

Abstract
Background
Gene duplication is believed to be the classical way to form novel genes, but overprinting may be an important alternative. Overprinting allows entirely novel proteins to evolve de novo, i.e., formerly non-coding open reading frames within functional genes become expressed. Only three cases have been described for Escherichia coli. Here, a fourth example is presented.
Results
RNA sequencing revealed an open reading frame weakly transcribed in cow dung, coding for 101 residues and embedded completely in the −2 reading frame of citC in enterohemorrhagic E. coli. This gene is designated novel overlapping gene, nog1. The promoter region fused to gfp exhibits specific activities and 5’ rapid amplification of cDNA ends indicated the transcriptional start 40-bp upstream of the start codon. nog1 was strand-specifically arrested in translation by a nonsense mutation silent in citC. This Nog1-mutant showed a phenotype in competitive growth against wild type in the presence of MgCl2. Small differences in metabolite concentrations were also found. Bioinformatic analyses propose Nog1 to be inner membrane-bound and to possess at least one membrane-spanning domain. A phylogenetic analysis suggests that the orphan gene nog1 arose by overprinting after Escherichia/Shigella separated from the other γ-proteobacteria.
Conclusions
Since nog1 is of recent origin, non-essential, short, weakly expressed and only marginally involved in E. coli’s central metabolism, we propose that this gene is in an initial stage of evolution. While we present specific experimental evidence for the existence of a fourth overlapping gene in enterohemorrhagic E. coli, we believe that this may be an initial finding only and overlapping genes in bacteria may be more common than is currently assumed by microbiologists.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-015-0558-z) contains supplementary material, which is available to authorized users.
Keywords: Overprinting, Overlapping gene, de novo evolution, Coding reserve, Orphan, EHEC, nog1/citC
Background
A widely established model to explain the evolutionary origin of novel genes is gene duplication [1–4]. However, recent phylogenetic evidence suggests that de-novo formation might be an alternative, important source for the de novo origin of orphan genes [5]. This is corroborated by findings that long non-coding RNA may serve as a novelty pool and that ribosomes indeed translate novel ORFs [6, 7]. It is hypothesized that this mechanism might produce novel domains or folds, which are added to existing genes or assembled to new genes [8, 9].
In eukaryotes, large parts of the genome do not harbor protein-coding genes, potentially providing DNA raw material for novel genes [10, 11]. In contrast, prokaryotic genomes are densely packed with genes and inter-genic space is quite limited. Therefore, as early as 1977, Grassé proposed a mechanism for the evolution of novel genes termed “overprinting” [12], which some years later was substantiated by Ohno [13]. According to this hypothesis, a previously non-coding sequence, overlapping an existing gene in an alternate reading frame, is transformed into a coding sequence by the creation of a new promoter next to a suitable ribosome binding site and a start codon. Alternatively, a gene may elongate through the emergence of an alternative start codon further upstream or the loss of its original stop codon, leading to an overlap with an adjacent gene. This mechanism of overprinting is an option to solve the de-novo evolution problem for prokaryotes.
Trivial overlaps of only a few base pairs are found in about 30 % of the bacterial genes [14, 15]. The likely benefit is a translational coupling of both genes, since the stop codon of the upstream located gene overlaps with the start codon of the downstream gene [16]. In non-trivially overlapping genes the protein coding regions are embedded completely or substantially in the annotated “mother gene”, which by definition occupies reading frame +1, and are encoded by one of the five alternate reading frames. Non-trivially overlapping genes are generally assumed to be very rare. This assumption is due to a severe information content constraint since single mutations often affect the protein function of both overlapping genes. Thus, such an arrangement is believed to be less likely to be beneficial for the organism carrying the overlapping gene pair [14].
The majority of non-trivial overlapping genes have been described in viruses [17–20] and their emergence was attributed to a hypothetical selection pressure acting on the size of the viral genome, exerted by spatial limitations of the capsid [21]. In sharp contrast, in prokaryotes only very few overlapping gene pairs are known. In the extremely well-researched Escherichia coli, as far as we know, only three overlapping gene pairs have been described: htgA/yaaW [22, 23], yghW/morA [24], and tnpA/astA [25, 26].
