

Abstract
The neurobiology of post-traumatic stress disorder (PTSD) remains unclear. Intense stress promotes PTSD, which has been associated with exaggerated startle and deficient sensorimotor gating. Here, we examined the long-term sequelae of a rodent model of traumatic stress (repeated predator exposure) on amygdala systems that modulate startle and prepulse inhibition (PPI), an operational measure of sensorimotor gating. We show in rodents that repeated psychogenic stress (predator) induces long-lasting sensitization of basolateral amygdala (BLA) noradrenergic (NE) receptors (α1) via a corticotropin-releasing factor receptor 1 (CRF-R1)-dependent mechanism, and that these CRF1 and NE α1 receptors are highly colocalized on presumptive excitatory output projection neurons of the BLA. A profile identical to that seen with predator exposure was produced in nonstressed rats by intra-BLA infusions of CRF (200 ng/0.5 μl), but not by repeated NE infusions (20 μg/0.5 μl). Infusions into the adjacent central nucleus of amygdala had no effect. Importantly, the predator stress- or CRF-induced sensitization of BLA manifested as heightened startle and PPI deficits in response to subsequent subthreshold NE system challenges (with intra-BLA infusions of 0.3 μg/0.5 μl NE), up to 1 month after stress. This profile of effects closely resembles aspects of PTSD. Hence, we reveal a discrete neural pathway mediating the enhancement of NE system function seen in PTSD, and we offer a model for characterizing potential new treatments that may work by modulating this BLA circuitry.
SIGNIFICANCE STATEMENT The present findings reveal a novel and discrete neural substrate that could underlie certain core deficits (startle and prepulse inhibition) that are observed in post-traumatic stress disorder (PTSD). It is shown here that repeated exposure to a rodent model of traumatic stress (predator exposure) produces a long-lasting sensitization of basolateral amygdala noradrenergic substrates [via a corticotropin-releasing factor (CRF)-dependent mechanism] that regulate startle, which is exaggerated in PTSD. Moreover, it is demonstrated that the sensitized noradrenergic receptors colocalize with CRF1 receptors on output projection neurons of the basolateral amygdala. Hence, this stress-induced sensitization of noradrenergic receptors on basolateral nucleus efferents has wide-ranging implications for the numerous deleterious sequelae of trauma exposure that are seen in multiple psychiatric illnesses, including PTSD.
Keywords: corticotropin-releasing factor, corticotropin-releasing hormone, noradrenergic, prepulse inhibition, schizophrenia, sensorimotor gating
Introduction
A deficit in information-filtering mechanisms such as sensorimotor gating is presumed to contribute to cognitive and affective impairment in neuropsychiatric disorders like schizophrenia and post-traumatic stress disorder (PTSD). These deficits can be quantified using prepulse inhibition (PPI), a paradigm in which startle responses are inhibited by the presentation of a weak prestimulus immediately before the startling event (Hoffman and Ison, 1980; Grillon et al., 1996; Braff et al., 2001). PPI deficits are observed in both illnesses, and stress is thought to exacerbate or precipitate symptomatology in them, possibly through a long-lasting hypersensitivity to perceived environmental threat (Norman and Malla, 1993; Walker and Diforio, 1997). Among the most widely implicated systems in stress-induced psychopathology are corticotropin-releasing factor (CRF) and norepinephrine (NE), both of which are activated by stress, and are hypothesized to be dysfunctional in PTSD and schizophrenia (Baker et al., 1999). Accordingly, stress, CRF, or NE receptor stimulation disrupts PPI (Conti et al., 2002; Risbrough et al., 2004; Groenink et al., 2008; Alsene et al., 2011; Sutherland and Conti, 2011; Bakshi et al., 2012).
CRF and NE systems interact reciprocally, but the extent to which this interaction modulates PPI is unclear (Gresack and Risbrough, 2011). The amygdala is a key site in which CRF and NE modulate stress-related processes, and it contains high levels of CRF, NE, and their receptors (Pickel et al., 1974; Rainbow et al., 1984; Pieribone et al., 1994; Chalmers et al., 1995; Pitkanen et al., 2000). Recently, the stimulation of NE receptors in amygdala was found to disrupt PPI (Alsene et al., 2011). Moreover, there is evidence for priming or plasticity in amygdala with repeated stimulation (Rainnie et al., 2004; Shekhar et al., 2005). Thus, one mechanism by which exposure to repeated stress could lead to enduring sensorimotor gating deficits in schizophrenia and PTSD is via stress-induced changes in amygdala CRF and NE systems.
We hypothesized that plasticity between these two systems within the amygdala could promote deficient PPI, such that previously ineffective manipulations would now disrupt PPI, and thereby model the phenomenon of a mild or innocuous stimulus triggering a pathological response when applied to a system perturbed by repeated psychological stress. To test this hypothesis, we repeatedly stimulated NE or CRF receptors within the amygdaloid basolateral nucleus (BLA) or central nucleus (CeA) and tested whether these manipulations altered PPI responses to subthreshold challenge with CRF or NE. Furthermore, we determined whether repeated exposure to a live predator, an animal model of traumatic psychological stress, would produce similar effects. Finally, we explored whether CRF receptor signaling is necessary for stress-induced plasticity in BLA NE systems. Such plasticity could potentially contribute to exaggerated responses to environmental stimuli long after the history of trauma has ended, similar to the enduring sensorimotor gating abnormalities seen in stress-sensitive disorders such as schizophrenia and PTSD (Grillon et al., 1996).
Materials and Methods
Subjects.
Male Sprague Dawley rats (300–325 g, Harlan Laboratories) were pair housed in clear cages in the vivarium (lights on for 12 h at 7:00 A.M.) with ad libitum access to food and water; experiments were performed between 10:00 A.M. and 3:00 P.M. Facilities/procedures followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin. As in the study by Bakshi and Kelley (1991), rats were given indwelling cannulae based on coordinates obtained from Paxinos and Watson [1998; BLA: anteroposterior (AP), −3.0 mm relative to bregma; lateromedial (LM), ±4.8 mm from midline; dorsoventral (DV), −5.8 mm from skull surface; CeA: AP, −2.1 mm relative to bregma; LM, ±3.85 mm from midline; DV, −5.4 mm from skull surface]. Rats recovered 1 week before undergoing testing.
Drugs and microinfusions.
CRF (Bachem and Peninsula) was dissolved in sterile distilled H2O (dH2O). l-(-)-Norepinephrine bitartrate, (±)-phenylephrine hydrochloride, (±)-isoproterenol hydrochloride, and yohimbine hydrochloride (Sigma-Aldrich) were dissolved in isotonic saline. NBI27914 hydrochloride (Tocris Bioscience) was dissolved with sonication in a vehicle of sterile dH2O containing 5% DMSO and 5% cremaphor (Sigma-Aldrich). Using infusion methodology as described in Perry et al. (2009), drugs were given in BLA (final DV coordinate, −8.3 mm from skull surface) or CeA (final DV coordinate, −7.9 mm from skull surface).
Predator stress.
Rats were placed individually in a protective metal wire cage (7 × 8 × 9 inches) that was secured to the floor of the homecage of a ferret, a natural predator of rats, for 5 min. This procedure allowed animals to see, hear, and smell each other, but did not permit physical contact.
Startle chambers.
Procedures for startle/PPI testing were the same as those in the study by Alsene et al. (2011). The session consisted of a 65 dB background noise, pulse-alone trials (40 ms, 120 dB broadband bursts), prepulse plus pulse trials (20 ms noises that were 3, 9, or 15 dB above the background noise and were presented 100 ms before the onset of the 120 dB pulse), and no-stimulus trials. Rats underwent testing with a sham infusion before the experiments began, and PPI and startle values from this test were used to create equally matched treatment groups. The startle response to the onset of the 120 dB burst was recorded for 100 ms for each pulse-alone and prepulse plus pulse trial. The following two measurements were calculated for each rat: startle magnitude was the average of the startle responses to all pulse-alone trials; and PPI was a percentage score for each prepulse plus pulse trial type, as follows: %PPI = 100 − ((startle response for prepulse plus pulse trial)/(startle response for pulse-alone trial)). Because no significant interactions were seen with prepulse intensity and any other factor in any experiment, a single composite %PPI score (average PPI collapsed across all three prepulse intensities) was calculated for each subject, and this was used for all analyses.
Histology.
Rats were perfused transcardially with 10% formalin (Sigma-Aldrich), 60 μm sections were taken and stained with cresyl violet, and placements were verified by an experimenter blind to the data; rats with missed placements were excluded from behavioral analyses, as reflected in the sample sizes.
Immunohistochemistry.
Forty micrometer coronal sections from BLA were processed (with washes between steps). For double-immunofluorescence labeling of α1 and CRF1 receptors (CRF1-Rs), slices were incubated in 5% donkey serum, then α1-R primary antibody (1:500, 72 h; PA1-047, Thermo Scientific), then Alexa Fluor 488 donkey anti-rabbit secondary antibody (1:200, 2 h; Life Technologies), then primary antibody for CRF1/2 receptor (1:500, 48 h; sc-1757, Santa Cruz Biotechnology), and then Alexa Fluor 594 donkey anti-goat secondary antibody (1:200, 2 h; Life Technologies). Sections were counterstained with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; Life Technologies) and were mounted with Prolong Anti-Fade Medium (Life Technologies). For colabeling α1-R and GAD67, GAD67 was labeled with GAD MAB5406 (1:1000, 24 h; Millipore) and Alexa Fluor 594 donkey anti-mouse secondary antibody. For double labeling with NeuN, mouse anti-NeuN MAB377 (Millipore) primary antibody and Alexa Fluor 594 donkey anti-mouse secondary antibody were used. For control sections, antibodies were preabsorbed with their respective peptides (α1 receptor (α1-R) peptide, PEP-216, Thermo Scientific; CRF1/2 receptor peptide, sc-1757p, Santa Cruz Biotechnology) and were taken through steps for immunofluorescence or immunoperoxidase labeling. For the secondary antibody-alone controls, the primary antibody was omitted. With the CRF1/2 receptor antibody in the fluorescence labeling, we clearly observed histological features consistent with cell-surface receptors (i.e., features with a “ring-shaped” distribution of cytoplasmic labeling) throughout the BLA. We considered these structures to be presumptive CRF1 receptors for the following reasons: (1) they colocalized with DAPI and NeuN labeling (hence, they were neuronal structures); or (2) they were eliminated after preabsorption with CRF-R1 peptide. We also observed additional labeling that did not appear to be receptor-like and that was not eliminated by antibody preabsorption. These structures strongly resembled blood vessels; given that the CRF1-R1 antibody used in our studies also labels CRF2 receptors, which are known to be expressed in “brain arterioles” (Lovenberg et al., 1995), it is reasonable to interpret this other labeling as blood vessels.
For peroxidase labeling, the same primary antibodies were used; endogenous peroxidase activity was inhibited with a quenching solution, then sections were incubated with primary antibody and exposed to goat anti-rabbit biotinylated secondary antibody (Vector Laboratories), an avidin—biotin-peroxidase complex (ABC complex, Vector Laboratories); and were stained with diaminobenzidine (Vector Laboratories), to yield a brown precipitate, or with nickel enhancement, to yield black precipitate in some cases. For double labeling with GAD, the steps followed were the same as those above except that horse anti-mouse secondary antibody was used for GAD labeling. Sections were exposed to Vector SG Chromagen (Vector Laboratories), mounted, dried, taken through graded alcohols, cleared in xylene, and coverslipped with Permount (Fisher).
For retrograde labeling of BLA from NAcc (nucleus accumbens, a prominent efferent target of the BLA) neurons, TransFluoSpheres Microspheres T8870 (0.4 μm diameter, Life Technologies) linked to Far Red fluorescent dye and diluted at 1:1 were delivered into NAcc (in mm from bregma: AP, +1.6, LM, +1.4, DV, −7) in 0.2 μl over 2 min (injectors in place for 10 more min; Meloni et al., 2006). After 9 d, sections through BLA were taken through immunofluorescence labeling steps for CRF1 receptors. Bright-field images were captured with a Leica DMI 600B Microscope, using Image-Pro AMS version 6.0 software. All fluorescence images were captured using a Nikon A1 Confocal Microscope at the W.M. Keck Laboratory for Biological Imaging and at the Waisman Center Cellular and Molecular Neuroscience Core of the University of Wisconsin, Madison. Alexa Fluor 488, Alexa Fluor 594, and TransFluoSpheres were excited with filters BP 500–550 nm, BP 570–620 nm, or BP 663–738 nm to elicit Green, Red, and Far Red Fluorophores (pseudo-colored blue for the purposes of this experiment), respectively.
