina. Curr. Genet. 48, 270–275 (2005). [DOI] [PubMed] [Google Scholar]
- Osiewacz H. D., Skaletz A. & Esser K. Integrative transformation of the ascomycete Podospora anserina: identification of the mating-type locus on chromosome VII of electrophoretically separated chromosomes. Appl. Microbiol. Biotechnol. 35, 38–45 (1991). [DOI] [PubMed] [Google Scholar]
- Wittig I., Braun H. P. & Schägger H. Blue native PAGE. Nat. Protoc. 1, 418–428 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.