Here we report on direct experimental evidence indicating the presence of a novel overlapping gene pair in enterohemorrhagic E. coli O157:H7 str. EDL933 (EHEC) which was found during the investigation of the transcriptomic response of EHEC to a number of environmental conditions [27]. The novel overlapping gene nog1 is completely embedded −2 antisense in its mother reading frame citC which is part of the operon citCDEFXG. This operon is responsible for citrate fermentation. citC is induced anaerobically and encodes the citrate lyase ligase which activates the γ-subunit of the citrate lyase [28]. In addition to a functional analysis of nog1, we provide evidence that this overlapping gene may be restricted to the Escherichia/Shigella/Salmonella clade and probably arose recently by genetic overprinting.
Methods
Bacterial strains and plasmids are listed in Additional file 1: Table S1.
Construction of gfp fusions and fluorescence measurement
The region 365 bp and 361 bp upstream of the start codons of citC and nog1 respectively was amplified from genomic EHEC-DNA (NC_002655, [29]) using primer GGC GGT CGA Ccg gtg cct ttt aac acc aga tc (Z0762 + 667R-Sall) and ACA GAA TTC gaa ctg ata aac ctc gcc tat g (Z0762+325F-EcoRI) and the primers GGC GGT CGA Caa aga tac gca gcg gaa atg c (Z0762-362F-SalI) and ACA GAA TTC tgg gag aaa ggg ggg tga tcg a (Z0762-3R-EcoRI) respectively. The PCR products were digested with the appropriate enzymes (cut sites italic above) according to the manufacturer (NEB) and ligated in pProbe-NT [30] using T4-ligase (NEB). EHEC was transformed with the plasmids and was grown under shaking at 37 °C in the dark in LB medium [31] supplemented with 25 μg/ml kanamycin. For aerobic growth, 1:100 diluted overnight cultures were used to inoculate 10 ml 1:2 diluted LB with 25 μg/ml kanamycin and cells were grown at 37 °C for 4 h until the culture reached OD600nm = 1 in plain LB medium. Induction of the promoter fusion was tested using 100 mM 1,2-propanediol, 20 mM CsCl, 1.25 mM CuCl2, 25 mM cycloheximide, 1 mM dithiothreitol, 78 μM erythromycin, 200 mM formamide, 4 mM HCOOH, 2 mM glutamine, 200 mM MgCl2, 320 μM menadione, 400 mM NaCl, 10 mM 1-methylimidazole, 10 mM propanedioic acid, 62.5 mM salicin, 16 mM Na3VO4, and 10 mM Na2B4O7.
For anaerobic growth, bacteria were grown in 15 ml medium as above in tightly closed 15-ml falcons. Anaerobiosis was tested using resazurin in separate tubes. All cultures, including an empty-vector control, were grown for approximately 7 h to OD600nm = 0.3 in the dark. To allow GFP to mature to fluorescence, anaerobically grown cultures were aerated for 15 min by shaking in larger bottles. Bacteria were washed once with 1 ml PBS (6600 × g, 2 min) and diluted to OD600nm = 0.6. Two-hundred μl of this suspension was measured for fluorescence using a black microtiter plate and a plate reader (Wallac Victor3, Perkin Elmer Life Science, excitation 485 nm, emission 535 nm, measuring time 1 s). The mean value of four replicate wells was calculated and the experiment was repeated three times.