Experimental design and statistical analyses.
Separate groups of rats (N = 7–12/study) were used for behavioral each study; each study was designed as a completely within-subjects protocol. All rats underwent stereotaxic surgery for the implantation of guide cannulae (aimed at either the BLA or CeA), recovered with daily handling and health checks for a week, and then were acclimated to the startle chambers twice before testing. Each behavioral study (Experiments 1–5) was then performed using the general timeline in Figure 1.
Figure 1.

General timeline used for all behavioral experiments. After recovery from surgery, rats underwent a mock infusion (injectors lowered without infusate), followed by the repeated treatment regimen (in purple; drug infusion, ferret exposure, or both), another mock infusion, a challenge drug infusion (dark blue), and then an additional mock infusion. Data from these latter two mock infusions were averaged and termed mock 2; mock 2 was used for all statistical comparisons. Amount of time between consecutive test days is indicated in hours along the timeline. Each test day (vertical line) included prepulse inhibition/startle testing immediately after the treatment (mock, repeated regimen, or challenge).
Hence, in each study, rats first received a mock injection (injectors lowered but no infusate delivered; mock injections were used in lieu of vehicle infusions to minimize the number of intraparenchymal infusions per rat to preserve the integrity of the tissue in the area of the injection site—this was particularly important for experiments in which the repeated treatment involved drug infusions). Then, rats underwent a repeated treatment regimen consisting of three presentations of the manipulation (for a list of what the repeated treatment was for each study, see Table 1). The repeated treatment was followed by another mock injection, and subsequently by a challenge drug injection (for what the challenge injection was for each study, see Table 1), and then a final mock injection. Startle and PPI were measured after every event displayed on the above timeline (i.e., mock injections, repeated treatments, and challenge injections), and 1–3 d (rats remained in homecages in the vivarium but were handled gently by the experimenter) separated consecutive events on the above timeline. Because no differences were found in any experiment between the two mock injections that “bracketed” the challenge injection (see Fig. 1; both of these are valid comparisons for the challenge injection since each of them is time matched to it), data from these 2 d were averaged and designated as “mock2” for the figures, tables, and analyses.
Table 1.
Experimental design for behavioral studies (experiments 1–5)
| Repeated treatment | Challenge injection | |
|---|---|---|
| Experiment 1 (BLA) | Ferret exposure (N = 8) | 0.3 μg NE, 4 d after ferret exposure |
| 0.3 μg NE, 11 d after ferret exposure | ||
| 0.3 μg NE, 18 d after ferret exposure | ||
| 1 mg/kg yohimbine, 28 d after ferret exposure | ||
| Experiment 2 (BLA) | 200 ng CRF (N = 7) | 0.3 μg NE |
| 20 μg NE (N = 12) | 200 ng CRF | |
| Vehicle (N = 8) | 0.3 μg NE | |
| Experiment 3 (CeA) | 200 ng CRF (N = 9) | 0.3 μg NE |
| 20 μg NE (N = 12) | 200 ng CRF | |
| Vehicle (N = 8) | 0.3 μg NE | |
| Experiment 4 (BLA) | NBI 1000 ng before ferret exposure (N = 8) | 0.3 μg NE |
| NBI 1000 ng after ferret exposure (N = 8) | 0.3 μg NE | |
| Ferret exposure (N = 11) | NBI 1000 ng before 0.3 μg NE | |
| Experiment 5 (BLA) | Ferret exposure (N = 8) | 30 μg PHEN |
| Ferret exposure (N = 9) | 30 μg ISO |
Five experiments were performed; each experiment except for Experiment 1 contained separate groups of rats that received different treatment regimens. The brain region into which injections were made for different groups of rats is indicated next to each experiment number. Repeated treatment = the manipulation (either ferret exposure and/or intracranial drug infusion) each group of rats underwent repeatedly (three exposures total, with consecutive exposures separated by 3d) as well as the number of rats per group; Challenge injection = the drug that was tested in each group of rats after the completion of the repeated treatment regimen (for all experiments except Experiment 1, the challenge injection took place 4 d after the termination of the repeated treatment for that particular group of rats). In Experiment 1, the initial challenge injection took place 4 d after repeated treatment, but there were additional challenge injections subsequently. Unless indicated otherwise, all doses are 0.5 μl/side. NBI, NBI27914. For Experiment 4, the first group of rats received intra-BLA infusion of NBI immediately before each ferret exposure; the second group received it 30 min after each ferret exposure; the third group did not receive it with ferret exposures, but instead received NBI into BLA immediately before the NE challenge injection.
This design was selected because it permitted direct comparisons of startle/PPI levels from before the repeated treatment with those from after the repeated treatment (i.e., comparison of the initial mock injection with mock2); this comparison provides a measure of whether the repeated treatment enacts a carryover effect on baseline startle/PPI levels. Moreover, this design allows for assessment of the short-term effects of each type of repeated treatment (i.e., first presentation of the repeated treatment versus the initial mock injection, which represents the no-treatment condition) as well as potential alterations in repeated treatment effects with each presentation (i.e., the first presentation of the repeated treatment vs the third presentation of the repeated treatment). Thus, possible habituation or sensitization of responses with each type of repeated treatment can be detected. Finally, interactions between neuropharmacological systems can be assessed by examining startle/PPI levels in response to the challenge injection, which was always different from the repeated treatment used in each particular study (i.e., did the challenge produce a different effect in rats that underwent a particular repeated treatment regimen than in rats that did not receive that repeated treatment regimen?).
These designated research questions were instantiated into appropriately matched statistical analyses as follows. An omnibus within-subjects ANOVA was conducted initially for each study to determine whether any of the treatments had any effects; for every study, separate omnibus ANOVAs analyzed startle and PPI data. Significant effects indicated by these ANOVAs were followed up subsequently with analysis of simple main effects using a one-factor ANOVA that included the initial mock injection and the three repeated treatment days; this ANOVA indicated whether the repeated treatment regimen produced short-term behavioral effects. Significant ANOVAs were further analyzed with Bonferroni-corrected t tests to minimize the occurrence of a type 1 error. Planned contrasts between mock2 and the challenge injection were used to assess the effects of the crossover treatment in each study; comparison of mock1 (the mock infusion before the repeated stress/infusion regimens) and mock2 evaluated conditioned or nonspecific carryover effects from the repeated regimens on baseline startle/PPI. For Experiment 1, several additional time points (later challenge injections far removed from the repeated ferret regimen) were assessed by comparison to their corresponding mock injections. Finally, separate sets of experimentally naive rats were used for Experiment 6, in which all anatomical labeling was conducted as described below.
Results
Omnibus ANOVA outcomes for each study are presented in Table 2 (for PPI) and Table 3 (for startle); these were each based on a completely within-subjects design for each study (please see Experimental design section in Materials and Methods) in which each subject received all the manipulations within a particular group of rats. For the sake of brevity, these omnibus ANOVAs are not repeated throughout the following sections, which instead focus on the follow-up analyses of simple main effects and Bonferroni-corrected comparisons of means.
Table 2.
Omnibus ANOVA results for PPI in behavioral studies (experiments 1–5)
| Study | Omnibus ANOVA for PPI | |
|---|---|---|
| Experiment 1 | Repeated ferret exposure | F(10,70) = 4.1, p < 0.005* |
| Experiment 2 | Repeated CRF infusions in BLA | F(5,30) = 2.5, p < 0.049* |
| Repeated NE infusions in BLA | F(5,55) = 6.1, p < 0.005* | |
| Repeated vehicle infusions in BLA | F(5,35) = 0.9, p < 0.514 | |
| Experiment 3 | Repeated CRF infusions in CeA | F(5,40) = 1.8, p < 0.117 |
| Repeated NE infusions in CeA | F(5,55) = 1.3, p < 0.251 | |
| Repeated vehicle infusions in CeA | F(4,28) = 0.4, p < 0.794 | |
| Experiment 4 | NBI before each repeated ferret exposure | F(5,35) = 0.5, p < 0.705 |
| NBI after each repeated ferret exposure | F(5,35) = 4.3, p < 0.003* | |
| Repeated ferret exposure and then NBI with NE challenge | F(5,50) = 7.5, p < 0.005* | |
| Experiment 5 | Repeated ferret exposure and then PHEN injection | F(5,35) = 3.3, p < 0.013* |
| Repeated ferret exposure and then ISO injection | F(5,40) = 4.4, p < 0.003* |
A single-factor within-subjects ANOVA of %PPI was performed for each group of rats in order to determine whether any manipulation (either the repeated treatment regimen or the subsequent challenge injection) affected %PPI in any study. Based on significant omnibus ANOVAs within each separate group of rats, subsequent analyses of simple main effects and comparisons of individual means were performed, as reported in the Results section of the text. Study, The specific group of rats within a particular experiment; Omnibus ANOVA for PPI, F ratio and p value for the ANOVA for that particular group of rats; NBI, NBI27914.
*Indicates a significant (p < 0.05) result from the omnibus ANOVA.
Table 3.
Omnibus ANOVA results for startle magnitude in behavioral studies (experiments 1–5)
| Study | Omnibus ANOVA for startle | |
|---|---|---|
| Experiment 1 | Repeated ferret exposure | F(10,70) = 6.7, p < 0.005* |
| Experiment 2 | Repeated CRF infusions in BLA | F(5,30) = 1.2, p < 0.314 |
| Repeated NE infusions in BLA | F(5,55) = 0.6, p < 0.710 | |
| Repeated vehicle infusions in BLA | F(5,35) = 1.0, p < 0.412 | |
| Experiment 3 | Repeated CRF infusions in CeA | F(5,40) = 1.3, p < 0.278 |
| Repeated NE infusions in CeA | F(5,55) = 0.6, p < 0.724 | |
| Repeated vehicle in CeA | F(4,28) = 0.6, p < 0.692 | |
| Experiment 4 | NBI before each repeated ferret exposure | F(5,35) = 1.3, p < 0.262 |
| NBI after each repeated ferret exposure | F(5,35) = 0.5, p < 0.729 | |
| Repeated ferret exposure and then NBI with NE challenge | F(5,50) = 2.8, p < 0.026* | |
| Experiment 5 | Repeated ferret exposure and then PHEN injection | F(5,35) = 0.3, p < 0.916 |
| Repeated ferret exposure and then ISO injection | F(5,40) = 2.7, p < 0.033* |
A single-factor within-subjects omnibus ANOVA of startle magnitude was performed for each group of rats in order to determine whether any manipulation (either the repeated treatment regimen or the subsequent challenge injection) affected startle in any study. Based on significant omnibus ANOVAs within each separate group of rats, subsequent analyses of simple main effects and comparisons of individual means were performed, as reported in the Results section of the text. Study, The specific group of rats within a particular experiment; Omnibus ANOVA for startle, the F ratio and p value for the ANOVA for that particular group of rats; NBI, NBI27914.
*Indicates a significant (p < 0.05) result from the ANOVA.