Determination of the transcription start site by 5’-RACE
5’-RACE was performed using the “5’RACE System for Rapid Amplification of cDNA Ends Version 2.0” (Invitrogen) according to the manufacturer. Wild type strain was grown in 1:2 diluted LB medium with the addition of 320 μM menadione to OD600nm = 0.5. RNA was isolated with Trizol. For 5’-RACE, the primers CAA CAT GCA CCT TCA GGA T (Z0762+59R) and TGG CGG AAA TCG CCC AAT TCC TGC AT (Z0762+140R) were used. After gel electrophoresis, the strongest band was cleaned (Invisorb® Fragment CleanUp, STRATEC, Berlin) and used as a template for subsequent amplification and sequencing (LGC Genomics, Berlin) using the nested primer GAG CGT TGA CAC CAC AGT CGA AGT AT (Z0762+177R).
Overexpression of Nog1 C-terminal fused with GFP
The open reading frame of nog1 was amplified via PCR using primers Z0762+323R-SphI (TTG CAT GCC GTG GCT AAT GTC AGC GCC AG) and Z0762+20F-KpnI (C CGG TAC CCG GTT TGC AAC ATT GAA CAA CA). The amplicon was cloned in the SphI and KpnI restriction sides of pEGFP (CLONTECH laboratories). The plasmid was sequenced for verification. Ten ml LB with 120 μg/ml ampicillin was inoculated with 100 μl overnight culture of TOP10 transformed with the empty vector and the vector containing the open reading frame of nog1. Bacteria were grown to OD600nm = 0.3 – 0.5 at 37 °C shaking. 1 mM IPTG was added and bacteria were grown for two more hours. Bacterial culture was washed with 2 ml PBS and subsequently suspended in PBS with OD600nm = 1. Two-hundred μl bacterial suspension was transferred in one well of a black microtiter plate and measured using a plate reader (Wallac Victor3, Perkin Elmer Life Science, excitation 485 nm, emission 535 nm, measuring time 1 s). The mean value of four replicate wells was calculated and the experiment was repeated three times.
Overexpression of Nog1 and Western blot
The open reading frame of nog1 and its upstream region was amplified via PCR using primer GAT CCC ATG GCG GTG CCT TTT AAC ACC AGA TC (Z0762+667R-NcoI) and GAG CGA ATT CG TTT GCA ACA TTG AAC AAC ATT (Z0762+21F-EcoRI). The amplicon was cloned in the NcoI and EcoRI restriction sites of pBAD Myc-His C (Invitrogen) resulting in pBAD-nog1::Myc-His. The plasmid was sequenced for verification and EHEC were transformed with pBAD-nog1::Myc-His or with pBAD Myc-His C as control. One-and-a-half L of 1:2 diluted LB medium supplemented with 120 μg/ml ampicillin and 320 μM menadione were inoculated 1:250 with an overnight culture of the transformants. Bacteria were grown in six 1-L bottles (250 ml medium each), shaking at 37 °C to OD600nm = 0.5. Bacteria were pelleted (10 min, 3500 × g) and resuspended in 15 ml lysis buffer included in the QIAexpress® Ni-NTA Fast Start kit (Qiagen). The cells were sonicated six times for 15 s (interval time 0.5 s at 25 % total power, ultrasonic converter tip UW 2200 powered by HD 2200, Bandelin electronics, Berlin) and proteins were purified under native conditions according to the manufacturer’s protocol. From the empty control, a mock sample was ‘purified’. Proteins were precipitated with acetone and Laemmli-buffer was added. The sample was heated for 5 min at 95 °C and loaded on a 15 % SDS-gel. One well was loaded with 5 μl of the PageRuler Prestained Protein Ladder (Fermentas) for size determination. After SDS-PAGE, the proteins were transferred to a PVDF membrane (Amersham/Millipore). Transfer to the membrane was carried out in blotting buffer (50 mM Tris, 39 mM glycine, 0.039 % SDS in 20 % methanol) for 10 min at 150 mA. Next, the membrane was blocked for 1 h in TBS-T (10 mM Tris, 150 mM NaCl, pH8, 0.1 % Tween 20) supplemented with 1 % BSA at room temperature. The membrane was then incubated with 5 ng/μl BD Pharmingen mouse anti-human c-myc-antibody (BD Biosciences) in TBS-T plus 1 % BSA. After washing twice in TBS-T for 10 min, the membrane was incubated for 1 h at room temperature with the second antibody (alkaline phosphatase (AP) anti-mouse chimera, 6 ng/μl) in TBS-T plus 1 % BSA. The membrane was washed twice for 10 min with TBS-T and equilibrated in AP-buffer (100 mM Tris, 150 mM NaCl, 5 mM MgCl2, pH 9.5). For the final detection of the fusion proteins, the membrane was incubated with 0.7 mg/ml BCIP and 0.07 mg/ml NBT solution in AP buffer.