Experiment 1: Repeated predator exposure causes long-lasting sensitization of NE systems in BLA that promote increased sensitivity to PPI deficits and startle hyper-reactivity
To determine whether sensitization to the PPI-disruptive effects of NE would occur in BLA with repeated stress exposure, a subthreshold dose of intra-BLA NE was given to rats with a history of stress. Because the intent was to model in rats the intense psychogenic stress that is hypothesized to trigger cognitive deterioration in schizophrenia and PTSD (Walker and Diforio, 1997; Yehuda, 2004), a predator stress paradigm was used. This paradigm is considered to capture many of the precipitating features of psychological trauma and is used to study PTSD-like effects in animal models (Cohen et al., 2006; Adamec et al., 2008; Zoladz et al., 2008). Thus, rats (N = 8) with BLA cannulae underwent three exposures to a ferret (one exposure per day); a mock injection into BLA (injectors lowered without infusion) occurred before and after the stress regimen. Poststress “NE challenges” (0.3 μg) were then performed 4, 11, and 18 d after stress termination, with accompanying mock infusions 1 d before each NE infusion. A final challenge with a subthreshold dose of yohimbine (1 mg/kg, i.p., 10 min before testing) was performed at 28 d after stress. Yohimbine (presumably through its NE transmission-enhancing properties via blockade of α2 NE autoreceptors) is well known for triggering symptom relapse in PTSD, and at high doses it disrupts PPI, but 1 mg/kg is considered a subthreshold dose for PPI (Morgan et al., 1995; Southwick et al., 1999; Powell et al., 2005). We confirmed this in a separate study in control rats that were tested with vehicle or yohimbine 1 mg/kg, i.p. (10 min before testing) in a counterbalanced design over 2 test days (N = 8; %PPI with yohimbine = 40 ± 3.3, %PPI with vehicle = 40 ± 5.6; F(1,7) = 0.01, p = NS). Startle was increased by this dose of yohimbine (281 ± 24% for vehicle and 412 ± 38% for yohimbine; F(1,7) = 8.4, p < 0.03); but, as indicated above, no effect was seen on PPI, confirming that in nonstressed rats this dose of yohimbine does not affect PPI.
Omnibus within-subjects ANOVA indicated that there were treatment effects for both PPI and startle magnitude in this study (Experiment 1, shown in Tables 2 and 3, respectively). To pinpoint the source of these overall treatment effects, subsequent one-factor ANOVAs for the repeated ferret regimen and then Bonferroni-adjusted t tests were conducted, as detailed below.
Short-term effects of repeated ferret exposure on PPI
ANOVA revealed no significant main effect of repeated ferret exposure (F(3,21) = 1.8, p = NS), showing that predator stress on its own did not alter PPI on any day (Fig. 2A).
Figure 2.

Effects on %PPI of repeated (three presentations) ferret exposure or of various intra-BLA treatments and subsequent subthreshold challenge injections. A, Repeated ferret exposure (termed 1st Ferret, 2nd Ferret, and 3rd Ferret) and subsequent intra-BLA challenges with NE (0.3 μg; which took place 4, 11, and 18 d after the termination of ferret exposure) or systemic yohimbine (1 mg/kg) with accompanying control mock injections (mock2 to mock4). Note that the final challenge (yohimbine) took place 28 d after ferret exposures had ended. B, Repeated intra-BLA CRF infusions (termed first CRF, second CRF, and third CRF; all were 200 ng) with subsequent intra-BLA NE challenge injection (0.3 μg) 4 d after repeated CRF infusions ended. C, Repeated intra-BLA NE infusions (termed first NE, second NE, and third NE; all were 20 μg) with subsequent intra-BLA CRF challenge injection (200 ng) 4 d after repeated NE infusions ended. D, Repeated intra-BLA vehicle (termed first vehicle, second vehicle, and third vehicle) with subsequent intra-BLA NE challenge injection (0.3 μg) 4 d after repeated vehicle infusions ended. Values are reported as the mean ± SEM. *p < 0.05, **p < 0.01, compared with corresponding mock infusion; +p < 0.06, $p < 0.05, compared with the first NE infusion.
Effect on PPI of subsequent intra-BLA NE challenges
There were no differences between the mock1 and mock2 test days, indicating no conditioned effects following repeated ferret exposure, but the comparison of mock2 to the first NE challenge (4 d after ferret exposure had ended) revealed a profound (nearly 50%) reduction in PPI (p < 0.001) in response to a low dose of NE (0.3 μg) that had no effect when given to rats without a history of stress (Fig. 2D). Additional NE challenges were conducted (with corresponding mock infusions as controls) to determine how long this NE system hypersensitivity endured (Fig. 2A). The second NE challenge, which took place 11 d after stress termination, revealed a similar PPI deficit (p < 0.05), as did the third NE challenge (at 18 d, p < 0.05) and the challenge with a subthreshold dose of systemic yohimbine (p < 0.05), which occurred 28 d after stress. Thus, repeated presentation of a model of psychological trauma (live predator exposure) causes enduring sensitization of NE systems in the BLA such that subsequent manipulations that cause susbthreshold/low-level NE transmission now result in marked PPI deficits. This is similar to the profile of poststress PPI deficits in PTSD patients that are often seen years after the traumatic stress has ended.
Startle
Values for the second NE challenge that took place 11 d poststress were significantly higher (p < 0.01) than those for the corresponding mock infusion; this was true also for the third NE challenge (18 d poststress) and the yohimbine challenge (28 d poststress) compared with their respective mock infusions (p < 0.01), indicating that enduring startle hyper-reactivity (like that seen in PTSD) that is BLA-NE receptor-mediated also emerges after a history of repeated predator stress. Note that these NE challenges did not affect startle in nonstressed control rats (Table 4, Experiment 2), and did not affect startle when rats were probed just 4 d after stress termination (i.e., the first NE challenge when PPI was disrupted by the subthreshold NE dose). Hence, PPI and startle effects are dissociable from each other. Exaggerated startle and PPI deficits to a low level of NE receptor stimulation after a history of predator stress appear to be two separate phenomena, both of which resemble the pathophysiology of PTSD (i.e., delayed but long-lasting hypersensitivity to low-grade stimuli causing both enhanced startle and deficient PPI).
Table 4.
Effects of various treatments on baseline startle magnitude after repeated treatment
| mock1 | 1st ferret | 2nd ferret | 3rd ferret | mock2 | 1st NE challenge | mock3 | 2nd NE challenge | mock4 | 3rd NE challenge | Yohimbine | ||
| Experiment 1 (ferret exposure) | 481 ± 70 | 402 ± 40 | 359 ± 51 | 352 ± 51 | 419 ± 41 | 421 ± 50 | 351 ± 50 | 561 ± 70* | 489 ± 59 | 621 ± 68* | 560 ± 91* | |
| mock1 | infusion1 | infusion2 | infusion3 | mock2 | Challenge | |||||||
| Experiment 2 | ||||||||||||
| CRF (200 ng) | 452 ± 64 | 422 ± 69 | 432 ± 71 | 428 ± 46 | 442 ± 66 | 525 ± 86 (NE 0.3 μg) | ||||||
| NE (20 μg) | 392 ± 97 | 358 ± 51 | 364 ± 47 | 329 ± 32 | 324 ± 35 | 349 ± 31 (CRF 200 ng) | ||||||
| Vehicle | 367 ± 49 | 386 ± 45 | 377 ± 42 | 424 ± 47 | 366 ± 42 | 430 ± 52 (NE 0.3 μg) | ||||||
| mock1 | 1st ferret | 2nd ferret | 3rd ferret | mock2 | NE challenge | |||||||
| Experiment 4 | ||||||||||||
| NBI before ferret exposure | 292 ± 45 | 276 ± 51 | 241 ± 26 | 326 ± 59 | 295 ± 31 | 309 ± 35 | ||||||
| NBI after ferret exposure | 371 ± 54 | 318 ± 53 | 312 ± 27 | 304 ± 34 | 303 ± 34 | 315 ± 34 | ||||||
| Ferret exposurea | 360 ± 55 | 319 ± 33 | 319 ± 37 | 311 ± 33 | 343 ± 38 | 419 ± 40* | ||||||
| mock1 | 1st ferret | 2nd ferret | 3rd ferret | mock2 | Challenge | |||||||
| Experiment 5 | ||||||||||||
| Ferret exposure | 452 ± 74 | 411 ± 58 | 409 ± 57 | 456 ± 56 | 431 ± 58 | 427 ± 57 (PHEN 30 μg) | ||||||
| Ferret exposure | 266 ± 26 | 331 ± 42 | 334 ± 39 | 304 ± 31 | 336 ± 30 | 259 ± 28* (ISO 30 μg) | ||||||
All experiments listed in this table were with intra-BLA infusions. Values are reported as the mean ± SEM. Except for yohimbine, which was 1 mg/kg given systemically, all drug doses are for an intra-BLA infusion in a volume of 0.5 μl/side. NBI, NBI27914, 1000 ng.
aIntra-BLA vehicle infusions occurred in this group with each ferret exposure, and NBI was injected into BLA immediately prior to intra-BLA NE challenge infusion.
*p < 0.05, compared to corresponding mock infusion.
Experiment 2: Repeated infusion of CRF into BLA leads to subsequent sensitization of NE-mediated PPI deficits
To determine the neuropharmacological basis for the stress-induced BLA plasticity observed above, an experiment was conducted in which rats underwent the same repeated testing design but with CRF, NE, or vehicle infusions instead of stress (i.e., would repeated CRF or repeated NE infusions reproduce the profile seen with repeated stress in the BLA?). The sequence of tests was as follows: a mock infusion (mock1); three drug infusions (3 d separating consecutive infusions) of either repeated CRF (200 ng, N = 7), NE (20 μg, N = 12), or vehicle (N = 8); another mock infusion; a drug challenge with NE (0.3 μg) given to the repeated CRF and vehicle rats and CRF (200 ng) given to the repeated NE rats; and a final mock infusion. PPI was measured immediately after the infusion on each day.
Omnibus within-subjects ANOVAs indicated that there were treatment effects for PPI in both the repeated CRF group and the repeated NE group (Table 2). To identify the source of these effects (i.e., did the effect arise from the repeated infusions or did it come from the subsequent challenge injections?), subsequent one-factor ANOVAs for the repeated infusion regimen and then Bonferroni-adjusted t tests were conducted, as detailed below. No effects in omnibus ANOVAs were seen for startle in any of the three groups (Table 3).
Short-term effects of repeated drug infusions on PPI
ANOVA revealed no significant main effect of repeated CRF treatment on PPI (F(3,18) = 0.8, p = NS), showing that none of the CRF infusions acutely altered PPI in BLA across infusion days (Fig. 2B). Repeated NE treatment, however, did have a significant main effect (F(3,33) = 5.4, p < 0.004; Fig. 2C), and post hoc comparisons showed that each NE infusion significantly lowered PPI compared with mock1 (p < 0.05). The magnitude of this NE-induced PPI deficit diminished with repeated infusions, as PPI values for the third NE infusion were significantly higher than those for the first (p < 0.05); a similar trend was nearly significant for the second NE infusion versus the first (p < 0.06). Thus, it appeared that tolerance to the PPI-disruptive effects of NE developed with repeated high-dose NE infusions into BLA. Repeated vehicle infusion had no effect, indicating that a repeated treatment regimen is not in and of itself sufficient to alter PPI (F(3,21) = 0.5, p = NS).
Effect on PPI of subsequent intra-BLA drug challenges
There were no differences between the mock1 and mock2 test days in any group, indicating that there were no conditioned or carryover effects following any repeated infusion regimen (Fig. 2B–D). When the effects of drug challenges were evaluated by comparing mock2 to drug challenge days in each group, however, a marked reduction in PPI (p < 0.05) was seen when low-dose NE was given to rats that had received repeated CRF infusions into BLA (Fig. 2B). Notably, this was a subthreshold NE dose (0.3 μg) that had no effect when given to the repeated vehicle infusion group (Fig. 2D), indicating that this dose does not affect PPI in rats without a history of intra-BLA CRF infusions. In contrast, CRF challenge in the rats receiving long-term treatment with NE did not have an effect on PPI (Fig. 2C). Thus, repeated CRF receptor stimulation in the BLA produces cross-sensitization to NE. This profile was identical to that seen with the repeated ferret stress in Experiment 1 (Fig. 2A).
Startle
There were no effects on startle magnitude in any group on any test day (all F ratios, <0.8; all p values, >0.5). This was consistent with the results of Experiment 1, in which PPI deficits with subthreshold NE appeared with the first NE challenge, but startle enhancement was seen only with the subsequent challenges. Later time points with additional subthreshold NE infusions were not examined in Experiment 2 because these rats already had received three BLA infusions during the repeated regimen.
Experiment 3: NE sensitization is not seen in CeA
To determine the anatomical specificity of repeated CRF-induced plasticity in the amygdala with regard to PPI/startle, an identical experiment was conducted in which infusions of CRF (N = 9), NE (N = 12), or vehicle (N = 8) were made into CeA instead of BLA. No effects in omnibus ANOVAs were seen for PPI or startle in any of the three groups (Tables 2, 3, respectively). Nevertheless, for the sake of parallel analyses of the preceding BLA studies, subsequent analyses of simple main effects and comparisons of means using Bonferroni-corrected t tests were performed. Not surprisingly, none of these subsequent analyses revealed any significant effects in any of the groups of rats. These analyses are detailed below.
Figure 3.
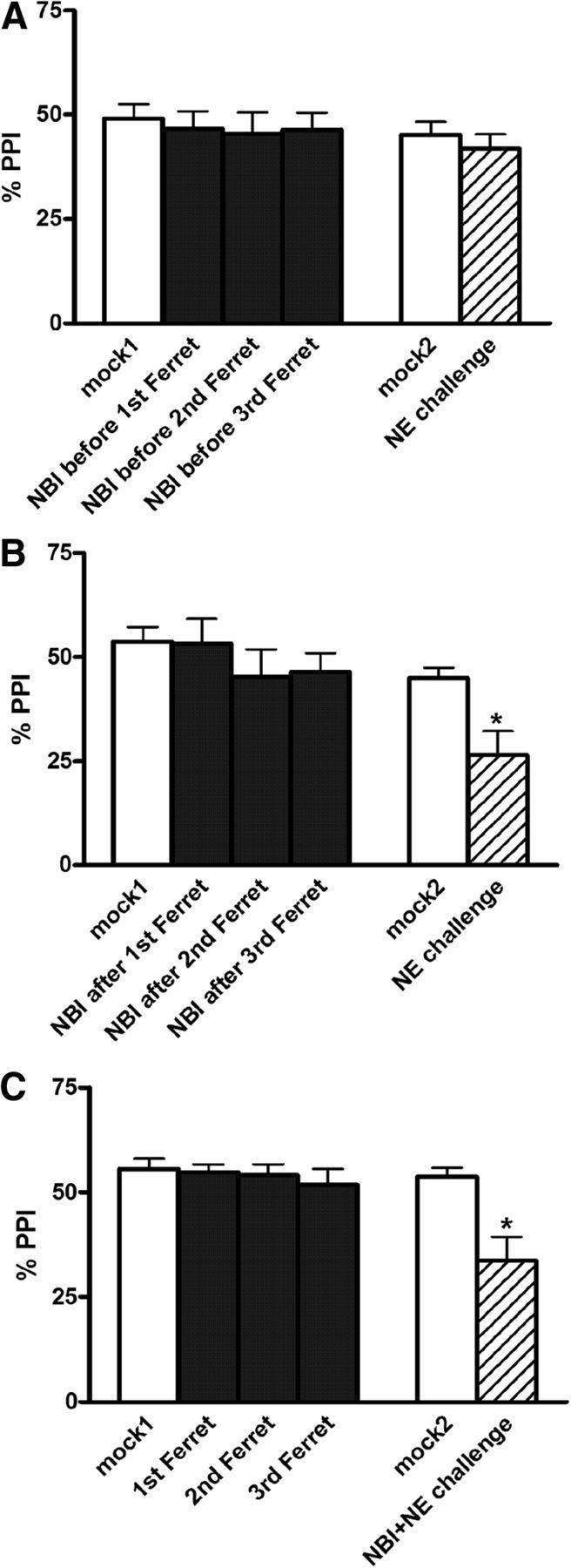
Effects on %PPI of CRF1-R antagonist (NBI27914, 1 μg) and repeated ferret exposure (three exposures total in each group: 1st Ferret, 2nd Ferret, and 3rd Ferret). A–C, CRF1-R antagonist was injected into the BLA immediately before (A) or 30 min after (B) each ferret exposure, or immediately before the intra-BLA NE challenge (C; 0.3 μg of NE). Values are reported as the mean ± SEM. NE challenges took place 4 d after the termination of the repeated ferret exposures. NBI, NBI27914. *p < 0.05, compared with mock2.
Short-term effects of repeated drug infusions on PPI
ANOVAs revealed no significant main effect of repeated CRF (F(3,24) = 0.687, p = NS), repeated NE (F(3,33) = 1.8, p = NS), or repeated vehicle (F(3,21) = 0.04, p = NS) on PPI, suggesting that none of the repeated treatments altered PPI acutely in CeA on any infusion day.
Effect on PPI of subsequent intra-CeA drug challenges
Similarly, no significant differences were found between the two mock infusion days or between mock2 and drug challenge days in any group (note that in CeA, this was despite using the 20 μg dose of NE for the challenge, allowing for maximal opportunity to detect any NE-induced effect). Because no effects were seen with even this high a dose, it was deemed unnecessary to test the low subthreshold 0.3 μg dose in the vehicle-treated rats, as had been done in BLA. In other words, in CeA a 20 μg dose of NE is subthreshold for PPI, and it did not alter PPI even in rats that previously had received repeated CRF infusions into CeA. Together, these findings indicate that the repeated CRF-induced sensitization of NE systems is anatomically specific to the BLA, as manipulations of the adjacent CeA had no effect.
Startle
There were no effects on startle magnitude in any group on any test day (all F ratios, <1.6; all p values, >0.2). For brevity, these negative results are not presented graphically.
Experiment 4: BLA CRF1 receptors are necessary for predator stress-induced NE system sensitization in BLA
To determine whether endogenous CRF release in the BLA mediated the predator stress-induced plasticity of BLA NE receptors, we conducted three additional experiments with infusions of the CRF1-selective receptor antagonist NBI27914 into the BLA. In separate groups of rats, NBI27914 was infused immediately before each ferret exposure (N = 8), 30 min after each ferret exposure (N = 8), or immediately before the poststress intra-BLA NE (0.3 μg) challenge (N = 11). These groups respectively tested BLA CRF1 receptor involvement in the development, consolidation, and expression of the stress-induced BLA neuroplasticity.
Omnibus within-subjects ANOVAs indicated that there was a treatment effect for PPI in the groups that received NBI27914 either after each ferret exposure or immediately before the NE challenge, but not in the group that received it immediately before each ferret exposure (Table 2). To identify the source of these effects (i.e., did the effect arise from the repeated infusions or did it come from the later challenge injections?), subsequent one-factor ANOVAs for the repeated infusion regimen and then Bonferroni-adjusted t tests were conducted, as detailed below.
Short-term effects of repeated ferret exposure on PPI
ANOVAs indicated no main effect of repeated treatments on acute PPI levels in the group that received NBI27914 before each ferret exposure (F(3,30) = 2.1, p = NS), in the group that received NBI27914 30 min after each stress exposure (F(3,21) = 0.9, p = NS), or in the group that underwent ferret exposures with vehicle infusions only (F(3,30) = 0.4, p = NS; this is the group that subsequently received NBI27914 immediately before the NE challenge; see below). These findings once again demonstrated that there is no immediate effect on PPI of repeated ferret exposure, and that NBI27914 infusion into BLA also does not affect PPI (Fig. 3A–C).
Subsequent intra-BLA NE challenge
As with all previous experiments, there were no differences in PPI for mock1 versus mock2 test days in any of these groups, indicating that basal PPI levels remain unaffected by the repeated stress/infusion/testing regimens. It is notable, however, that the low-dose NE challenge had absolutely no effect on PPI in rats that had received the CRF1-R antagonist immediately before each ferret exposure (Fig. 3A), demonstrating that CRF1 receptors in the BLA are required for the development of this stress-induced BLA NE neuroplasticity. Prevention of some long-term effects of a single predator stress have been reported previously with systemic CRF1-R antagonist (Adamec et al., 2010); our present results, for the first time, suggest a possible anatomical site for this phenomenon (the BLA). In contrast, the NE-induced PPI deficit was not altered by the administration of NBI27914 30 min after each ferret exposure (p < 0.05 for mock2 vs NE challenge; Fig. 3B) or immediately before the NE challenge (p < 0.01 for mock2 vs NE challenge; Fig. 3C), suggesting that BLA CRF1 receptors are not necessary for the consolidation or acute expression of this stress-induced BLA NE neuroplasticity for PPI.
Startle
A small but statistically significant (p < 0.05) increase in startle magnitude was seen when comparing the NE challenge day with the mock2 data in the group that received repeated ferret exposure and then NBI27914 immediately before the NE challenge. No other effects were seen in any group (all F ratios, <2.0; all p values, >0.1).
Experiment 5: Predator stress-induced sensitization of BLA NE systems may involve a functional uncoupling of α1 and β receptors
The primary postsynaptic heteroreceptors within the NE family are α1 and β receptors, which are both expressed densely within the BLA (Pupo and Minneman, 2001). Thus, to determine whether the stress-induced plasticity of BLA NE systems was due to altered functioning of α1 receptors, β receptors, or both, one additional study was performed in which rats underwent the same repeated predator stress procedure described above, but were challenged subsequently with either the α1 NE receptor agonist phenylephrine (PHEN; 30 μg/0.5 μl; N = 8) or the β receptor agonist isoproterenol (ISO; 30 μg/0.5 μl; N = 9) into the BLA. To determine how each of these drug doses affected PPI after their administration into BLA in stress-naive rats, we conducted an initial experiment in which rats (N = 11) received (in a counterbalanced order over 3 test days) vehicle (%PPI = 40 ± 5), PHEN (%PPI = 33 ± 4), or ISO (%PPI = 35 ± 3) immediately before testing. No main effect of drug was seen on PPI (F(2,20) = 1.2, p = NS), although a nonsignificant trend for ISO to reduce baseline startle was seen (F(2,20) = 3.0, p < 0.08). These findings indicate that neither PHEN nor ISO at this dose affected PPI when infused individually into BLA; hence, 30 μg/0.5 μl is a subthreshold/ineffective dose for altering PPI when each drug is given alone into BLA.
Omnibus within-subjects ANOVAs indicated that there was a treatment effect for PPI in both groups (Table 2). To identify the source of these effects (i.e., did the effect arise from the repeated infusions or did it come from the later challenge injections?), subsequent one-factor ANOVAs for the repeated ferret regimen and then Bonferroni-adjusted t tests were conducted, as detailed below.
Short-term effects of repeated ferret exposure on PPI
As seen previously, there was no main effect of repeated ferret exposure on PPI acutely in either group (F(3,21) = 0.4, p = NS for rats that subsequently received phenylephrine; F(3,24) = 0.4, p = NS for rats that subsequently received isoproterenol). For the sake of visual simplicity, these data are combined (there were no significant differences between these groups) in the graph (Fig. 4).
Figure 4.

Effects on %PPI of intra-BLA infusion of the α1 noradrenergic receptor agonist PHEN or the β-receptor agonist ISO. PHEN (30 μg) or ISO (30 μg) were tested 4 d after the termination of the repeated ferret exposures (three exposures total in each group: 1st Ferret, 2nd Ferret, and 3rd Ferret). Values are reported as the mean ± SEM. *p < 0.05, compared with mock2.
Effect on PPI of subsequent intra-BLA drug challenge
No differences were seen between mock1 and mock2 test days, once again indicating no conditioned effects of repeated stress exposure. A large reduction in PPI was seen, however, in response to intra-BLA infusion of either phenylephrine (p < 0.01) or isoproterenol (p < 0.05; challenges compared with their respective mock2 days). In nonstressed rats, simultaneous infusion of phenylephrine and isoproterenol into BLA disrupts PPI (Alsene et al., 2011) but has no effect if the drugs are given individually at the presently used dose (see above). This profile suggests that, under nonstress baseline conditions, coactivation of both α1 and β NE receptors in BLA may be required to disrupt PPI via BLA NE transmission, and that stimulating either one alone is not sufficient. Hence, the present result that either drug disrupts PPI on its own in ferret-exposed rats is noteworthy and indicates that the repeated ferret exposure regimen alters the dynamics between α1 and β receptors in BLA such that poststress stimulation of either receptor individually is now sufficient to disrupt PPI via this site. This putative mechanism could contribute to the enhanced sensitivity to low-dose (subthreshold) NE that is seen after the repeated predator stress procedure here.