Construction of translationally arrested mutants
Chromosomal DNA was modified for the Δnog1 and ΔcitC mutants using plasmid pMRS101 [32]. For Δnog1, two fragments of this gene were amplified using primers CAT TTT CAT GAA GGA ATT GGG (Z0762+152RmutS) and ata cta gtA TTT CAC GCC GAA ATA CTC C (Z0762-59F-SpeI) and the primer CCC AAT TCC TTC ATG AAA ATG (Z0762+152FmutS) and gcg ggc ccA ACA GCG CCT CGT ATT CGG T (Z0762+382R-ApaI). Mutated bases in the primers and the added restriction sites are marked bold and italic respectively. The two nog1-fragments are located up- and downstream of the desired mutation and overlap in this area. The fragments were used in a third PCR using primer Z0762-59F-SpeI and Z0762+382R-ApaI to recreate a complete fragment with the mutation. This was conducted accordingly for ΔcitC using the primer pairs TGT TAT CGA TCT TCA ACG AAT GT (Z0762+38RmutA) and ata cta gtT AAA TCA ATT AAA TCA CTT A (Z0762-171F-SpeI), and primer pair ACA TTC GTT GAA GAT CGA TAA CA (Z0762+38FmutA) and gcg ggc ccG ATT CAC TGA TAG CAA CGC A (Z0762+268R-ApaI). The final PCR products were cloned in the SpeI and ApaI sites of pMRS101, and transformants grown in LB medium plus 100 μg/ml ampicillin. The high-copy number ori was removed by restriction with NotI and subsequent self-ligation. The remaining plasmid contains a second low-copy pir-dependent ori. This plasmid was introduced into E. coli CC118λpir via electroporation, cells were grown in LB medium plus 30 μg/ml streptomycin, and the plasmid sequence was verified by sequencing using either primer CTT ATC GAT GAT AAG CTG TC (pMRS101+458R) or TCA ATC ATG CGA AAC GAT CC (pMRS101+184F). Plasmids were transferred to E. coli SM10λpir, which enables conjugation, and integrated in EHEC NalR via plate mating: Each 500 μl overnight culture was mixed, plated on LB agar, and incubated for 24 h at 30 °C. Cells were resuspended in LB and plated on LB-plates containing 30 μg/ml streptomycin and 20 μg/ml nalidic acid. The correct insertion of the suicide plasmid was confirmed using primers pMRS101+184 F and CTT GCG GGT TGT CCC GAG CC (Z0762-272F) for Δnog1 or ΔcitC from genomic DNA of the trans-conjugants. To facilitate a second cross-over, cultures were grown in plain LB to OD600nm = 0.8 and counter-selected on sucrose agar (modified LB-agar without NaCl, supplemented with 10 % sucrose). A PCR fragment using primers TTC AGT CGC GTG GCG CTG TT (Z0762+460R) and CTT GCG GGT TGT CCC GAG CC (Z0762-272F) obtained from the chromosome was sequenced to identify the desired strand-specific mutants of either nog1 or citC.
Growth curves of individual strains
Overnight cultures of EHEC wild type, ΔcitC and Δnog1 were used to inoculate 100 ml 0.5 × LB medium supplemented with 200 mM MgCl2 in 250-ml Schott bottles at an OD600nm = 0.03. Bacteria were grown at 37 °C under shaking (150 rpm) and the OD600nm was recorded at given time points.
Competitive growth assays
Overnight cultures in LB were adjusted to OD600nm = 1 and the two test strains were mixed in equal numbers. Control samples were taken immediately (t = 0). Half-strength LB medium (supplemented as indicated, e.g. 150 mM MgCl2, or the substances used before for the promoter induction) was inoculated 1:30,000 using the bacterial mixture. Bacteria were grown under shaking at 150 rpm for 18 h at 37 °C, harvested by centrifugation (3 min, 16,000 × g) and boiled for 5 min at 95 °C to release the DNA. Each sample was used as template for PCR with a locus specific primer pair GTT TGC AAC ATT GAA CAA CAT TCG (Z0762+21F), and GAC TGT GGT GTC AAC GCT CAA ATC (Z0762+172R), for Δnog1 or ΔcitC versus wild type. PCR products were purified and sequenced (AGOWA, Berlin) using primer Z0762+21F for Δnog1 and primer Z0762+172R for ΔcitC. For competitive growth experiments between Δnog1 and ΔcitC, PCR products were generated using primer TTC AGT CGC GTG GCG CTG TT (Z0762+460R) and CTT GCG GGT TGT CCC GAG CC (Z0762-272F). The PCR product was sequenced using primer Z0762+460R. After sequencing, the ratios between peak heights from the Sanger sequencing of the alleles were determined and expressed as a percentage of each strain. This experiment was repeated at least three times.
To complement the translational arrested mutant Δnog1, nog1 was cloned on the arabinose-inducible plasmid pBAD Myc-His C (Invitrogen) using a PCR-product generated with the following primers gat ccc atg gca gtg gct aat gtc agc gcc ag (Z0762+304R-NcoI) and gag cga att ctc agt ttg caa cat tga aca ac (Z0762+18F-EcoRI). The amplicon was cut with NcoI and EcoRI and ligated into the plasmid using T4-ligase. The plasmid sequence was verified by sequencing. The translational arrested mutant was transformed either with an empty plasmid (control) or with the overexpression plasmid pBAD-nog1. A wild type transformed with the empty vector was used as control. These control bacteria and complemented mutants of nog1 were grown in competitive assays as described above in medium supplemented with 120 μg/ml ampicillin (for plasmid selection) and 0.2 % arabinose (for nog1 induction). The percentage of “complemented mutants” versus “control bacteria” was determined as described above after 18 h of competitive growth.
Metabolomics
For a full description of the metabolome measurements see Additional file 2: File S1. In short, the following strategies have been applied.
Metabolomes by GC-MS
The methanol-soluble part of the metabolome was determined from six biologically independent cultures of mutant and wild-type, respectively. Briefly, flash frozen bacteria were extracted derivatized for metabolite analysis via GC-MS in a two-step procedure as described [33]. For metabolite profiling a HP Agilent 7890 gas chromatograph was used to perform GC analysis and coupled to an Agilent 5975 Quadrupole mass spectrometer (Agilent Technologies, Böblingen) for mass determination. The software MetaboliteDetector (version 2.06) was used for processing. Differences in the metabolome between wild type and the mutants were tested for significance using Student’s test (T-test; p ≤ 0.05).
Metabolomes by ICR-FT/MS
Sample preparation was carried out as above for three biological replicates of each wild type, Δnog1 and ΔcitC mutants. Metabolite profiling was conducted using Ion cyclotron resonance Fourier transform Mass spectrometry (ICR-FT/MS) on a Bruker solariX equipped with a 12 T magnet (Bruker Daltonics, Bremen). Putative metabolites were annotated using the MassTRIX webserver [34]. Statistical analysis was carried out in MS Excel 2010 and Genedata Expressionist for MS 7.6 (Genedata, Martinsried) using Welch’s T test (p ≤ 0.05) [35, 36].