Startle
There were no effects on startle magnitude with repeated ferret exposure in either group, and no differences between mock1 and mock2 startle values (F ratios, <2.1; p values, >0.11), but there was a small reduction in startle (p < 0.05) in rats that were challenged with isoproterenol; no effect was seen in the phenylephrine challenge group.
Histology
Figure 5 depicts the location of injector tip placements in the CeA and BLA for all startle/PPI experiments (Experiments 1–5). A total of two rats for the CeA and five rats for the BLA had erroneous placements; the final sample sizes reflect the removal of these rats from analyses.
Figure 5.

A–D, Representative injector tip locations within the CeA (A) and BLA (C) of the amygdala, which are indicated by arrows; chartings depicting the location of infusions of CRF (squares), NE (triangles), or vehicle (circles) into CeA (B) and BLA (D). VM, Ventromedial thalamus; opt, optic tract; Pe, periventricular hypothalamus.
Experiment 6: CRF1 and α1 NE receptors are highly colocalized on output projection neurons of the BLA that innervate the nucleus accumbens
To better understand the neuronal mechanism for the interaction between CRF1 and NE receptors demonstrated above, an additional experiment was performed to determine the cellular localization of these receptors within the BLA. Although previous studies indicate abundant expression of both CRF1 and NE receptors in this site, surprisingly there are no reports examining the expression of either one in relation to the other. Thus, this experiment explored the relative localizations of these receptors on individual cells within the BLA using immunofluorescence and immunoperoxidase labeling. Note that the CRF receptor antibody used in this experiment recognizes both CRF1 and CRF2 receptors; however, because the BLA is known to contain virtually no CRF2 receptors (Chalmers et al., 1995), in BLA this antibody would exclusively label CRF1 receptors. Initial studies with an α1 receptor antibody and with a β receptor antibody indicated superior immunostaining with the α1 receptor antibody. Therefore, all multilabeling studies described below were conducted with the α1-R antibody. Since the above findings with the NE receptor-specific agonists indicate that both α1 and β receptors in the BLA are functionally altered by repeated traumatic stress exposure, examination of α1-R localization was deemed to be an appropriate starting point for these studies.
Initially, triple labeling for CRF1-R, α1-R, and DAPI (which stains cellular nuclei) was conducted with BLA sections. CRF1 and NE α1 receptors were found to be highly colocalized within the BLA, with quantification indicating almost 96% colocalization (Fig. 6A–A4). Cell counts in two cases by an observer who was blind to the experimental design revealed that 95 of 99 (case 1) and 106 of 111 (case 2) α1-R-expressing cells also expressed CRF1-R; conversely, 95 of 98 (case 1) and 106 of 113 (case 2) CRF1-R-expressing cells also expressed α1-R. Based on these positive findings, a second study in which colabeling for NeuN (a neuronal marker) and α1 receptors was conducted to verify that the cells that expressed CRF1 and α1 NE receptors were indeed neurons (as opposed to glial cells). For this study, just the α1-R antibody was used for the sake of simplicity; because of the nearly complete colocalization of CRF1 and α1 receptors that was found, it was expected that either receptor antibody by itself would yield the same pattern of expression and be sufficient to answer these additional cellular characterization questions. Labeling for NeuN and α1 receptors showed exclusive localization of α1 receptors on NeuN-containing cells, indicating that these receptors are localized on neuronal cells (as opposed to glial cells; Fig. 6B–B2). The preabsorbed and secondary antibody-alone controls showed an absence of labeling for α1 and CRF1 receptors, indicating that staining represented specific labeling of receptors, and not nonspecific artifacts.
Figure 6.

A high degree of colocalization of CRF1 and α1 receptors is seen in BLA neurons. A–A3, Immunofluorescence labeling of CRF1 receptors (A), α1 receptors (A1), DAPI-labeled cells (A2), and their merge (A3) showing the coexpression of red, green, and blue in the same individual cells. A4 shows a high-magnification image of the cell identified with an arrow. B–B2, Double-immunofluorescence labeling in BLA showing α1 receptor (B), NeuN (B1), and their merge (B2). Arrows indicate individual neurons.
A second set of studies then further characterized the nature of the BLA neurons that were found to express the CRF1/α1 receptors. Because the BLA is known to contain inhibitory GABA interneurons that modulate local neural circuitry activity (McDonald and Mascagni, 2004; Aroniadou-Anderjaska et al., 2007), we explored whether α1 receptors are present on GABA-containing cells. Slices containing the BLA were taken through double immunoperoxidase labeling steps for GAD (an enzymatic marker of GABAergic neurons) and α1 receptors. Double labeling with α1-R and GAD67 antibody revealed that α1 receptors are not present on GAD-containing neurons, indicating that these receptors do not appear to colocalize to an appreciable extent with GABA-containing neurons in the BLA (Fig. 7A–B1). Interestingly, we found that α1 receptors receive a substantial number of synapse-like varicosities that were immunolabeled for GAD.
Figure 7.

BLA neurons that express α1 NE receptors are not GABAergic presumptive interneurons. A, A1, Double-immunofluorescence labeling of GAD67 (A) and α1 NE receptors (A1) from the same section/slice in BLA. B, B1, Immunoperoxidase labeling of GAD67 (brown) and α1 NE receptors (blue) in BLA. Arrows indicate individual cells.
Finally, when it was found that α1 receptors did not colocalize with GAD, we determined whether instead they were located on output projection neurons of the BLA. Because the nucleus accumbens (NAcc) is a prominent projection site of the BLA (Kelley et al., 1982; Pitkanen et al., 2000), we addressed this question by determining whether intra-accumbens infusion of retrogradely transported latex microspheres would lead to microsphere deposition in CRF1/α1 receptor-containing cells of the BLA. Verification of the injection site showed that the microsphere deposition was restricted to the NAcc and did not spread to neighboring structures (Fig. 8A–C). Visualization of BLA-containing tissue showed microsphere localization in these sections, indicating that the microspheres were taken up by the neurons in the NAcc and transported back to the BLA (Fig. 8E). Immunolabeling for CRF1 receptors in the BLA slices revealed that these receptors colocalized with the microbeads (Fig. 8D,F), indicating that these receptors are present on projection neurons of BLA, including those that innervate the NAcc. Although the microspheres were infused unilaterally in the NAcc, we found bilateral labeling in the BLA, but with a higher degree of labeling on the ipsilateral BLA rather than the contralateral BLA. Note that a diagram integrating all of the aforementioned anatomical findings with the behavioral results is presented in Figure 9 and serves as a working model for the putative mechanisms by which repeated predator stress might influence basolateral amygdala, and ultimately promote enhanced sensitivity to the PPI-disruptive and startle-elevating effects of low levels of NE transmission.
Figure 8.
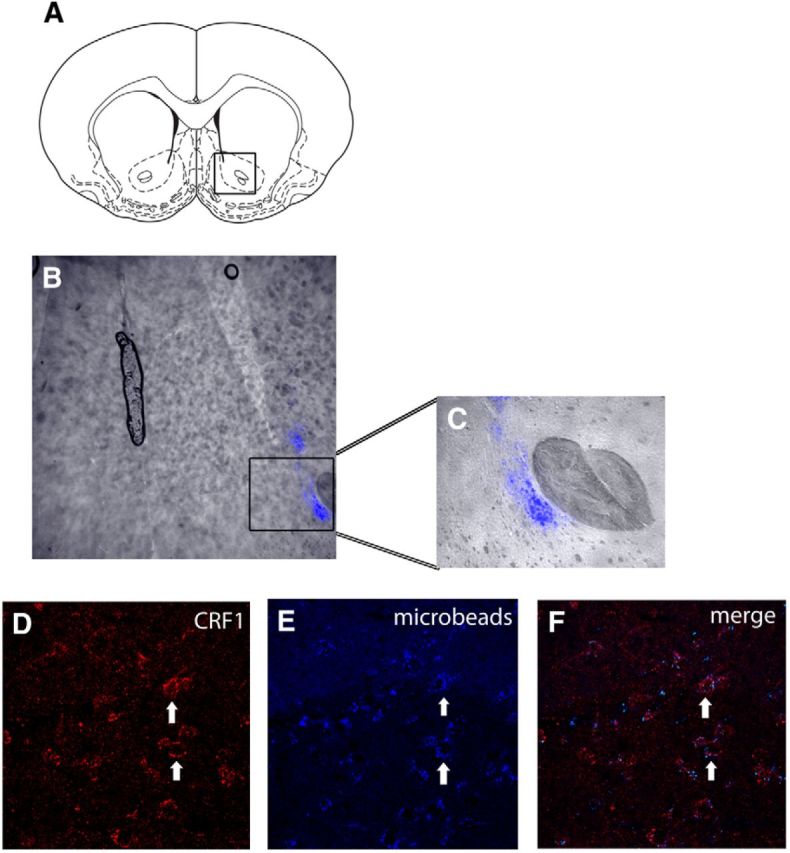
Retrogradely transported microsphere beads injected into nucleus accumbens (NAcc) are deposited in basolateral amygdala (BLA) neurons that express CRF1 receptors, indicating that repeated ferret exposure changes the functional sensitivity of BLA output projections neurons that innervate the NAcc. A–E, Line drawing of NAcc area for microsphere infusion (A); overlaid fluorescent image (B) and close-up (C) showing restricted deposition of retrograde tracer in NAcc; immunofluorescence labeling of CRF1 receptors in the BLA (D), BLA cells with fluorescence-labeled microspheres (E), and the merged image (F) showing CRF1 receptors colabeled with cells that are filled with the microspheres. Arrows indicated representative cells.
Figure 9.

Schematic of the working model for predator stress-induced effects in the basolateral amygdala. A, Before our studies, it was known only that α1 ( ) and CRF1 (
) and CRF1 ( ) receptors were both present in BLA, but their localization relative to each other and on different types of BLA cells (glia, orange; GABAergic inhibitory interneurons, blue; excitatory output neurons, green) was not known. B, We completed immunohistochemical and tract-tracing studies (Experiment 6; Figs. 6, 7, 8) to address these gaps in knowledge. First, we found that α1 and CRF1 receptors are colocalized on the same neurons and not on glial cells. Second, we failed to find these receptors on GABAergic cells. Third, we found that these receptors are present on BLA glutamatergic projection neurons that innervate the NAcc. Finally, we found that these output projection neurons that express CRF1 and α1 receptors are innervated by GABAergic neurons (presumptive inhibitory interneurons). C, Working model for behavioral sensitization induced by repeated predator exposure. Repeated exposure to predator stress sensitizes α1 receptors on BLA output neurons via initial activation of CRF1 receptors. Sensitized α1 receptors are hence abnormally responsive to low levels of NE, leading to increased glutamate release in downstream targets such as the NAcc, and thereby disrupting PPI. This putative mechanism of stress-induced BLA NE α1 receptor sensitization could thus contribute to startle abnormalities (exaggerated startle and disrupted PPI) that are seen in post-traumatic stress disorder.
) receptors were both present in BLA, but their localization relative to each other and on different types of BLA cells (glia, orange; GABAergic inhibitory interneurons, blue; excitatory output neurons, green) was not known. B, We completed immunohistochemical and tract-tracing studies (Experiment 6; Figs. 6, 7, 8) to address these gaps in knowledge. First, we found that α1 and CRF1 receptors are colocalized on the same neurons and not on glial cells. Second, we failed to find these receptors on GABAergic cells. Third, we found that these receptors are present on BLA glutamatergic projection neurons that innervate the NAcc. Finally, we found that these output projection neurons that express CRF1 and α1 receptors are innervated by GABAergic neurons (presumptive inhibitory interneurons). C, Working model for behavioral sensitization induced by repeated predator exposure. Repeated exposure to predator stress sensitizes α1 receptors on BLA output neurons via initial activation of CRF1 receptors. Sensitized α1 receptors are hence abnormally responsive to low levels of NE, leading to increased glutamate release in downstream targets such as the NAcc, and thereby disrupting PPI. This putative mechanism of stress-induced BLA NE α1 receptor sensitization could thus contribute to startle abnormalities (exaggerated startle and disrupted PPI) that are seen in post-traumatic stress disorder.