Bioinformatics methods
σ70 Promoters were searched for using BProm (Softberry Inc., New York), and terminators using WebGeSTer DB [37]. Sequences from other bacteria were searched with blastp or tblastn (NCBI, http://blast.ncbi.nlm.nih.gov/Blast.cgi, default parameters). All methods mentioned in the following were used with preset values. Domain search was conducted using CDD [38] with an e value cut-off of 10−2. The distribution of charged amino acids in Nog1 was analyzed manually. Prediction of disordered regions was conducted using GlobPlot [39] and Meta-Disorder [40] The authors of GlobPlot claim for a given prediction a specificity of 88 % and a sensitivity of 28 % [39]. For Meta-Disorder, the preset reliability index suggests to find about 52 % of the disordered residues and 68 % of those would be correct [40]. Low complexity regions were discovered using CDD [38] which implements SDUST with preset values [41]. The transmembrane and overall topology was predicted using hmmtop [42], TMHMM [43], PHDhtm [44], and BCL::Jufo9D [45]. The program hmmtop correctly predicts 89 % of the membrane spanning regions and 71 % of the correct topology [42]. TMHMM correctly predicts 97-98 % of the transmembrane helices. The specificity and sensitivity of this program is greater than 99 % if no signal peptide is present [43]. PHDhtm predicts the topology for helical transmembrane proteins at 86 % accuracy [44]. Finally, BCL::Jufo9D has a prediction accuracies of 73.2 % the secondary structure prediction, and 94.8 % for the transmembrane span prediction [45]. Beta-turns were predicted using NetTurnP [46]. Sensitivity is 76 % and specificity 79 %, respectively, in distinguishing turns from not-turns [46]. The protein secondary structure was established using SOPMA [47], PSIPRED [48], Pred2ary [49], and GOR IV [50] via the web server NPS@ [51]; PROFseq [52] via the PredictProtein server [53], BCL::Jufo9D [45], or using Porter [54]. SOPMA correctly predicts 69.5 % of amino acids for a three-state description (α-helix, β-sheet and coil; Q3) of the secondary structure [47]. The current PSIPRED 3.2 achieves an average Q3 score of 81.6 % [55]. For Pred2ary, the secondary structure prediction accuracy was given as 63 % [49], and GOR IV has a mean accuracy of 64.4 % for Q3 [50]. PROFseq has an overall accuracy of 71.6 % and a sustained Q3 of 88 % for 40 % of the residues [56]. BCL::Jufo9D has prediction accuracies of 70.3 % for nine possible states, and 73.2 % for the Q3 secondary structure prediction [45]. Finally, Porter's accuracy exceeds 79 % [54]. Protein binding sites were predicted using ISIS, which has total two-state accuracy of 68 % [57]. Protein statistics were calculated using ProtParam [58], signal protein sequences were searched for using SignalP [59], and the protein localization was predicted using LocTree3. This program reaches a six-state accuracy (6 localizations) of 89 ± 4 % for bacteria [60].
Tree construction
The evolutionary tree is based on a concatemer of the conserved genes 16S rRNA, atpD, adk, gyrB, purA and recA and is independent of citC/nog1. The gene sequences were obtained from the genome of each organism (for sequences and accession numbers, see Additional file 3: Table S2). All the above-mentioned genes of each organism were concatenated and aligned using muscle in Mega6.06 [61] with default parameters. The alignment was checked and since this original alignment had gaps (Additional file 4: File S2), all nucleotides in the respective column which had a gap were manually removed by deleting this column until no gap remained in the complete final alignment. In addition, positions (i.e., columns within the alignment) in which ambiguities were present, were manually deleted as well. There were a total of 7721 positions in the final dataset (Additional file 5: File S3). MEGA6.06 allows precomputation of the best nucleotide substitution model for Maximum Likelihood (ML) to match the data, including General Time Reversible, Hasegawa-Kishino-Yano, Tamura-Nei, Tamura 3-parameter, Kimura 2-parameter, and Jukes-Cantor. Non-uniformity of evolutionary rates among sites may or may not be modeled by using a discrete Gamma distribution (+G) with 5 rate categories and by assuming that a certain fraction of sites are evolutionarily invariable (+I). The model with the lowest Bayesian Information Criterion score is considered to best describe the substitution pattern [62], which is General Time Reversible with gamma distribution and invariable sites in this case. For the precomputation, the tree topology was automatically computed using Neighbor-Joining.