It is important to consider the specificity of the Santa Cruz Biotechnology antibody (sc-1757) used in the present study. In Western blot analyses, this antibody labels a band corresponding to CRF1-R; this band is eliminated with antibody preabsorption with the CRF1-R antigen (Fig. 10; Xu et al., 2004; Bangasser et al., 2010). Moreover, receptor-like labeling with sc-1757 is eliminated in CRF1-R knock-out mice (Bangasser et al., 2010). Here, we observed “receptor-like” cytoplasmic labeling with sc-1757; these labeled structures colocalized with NeuN and DAPI, and were eliminated from the tissue by preabsorption of the primary antibody with CRF1-R antigen. Previous in situ hybridization studies revealed that BLA expresses high levels of CRF1-R mRNA (De Souza et al., 1985; De Souza, 1987). Although the CRF1-R antibody used in our studies also recognizes CRF2 receptors (Bangasser et al., 2010), BLA does not contain CRF2 receptors (Chalmers et al., 1995). Hence, we interpret the intra-BLA receptor-like labeling with sc-1757 as representing CRF1-R labeling. This conclusion is further supported by the fact that receptor-like labeling was present in the BLA but not the CeA (Fig. 11). Previous studies have shown that the CeA does not contain CRF receptors (Lovenberg et al., 1995).
Figure 10.
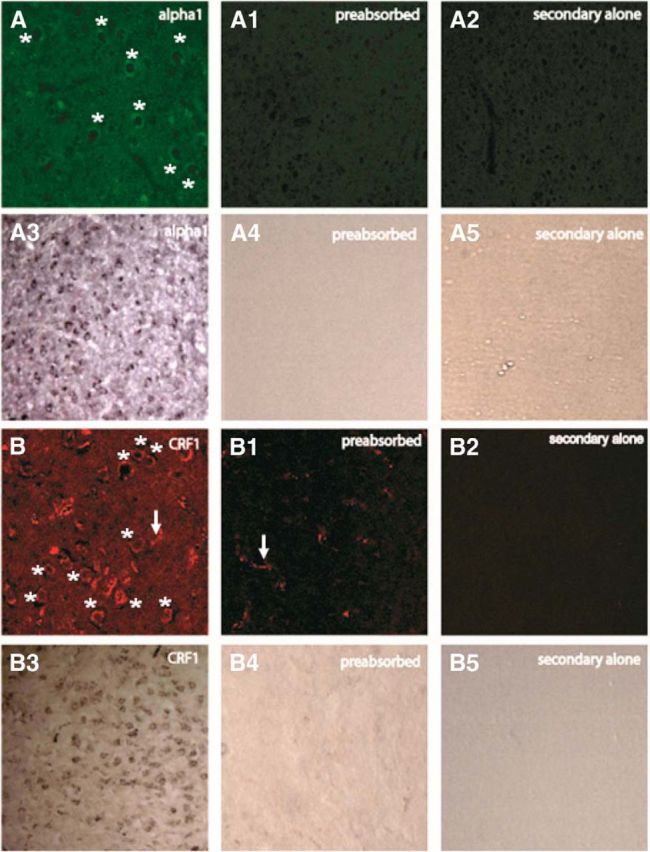
Preabsorption with α1 peptide and CRF1 peptide eliminates receptor-like labeling in the BLA, shown with parallel fluorescence and immunoperoxidase methods. A–A2, α1 Preabsorption and secondary alone controls with fluorescence labeling. A3–A5, Bright-field images of the preabsorption and secondary-alone controls for α1 receptor using an immunoperoxidase staining method. B–B2, CRF1 preabsorption and secondary-alone controls with fluorescence labeling. B3–B5, bright-field images of the preabsorption and secondary alone controls for CRF1 receptor, using an immunoperoxidase method. With the CRF1 receptor antibody preabsorption, we noticed some residual labeling resembling blood vessels (see B1), which is consistent with the presence of CRF2-R (CRF2-R is also recognized to some degree by the sc-1757 antibody; Lukkes et al., 2011). However, receptor-like, “ring-shaped” structures were eliminated by primary antibody preabsorption with CRFR1 antigen. Asterisks denote presumptive receptors; arrows denote presumptive blood vessels.
Figure 11.

CRF1 receptor labeling is present in the BLA but is absent in the CeA, a region known to be devoid of CRF1 receptors (Lovenberg et al., 1995), further suggesting that the antibody revealed true labeling of CRF1 receptors. Top, Fluorescence labeling with ring-shaped, receptor-like structures in the BLA is shown with asterisks. In both BLA and CeA, blood vessel-like structures were also noted, as shown with arrows. It is possible that these could be CRF2 labeling as these receptors are present in brain arterioles (Lovenberg et al., 1995). Bottom, Bright-field images of the BLA and CeA CRF1 receptor-like labeling conducted in parallel with an immunoperoxidase method. Again, note the absence of receptor-like labeling in the CeA.
The antibody also produced some artifactual labeling of structures resembling blood vessels. These structures were easily discriminable from presumptive receptor structures. Blood vessel-like labeling was not eliminated by primary antibody preabsorption. Because the CRF1-R antibody used in our study also recognizes CRF2 receptors (to some degree; Lukkes et al., 2011), which have been reported in brain “arterioles” (Lovenberg et al., 1995), it would not be unreasonable to assume that blood vessel-like artifacts reflected the labeling of CRF2 receptors. This observation of artifactual labeling is relevant to the findings of a previous study (Refojo et al., 2011), which purported to detect CRF1-R antibody labeling in CRF-R1 knock-out mice using the sc-1757 antibody (see Refojo et al., 2011, Supplemental Materials) and thereby concluded that this antibody did not label CRF receptors selectively. However, these authors do not provide detailed analyses (e.g., colocalization studies with neuronal markers) regarding whether or not this antibody labeling in their paper represents receptor-like structures or artifactual blood vessel-like structures. Overall, the preponderance of evidence, especially the antibody preabsorption studies shown in the present article, suggests that receptor-like BLA labeling with sc-1757 does in fact represent veritable CRF1-R receptor labeling.
Discussion
Repeated predator stress caused enduring hypersensitivity of BLA-based NE receptors, which was revealed functionally as exaggerated NE-induced startle and PPI disruption. The development of this sensitization requires CRF1-R signaling, because blockade of CRF1-R during (but not after) each predator exposure prevented the PPI deficit and startle hyper-reactivity to a subsequent subthreshold BLA NE infusion. This sensitization was long-lasting. Even 28 d poststress, systemic challenge with the α2-receptor antagonist yohimbine (a “pharmacological stressor” that causes NE release) caused exaggerated PPI deficits and startle hyper-reactivity in stress-exposed rats. Repeated intra-BLA CRF infusions (in nonstressed rats) recapitulated the predator stress profile. Repeated intra-BLA NE infusions, however, did not cause cross-sensitization to subsequent intra-BLA CRF challenge. Within-system sensitization was not observed in either case. Hence, repeated CRF receptor stimulation in BLA appears to represent the triggering event in a cascade that results in functional sensitization of BLA NE receptors. These findings for the first time demonstrate a discrete neural mechanism by which a model of traumatic stress causes long-lasting sensitization of specific systems previously implicated in PTSD (CRF and NE) and results in specific behavioral abnormalities that are seen in this illness (exaggerated startle and disrupted PPI). Traumatic stress thus causes CRF release in BLA and activates CRF1 receptors that are colocalized on output projection neurons with α1 NE receptors; repeated CRF1-R activation results in long-lasting functional sensitization of these α1-R, allowing for subsequent low-level NE release in BLA to now trigger startle hyper-reactivity and PPI deficits.
One hallmark feature of PTSD is hypersensitivity to mild stimuli (Yehuda, 2004), which is manifested as exaggerated startle and sometimes PPI deficits, long after the end of the traumatic event. Our finding that repeated ferret exposure (a putative rodent model of trauma) engendered NE receptor sensitization that lasted at least 28 d (∼2–3 human years) after stress termination provides a striking parallel. Indeed, challenge with a mild stimulus long after traumatic experiences have ended leads to the deterioration of PPI and enhanced startle in humans (Grillon et al., 1996, 1998, 2004). In PTSD, the reoccurrence of intrusive memories could be due to greater NE activity in locus ceruleus-innervated brain regions, including BLA (O'Donnell et al., 2004). Brain imaging studies suggest abnormalities of amygdala function in PTSD (Shin et al., 2006). Our results indicate the direct modulation of PTSD-like symptoms (reduced PPI and exaggerated startle) by a discrete neuronal substrate within a specific subregion of the amygdala. Our findings may also be relevant for understanding stress-induced triggering of symptoms in schizophrenia, considering that deficient PPI is a prominent feature of schizophrenia as well (Lukoff et al., 1984; Walker and Diforio, 1997; Braff et al., 2001; Betensky et al., 2008).
This model could enable the discovery of novel therapeutic targets for these illnesses by identifying molecules that transfer information from the CRF1-R to the α1-R in the BLA after traumatic stress and modulate the stress-induced sensitization of the latter. Manipulations that prevent the stress-induced plasticity of BLA NE receptors may thus represent putative new “prophylactic” treatments for PTSD, and those that counteract or dampen the functional NE receptor sensitization within BLA may be “palliative” treatments. Accordingly, α1 and β receptor antagonists reverse trauma-related emotional memories and nightmares in PTSD (van Stegeren et al., 1998; Pitman et al., 2002; Raskind et al., 2002), and propranolol reduces BLA reactivity (Hurlemann et al., 2010).
Predator stress “reorganized” the functional interdependence of α1 and β adrenergic receptor signaling. Intra-BLA infusion of agonists for either receptor alone produced PPI deficits after this traumatic stress history. Without stress exposure, the individual administration of these drugs had no effect; coadministration of these doses of α1 and β receptor agonists was needed to elicit a PPI deficit (Alsene et al., 2011). Therefore, repeated predator stress exposure modifies the functional dynamic between α1 and β NE receptors such that they gain the ability to independently promote PPI disruption after a history of stress exposure.
CRF1-Rs and α1-Rs were >90% colocalized within non-GABAergic projection neurons of the BLA. Previously, BLA was known to possess CRF1 and α1 receptors, but their relative location was unknown (Rainbow et al., 1984; De Souza et al., 1985; Radulovic et al., 1998; Wozniak et al., 2000). The present work is the first (to our knowledge) to identify the cellular colocalization of CRF1 and NE α1 receptors in BLA. This extensive colocalization provides a framework for understanding how these receptors interact to modulate startle and preattentional sensorimotor gating. Stress enhances BLA neuronal excitation, and CRF and α1 NE receptors modulate such activity (Rodriguez Manzanares et al., 2005; Shekhar et al., 2005; Aroniadou-Anderjaska et al., 2007). Electrophysiology data reveal a pattern that is consistent with our behavioral findings. First, repeated stress or repeated CRF receptor stimulation within BLA causes hyperexcitability of BLA pyramidal output neurons (Shekhar et al., 2005; Rosenkranz et al., 2010). Second, chronic stress exposure causes NE to “switch” from being an inhibitory influence on BLA neuronal activity to being excitatory on BLA neuronal activity (Buffalari and Grace, 2009). Finally, CRF and NE interact in other forms of BLA plasticity (Ferry and McGaugh, 2000). Our present findings provide a mechanism to integrate these results. Specifically, it may be that repeated traumatic stress via stimulation of BLA CRF1 receptors amplifies the electrophysiological response of BLA glutamatergic output projection neurons to α1-NE signaling, thereby engaging downstream BLA targets at a supernormal level. This abnormal recruitment of BLA targets then alters startle plasticity.
Predator stress-induced changes in the BLA could thereby alter neuronal network dynamics in multiple structures that receive projections from BLA. We show here that NAcc is one of the brain regions to which some BLA neurons expressing CRF1/α1 receptors project. NAcc neuronal properties are dependent upon BLA activation/inactivation state (Floresco et al., 2001; McGinty and Grace, 2009; Jones et al., 2010; Stuber et al., 2011; Papp et al., 2012), and this site modulates sensorimotor gating, with increased glutamatergic signaling in NAcc disrupting PPI (Kodsi and Swerdlow, 1994, 1995; Wan and Swerdlow, 1996; Alsene et al., 2011). Glutamatergic projections from the BLA synapse onto the medium spiny neurons of the NAcc that project to the ventral pallidum (VP; French and Totterdell, 2003; Papp et al., 2012). The VP, in turn, projects to the pedunculopontine tegmental nucleus (PPTg) to regulate PPI (Kodsi and Swerdlow, 1997). Hence, one route through which stress-induced α1 receptor hypersensitivity in BLA could ultimately alter PPI is via a NAcc–VP–PPTg circuit. Given the many psychiatric conditions exacerbated by stress and in which altered signaling within cortico-striato-pallidal pathways is hypothesized (Swerdlow, 2010), the present finding that traumatic stress (predator exposure) enacts long-lasting sensitization of a prominent node impinging on this circuit has far-reaching implications for the neural substrates of mental illness. Future studies can examine other BLA output sites such as prefrontal cortex.
Given that BLA CRF1 and α1 receptors appear to be extensively colocalized, CRF-induced α1-R sensitization may occur via actions within the individual neurons bearing them. The intracellular mechanisms by which CRF1-Rs and α1-Rs interact to produce NE sensitization are unknown, but ERK and MAPK signaling pathways may participate. Stress increases BLA CREB phosphorylation (Kreibich and Blendy, 2004), and CRF1-R stimulation engages CREB-dependent ERK and MAPK expression (Refojo et al., 2005). NE α1-R is linked to the same pathways; hence, they may represent the intracellular substrate through which repeated CRF1-R stimulation ultimately leads to functional sensitization of α1-R in BLA. Quirarte et al. (1997) and Roozendaal et al. (2002, 2006, 2008) showed that glucocorticoids regulate CRF–NE dynamics within the BLA for fear-related memory processes. Our laboratory and others, however, have shown for PPI that CRF- and stress-induced effects are independent of glucocorticoids (Groenink et al., 2008; Bakshi et al., 2012). Future studies can examine this issue directly. Also, it will be interesting to see what the variation in predator stress-induced sensitization of NE receptors is since there are individual differences in the propensity for the development of PTSD (Yehuda, 2004).
It seems unlikely that startle changes contributed to PPI effects in our studies. Changes in startle appear to be a distinct long-term consequence of repeated trauma exposure because PPI deficits were dissociable from baseline startle changes. Intracerebroventricular CRF disrupts PPI, and these effects are reversed by CRF-R antagonism (Conti et al., 2002; Risbrough et al., 2004). We did not find any PPI-modulatory effects of intra-amygdala CRF infusion. It is unlikely that the CRF dose was too low, as it produces other short-term behavioral effects (Jochman et al., 2005). Rather, CRF receptors in other brain regions such as the bed nucleus of the stria terminalis likely are responsible for the acute PPI- and startle-modulatory effects of intracerebroventricular CRF (Toth et al., 2014).
Our findings provide a new working model for the mechanisms by which traumatic stress exposure produces long-term alterations in startle reactivity and sensorimotor gating, both of which are dysfunctional in PTSD. Given the involvement of BLA CRF and NE receptors in anxiety-related processes, it is also possible that the mechanisms revealed in the present studies regarding stress-induced sensitization of BLA NE receptors is relevant for the enhancement of anxiety in the aftermath of trauma. Ultimately, the present findings could help to identify novel treatment strategies for traumatic stress-induced psychopathology.
Footnotes
This work was supported by Grants R01-MH-075980 (to V.P.B.) and T32 GM007507, and a NARSAD Young Investigator Award (to V.P.B.). We thank Dr. Karla Knobel for assistance with the fluorescence images.
The authors declare no competing financial interests.
References
- Adamec R, Holmes A, Blundell J. Vulnerability to lasting anxiogenic effects of brief exposure to predator stimuli: sex, serotonin and other factors-relevance to PTSD. Neurosci Biobehav Rev. 2008;32:1287–1292. doi: 10.1016/j.neubiorev.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamec R, Fougere D, Risbrough V. CRF receptor blockade prevents initiation and consolidation of stress effects on affect in the predator stress model of PTSD. Int J Neuropsychopharmacol. 2010;13:747–757. doi: 10.1017/S1461145709990496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsene KM, Rajbhandari AK, Ramaker MJ, Bakshi VP. Discrete forebrain neuronal networks supporting noradrenergic regulation of sensorimotor gating. Neuropsychopharmacology. 2011;36:1003–1014. doi: 10.1038/npp.2010.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Qashu F, Braga MF. Mechanisms regulating GABAergic inhibitory transmission in the basolateral amygdala: implications for epilepsy and anxiety disorders. Amino Acids. 2007;32:305–315. doi: 10.1007/s00726-006-0415-x. [DOI] [PubMed] [Google Scholar]
- Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD., Jr Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry. 1999;156:585–588. doi: 10.1176/ajp.156.4.585. [DOI] [PubMed] [Google Scholar]
- Bakshi VP, Kelley AE. Dopaminergic regulation of feeding behavior: II. Differential effects of amphetamine microinfusion into three striatal subregions. Psychobiology. 1991;19:233–242. [Google Scholar]
- Bakshi VP, Alsene KM, Roseboom PH, Connors EE. Enduring sensorimotor gating abnormalities following predator exposure or corticotropin-releasing factor in rats: a model for PTSD-like information-processing deficits? Neuropharmacology. 2012;62:737–748. doi: 10.1016/j.neuropharm.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangasser DA, Curtis A, Reyes BA, Bethea TT, Parastatidis I, Ischiropoulos H, Van Bockstaele EJ, Valentino RJ. Sex differences in corticotropin-releasing factor receptor signaling and trafficking: potential role in female vulnerability to stress-related psychopathology. Mol Psychiatry. 2010;15:877, 896–904. doi: 10.1038/mp.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betensky JD, Robinson DG, Gunduz-Bruce H, Sevy S, Lencz T, Kane JM, Malhotra AK, Miller R, McCormack J, Bilder RM, Szeszko PR. Patterns of stress in schizophrenia. Psychiatry Res. 2008;160:38–46. doi: 10.1016/j.psychres.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Light GA, Sprock J, Perry W, Cadenhead KS, Swerdlow NR. Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophr Res. 2001;49:171–178. doi: 10.1016/S0920-9964(00)00139-0. [DOI] [PubMed] [Google Scholar]
- Buffalari DM, Grace AA. Chronic cold stress increases excitatory effects of norepinephrine on spontaneous and evoked activity of basolateral amygdala neurons. Int J Neuropsychopharmacol. 2009;12:95–107. doi: 10.1017/S1461145708009140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci. 1995;15:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen H, Matar MA, Richter-Levin G, Zohar J. The contribution of an animal model toward uncovering biological risk factors for PTSD. Ann N Y Acad Sci. 2006;1071:335–350. doi: 10.1196/annals.1364.026. [DOI] [PubMed] [Google Scholar]
- Conti LH, Murry JD, et al. Effects of corticotropin-releasing factor on prepulse inhibition of the acoustic startle response in two rat strains. Psychopharmacology (Berl) 2002;161:296–303. doi: 10.1007/s00213-002-1025-2. [DOI] [PubMed] [Google Scholar]
- De Souza EB. Corticotropin-releasing factor receptors in the rat central nervous system: characterization and regional distribution. J Neurosci. 1987;7:88–100. doi: 10.1523/JNEUROSCI.07-01-00088.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza EB, Insel TR, Perrin MH, Rivier J, Vale WW, Kuhar MJ. Corticotropin-releasing factor receptors are widely distributed within the rat central nervous system: an autoradiographic study. J Neurosci. 1985;5:3189–3203. doi: 10.1523/JNEUROSCI.05-12-03189.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferry B, McGaugh JL. Role of amygdala norepinephrine in mediating stress hormone regulation of memory storage. Acta Pharmacol Sin. 2000;21:481–493. [PubMed] [Google Scholar]
- Floresco SB, Blaha CD, Yang CR, Phillips AG. Dopamine D1 and NMDA receptors mediate potentiation of basolateral amygdala-evoked firing of nucleus accumbens neurons. J Neurosci. 2001;21:6370–6376. doi: 10.1523/JNEUROSCI.21-16-06370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French SJ, Totterdell S. Individual nucleus accumbens-projection neurons receive both basolateral amygdala and ventral subicular afferents in rats. Neuroscience. 2003;119:19–31. doi: 10.1016/s0306-4522(03)00150-7. [DOI] [PubMed] [Google Scholar]
- Gresack JE, Risbrough VB. Corticotropin-releasing factor and noradrenergic signalling exert reciprocal control over startle reactivity. Int J Neuropsychopharmacol. 2011;14:1179–1194. doi: 10.1017/S1461145710001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Morgan CA, Southwick SM, Davis M, Charney DS. Baseline startle amplitude and prepulse inhibition in Vietnam veterans with posttraumatic stress disorder. Psychiatry Res. 1996;64:169–178. doi: 10.1016/S0165-1781(96)02942-3. [DOI] [PubMed] [Google Scholar]
- Grillon C, Morgan CA, 3rd, Davis M, Southwick SM. Effects of experimental context and explicit threat cues on acoustic startle in Vietnam veterans with posttraumatic stress disorder. Biol Psychiatry. 1998;44:1027–1036. doi: 10.1016/S0006-3223(98)00034-1. [DOI] [PubMed] [Google Scholar]
- Grillon C, Baas JP, Lissek S, Smith K, Milstein J. Anxious responses to predictable and unpredictable aversive events. Behav Neurosci. 2004;118:916–924. doi: 10.1037/0735-7044.118.5.916. [DOI] [PubMed] [Google Scholar]
- Groenink L, Dirks A, Verdouw PM, de Graaff M, Peeters BW, Millan MJ, Olivier B. CRF1 not glucocorticoid receptors mediate prepulse inhibition deficits in mice overexpressing CRF. Biol Psychiatry. 2008;63:360–368. doi: 10.1016/j.biopsych.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Hoffman HS, Ison JR. Reflex modification in the domain of startle: I. Some empirical findings and their implications for how the nervous system processes sensory input. Psychol Rev. 1980;87:175–189. doi: 10.1037/0033-295X.87.2.175. [DOI] [PubMed] [Google Scholar]
- Hurlemann R, Walter H, Rehme AK, Kukolja J, Santoro SC, Schmidt C, Schnell K, Musshoff F, Keysers C, Maier W, Kendrick KM, Onur OA. Human amygdala reactivity is diminished by the beta-noradrenergic antagonist propranolol. Psychol Med. 2010;40:1839–1848. doi: 10.1017/S0033291709992376. [DOI] [PubMed] [Google Scholar]
- Jochman KA, Newman SM, Kalin NH, Bakshi VP. Corticotropin-releasing factor-1 receptors in the basolateral amygdala mediate stress-induced anorexia. Behav Neurosci. 2005;119:1448–1458. doi: 10.1037/0735-7044.119.6.1448. [DOI] [PubMed] [Google Scholar]
- Jones JL, Day JJ, Wheeler RA, Carelli RM. The basolateral amygdala differentially regulates conditioned neural responses within the nucleus accumbens core and shell. Neuroscience. 2010;169:1186–1198. doi: 10.1016/j.neuroscience.2010.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley AE, Domesick VB, Nauta WJ. The amygdalostriatal projection in the rat—an anatomical study by anterograde and retrograde tracing methods. Neuroscience. 1982;7:615–630. doi: 10.1016/0306-4522(82)90067-7. [DOI] [PubMed] [Google Scholar]
- Kodsi MH, Swerdlow NR. Quinolinic acid lesions of the ventral striatum reduce sensorimotor gating of acoustic startle in rats. Brain Res. 1994;643:59–65. doi: 10.1016/0006-8993(94)90008-6. [DOI] [PubMed] [Google Scholar]
- Kodsi MH, Swerdlow NR. Prepulse inhibition in the rat is regulated by ventral and caudodorsal striato-pallidal circuitry. Behav Neurosci. 1995;109:912–928. doi: 10.1037/0735-7044.109.5.912. [DOI] [PubMed] [Google Scholar]
- Kodsi MH, Swerdlow NR. Regulation of prepulse inhibition by ventral pallidal projections. Brain Res Bull. 1997;43:219–228. doi: 10.1016/s0361-9230(96)00440-6. [DOI] [PubMed] [Google Scholar]
- Kreibich AS, Blendy JA. cAMP response element-binding protein is required for stress but not cocaine-induced reinstatement. J Neurosci. 2004;24:6686–6692. doi: 10.1523/JNEUROSCI.1706-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovenberg TW, Liaw CW, Grigoriadis DE, Clevenger W, Chalmers DT, De Souza EB, Oltersdorf T. Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain. Proc Natl Acad Sci U S A. 1995;92:836–840. doi: 10.1073/pnas.92.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukkes JL, Staub DR, Dietrich A, Truitt W, Neufeld-Cohen A, Chen A, Johnson PL, Shekhar A, Lowry CA. Topographical distribution of corticotropin-releasing factor type 2 receptor-like immunoreactivity in the rat dorsal raphe nucleus: co-localization with tryptophan hydroxylase. Neuroscience. 2011;183:47–63. doi: 10.1016/j.neuroscience.2011.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoff D, Snyder K, Ventura J, Nuechterlein KH. Life events, familial stress, and coping in the developmental course of schizophrenia. Schizophr Bull. 1984;10:258–292. doi: 10.1093/schbul/10.2.258. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F. Parvalbumin-containing interneurons in the basolateral amygdala express high levels of the alpha1 subunit of the GABAA receptor. J Comp Neurol. 2004;473:137–146. doi: 10.1002/cne.20101. [DOI] [PubMed] [Google Scholar]
- McGinty VB, Grace AA. Timing-dependent regulation of evoked spiking in nucleus accumbens neurons by integration of limbic and prefrontal cortical inputs. J Neurophysiol. 2009;101:1823–1835. doi: 10.1152/jn.91162.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloni EG, Gerety LP, Knoll AT, Cohen BM, Carlezon WA., Jr Behavioral and anatomical interactions between dopamine and corticotropin-releasing factor in the rat. J Neurosci. 2006;26:3855–3863. doi: 10.1523/JNEUROSCI.4957-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CA, 3rd, Grillon C, et al. Yohimbine facilitated acoustic startle in combat veterans with post-traumatic stress disorder. Psychopharmacology (Berl) 1995;117:466–471. doi: 10.1007/BF02246220. [DOI] [PubMed] [Google Scholar]
- Norman RM, Malla AK. Stressful life events and schizophrenia. I: a review of the research. Br J Psychiatry. 1993;162:161–166. doi: 10.1192/bjp.162.2.161. [DOI] [PubMed] [Google Scholar]
- O'Donnell T, Hegadoren KM, Coupland NC. Noradrenergic mechanisms in the pathophysiology of post-traumatic stress disorder. Neuropsychobiology. 2004;50:273–283. doi: 10.1159/000080952. [DOI] [PubMed] [Google Scholar]
- Papp E, Borhegyi Z, Tomioka R, Rockland KS, Mody I, Freund TF. Glutamatergic input from specific sources influences the nucleus accumbens-ventral pallidum information flow. Brain Struct Funct. 2012;217:37–48. doi: 10.1007/s00429-011-0331-z. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic Press; 1998. [DOI] [PubMed] [Google Scholar]
- Perry ML, Baldo BA, Andrzejewski ME, Kelley AE. Muscarinic receptor antagonism causes a functional alteration in nucleus accumbens mu-opiate-mediated feeding behavior. Behav Brain Res. 2009;197:225–229. doi: 10.1016/j.bbr.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickel VM, Segal M, Bloom FE. A radioautographic study of the efferent pathways of the nucleus locus coeruleus. J Comp Neurol. 1974;155:15–42. doi: 10.1002/cne.901550103. [DOI] [PubMed] [Google Scholar]
- Pieribone VA, Nicholas AP, Dagerlind A, Hökfelt T. Distribution of α1 adrenoceptors in rat brain revealed by in situ hybridization experiments utilizing subtype-specific probes. J Neurosci. 1994;14:4252–4268. doi: 10.1523/JNEUROSCI.14-07-04252.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Jolkkonen E, et al. Anatomic heterogeneity of the rat amygdaloid complex. Folia Morphol (Warsz) 2000;59:1–23. [PubMed] [Google Scholar]
- Pitman RK, Sanders KM, Zusman RM, Healy AR, Cheema F, Lasko NB, Cahill L, Orr SP. Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biol Psychiatry. 2002;51:189–192. doi: 10.1016/S0006-3223(01)01279-3. [DOI] [PubMed] [Google Scholar]
- Powell SB, Palomo J, et al. Yohimbine disrupts prepulse inhibition in rats via action at 5-HT1A receptors, not alpha2-adrenoceptors. Psychopharmacology (Berl) 2005;180:491–500. doi: 10.1007/s00213-005-2193-7. [DOI] [PubMed] [Google Scholar]
- Pupo AS, Minneman KP. Adrenergic pharmacology: focus on the central nervous system. CNS Spectr. 2001;6:656–662. doi: 10.1017/s1092852900001346. [DOI] [PubMed] [Google Scholar]
- Quirarte GL, Roozendaal B, McGaugh JL. Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci U S A. 1997;94:14048–14053. doi: 10.1073/pnas.94.25.14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic J, Sydow S, Spiess J. Characterization of native corticotropin-releasing factor receptor type 1 (CRFR1) in the rat and mouse central nervous system. J Neurosci Res. 1998;54:507–521. doi: 10.1002/(SICI)1097-4547(19981115)54:4%3C507::AID-JNR8%3E3.0.CO%3B2-E. [DOI] [PubMed] [Google Scholar]
- Rainbow TC, Parsons B, Wolfe BB. Quantitative autoradiography of beta 1- and beta 2-adrenergic receptors in rat brain. Proc Natl Acad Sci U S A. 1984;81:1585–1589. doi: 10.1073/pnas.81.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci. 2004;24:3471–3479. doi: 10.1523/JNEUROSCI.5740-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskind MA, Thompson C, Petrie EC, Dobie DJ, Rein RJ, Hoff DJ, McFall ME, Peskind ER. Prazosin reduces nightmares in combat veterans with posttraumatic stress disorder. J Clin Psychiatry. 2002;63:565–568. doi: 10.4088/JCP.v63n0705. [DOI] [PubMed] [Google Scholar]
- Refojo D, Echenique C, Muller MB, Reul JM, Deussing JM, Wurst W, Sillaber I, Paez-Pereda M, Holsboer F, Arzt E. Corticotropin-releasing hormone activates ERK1/2 MAPK in specific brain areas. Proc Natl Acad Sci U S A. 2005;102:6183–6188. doi: 10.1073/pnas.0502070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refojo D, Schweizer M, Kuehne C, Ehrenberg S, Thoeringer C, Vogl AM, Dedic N, Schumacher M, von Wolff G, Avrabos C, Touma C, Engblom D, Schütz G, Nave KA, Eder M, Wotjak CT, Sillaber I, Holsboer F, Wurst W, Deussing JM. Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science. 2011;333:1903–1907. doi: 10.1126/science.1202107. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, Hauger RL, Roberts AL, Vale WW, Geyer MA. Corticotropin-releasing factor receptors CRF1 and CRF2 exert both additive and opposing influences on defensive startle behavior. J Neurosci. 2004;24:6545–6552. doi: 10.1523/JNEUROSCI.5760-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez Manzanares PA, Isoardi NA, Carrer HF, Molina VA. Previous stress facilitates fear memory, attenuates GABAergic inhibition, and increases synaptic plasticity in the rat basolateral amygdala. J Neurosci. 2005;25:8725–8734. doi: 10.1523/JNEUROSCI.2260-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Quirarte GL, McGaugh JL. Glucocorticoids interact with the basolateral amygdala beta-adrenoceptor—cAMP/cAMP/PKA system in influencing memory consolidation. Eur J Neurosci. 2002;15:553–560. doi: 10.1046/j.0953-816x.2001.01876.x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Okuda S, Van der Zee EA, McGaugh JL. Glucocorticoid enhancement of memory requires arousal-induced noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci U S A. 2006;103:6741–6746. doi: 10.1073/pnas.0601874103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Schelling G, McGaugh JL. Corticotropin-releasing factor in the basolateral amygdala enhances memory consolidation via an interaction with the β-adrenoceptor-cAMP pathway: dependence on glucocorticoid receptor activation. J Neurosci. 2008;28:6642–6651. doi: 10.1523/JNEUROSCI.1336-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Venheim ER, Padival M. Chronic stress causes amygdala hyperexcitability in rodents. Biol Psychiatry. 2010;67:1128–1136. doi: 10.1016/j.biopsych.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar A, Truitt W, Rainnie D, Sajdyk T. Role of stress, corticotrophin releasing factor (CRF) and amygdala plasticity in chronic anxiety. Stress. 2005;8:209–219. doi: 10.1080/10253890500504557. [DOI] [PubMed] [Google Scholar]
- Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. 2006;1071:67–79. doi: 10.1196/annals.1364.007. [DOI] [PubMed] [Google Scholar]
- Southwick SM, Morgan CA, 3rd, Charney DS, High JR. Yohimbine use in a natural setting: effects on posttraumatic stress disorder. Biol Psychiatry. 1999;46:442–444. doi: 10.1016/S0006-3223(99)00107-9. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Sparta DR, Stamatakis AM, van Leeuwen WA, Hardjoprajitno JE, Cho S, Tye KM, Kempadoo KA, Zhang F, Deisseroth K, Bonci A. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature. 2011;475:377–380. doi: 10.1038/nature10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland JE, Conti LH. Restraint stress-induced reduction in prepulse inhibition in Brown Norway rats: role of the CRF2 receptor. Neuropharmacology. 2011;60:561–571. doi: 10.1016/j.neuropharm.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR. Integrative circuit models and their implications for the pathophysiologies and treatments of the schizophrenias. Curr Top Behav Neurosci. 2010;4:555–583. doi: 10.1007/7854_2010_48. [DOI] [PubMed] [Google Scholar]
- Toth M, Gresack JE, Bangasser DA, Plona Z, Valentino RJ, Flandreau EI, Mansuy IM, Merlo-Pich E, Geyer MA, Risbrough VB. Forebrain-specific CRF overproduction during development is sufficient to induce enduring anxiety and startle abnormalities in adult mice. Neuropsychopharmacology. 2014;39:1409–1419. doi: 10.1038/npp.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Stegeren AH, Everaerd W, et al. Memory for emotional events: differential effects of centrally versus peripherally acting beta-blocking agents. Psychopharmacology (Berl) 1998;138:305–310. doi: 10.1007/s002130050675. [DOI] [PubMed] [Google Scholar]
- Walker EF, Diforio D. Schizophrenia: a neural diathesis-stress model. Psychol Rev. 1997;104:667–685. doi: 10.1037/0033-295X.104.4.667. [DOI] [PubMed] [Google Scholar]
- Wan FJ, Swerdlow NR. Sensorimotor gating in rats is regulated by different dopamine-glutamate interactions in the nucleus accumbens core and shell subregions. Brain Res. 1996;722:168–176. doi: 10.1016/0006-8993(96)00209-0. [DOI] [PubMed] [Google Scholar]
- Wozniak M, Schramm NL, Limbird LE. The noradrenergic receptor subtypes. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: the fourth generation of progress. New York: Raven; 2000. [Google Scholar]
- Xu GP, Van Bockstaele E, Reyes B, Bethea T, Valentino RJ. Chronic morphine sensitizes the brain norepinephrine system to corticotropin-releasing factor and stress. J Neurosci. 2004;24:8193–8197. doi: 10.1523/JNEUROSCI.1657-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R. Risk and resilience in posttraumatic stress disorder. J Clin Psychiatry. 2004;65(Suppl 1):29–36. [PubMed] [Google Scholar]
- Zoladz PR, Conrad CD, Fleshner M, Diamond DM. Acute episodes of predator exposure in conjunction with chronic social instability as an animal model of post-traumatic stress disorder. Stress. 2008;11:259–281. doi: 10.1080/10253890701768613. [DOI] [PMC free article] [PubMed] [Google Scholar]