The final evolutionary history was inferred by using the ML method based on the General Time Reversible model. The tree with the highest log likelihood (−58141.5) is shown. Values within the tree indicate the percentage of 1000 bootstrap tests showing the same cluster. Bootstrap values below 50 % are not shown. Initial trees for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories, parameter = 0.6072). The rate variation model allowed for some sites to be evolutionarily invariable. The final tree is drawn to scale, with branch lengths measured in the number of substitutions per site.
Results
Discovery of an overlapping protein-coding ORF displaying protein-coding features
In previous experiments, EHEC was grown under eleven different growth conditions and strand specific transcriptomes were sequenced [27]. These conditions comprised LB medium at pH4, pH7, pH9, or at 15 °C; LB with addition of nitrite or trimethoprim-sulfamethoxazole; LB-agar surface, M9 minimal medium, spinach leaf juice, the surface of living radish sprouts, and cow dung. In the condition “cow dung”, we found a novel RNA to be induced about 14-fold compared to LB, based on RPKM values (Fig. 1a). Briefly, for the condition “cow dung”, 10 g cattle feces were inoculated for 6 h at 37 °C with EHEC pre-grown in LB [27]. This RNA covers an overlapping ORF which consists of 306 bp (position from 732757 to 733062 in EHEC genome, accession no. NC_002655) and is completely embedded in antisense to citC in frame -2 (position 733079 to 731934, the reading frame of citC being defined as +1; Fig. 1a). Thus, we wondered whether this novel RNA was maybe protein coding and undertook experiments to verify this hypothesis. To facilitate further reading, we introduce the suggested gene name nog1 here (novel overlapping gene).
Fig. 1.
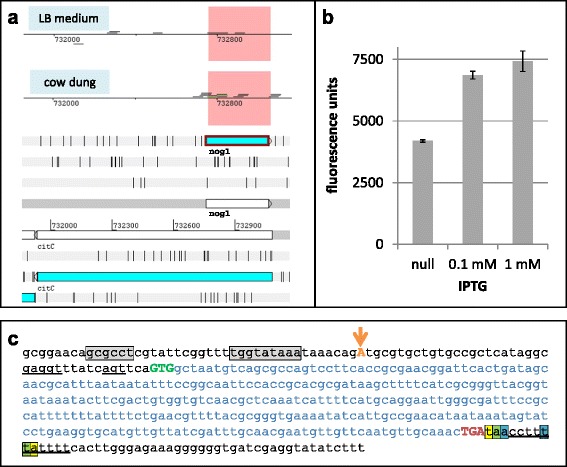
a Strand-specific transcription signals of nog1. Shown are the transcriptome data of EHEC (upper panel) grown aerobically in 1:10 diluted LB medium (upper line) or incubated in cow dung (lower line). The transcriptome sequencing reads are shown above or below the genome line for the forward and reverse strand respectively. Only a single read of citC is visible in the LB-condition. However, nog1 (pink shaded area) is induced about 14-fold in cow dung compared to LB, based on RPKM values. The lower panel shows the genomic architecture around citC, drawn using Artemis [93]. The open reading frames of nog1 and citC are indicated by blue arrows in the respective reading frames. b Fluorescence units of nog1 C-terminally fused with gfp after IPTG induction for 2 h using the indicated inducer concentrations. This experiment shows that nog1 is principally able to be translated by ribosomes. c Genetic organization of nog1. Grey, predicted promoter (BProm); orange letter, transcription start site according to the transcriptome sequencing data (Fig. 1a); orange arrow, transcription start site according to 5’-RACE; underlined, possible Shine-Dalgarno sequences; green, predicted start codon; blue, coding sequence; red, stop codon; bases highlighted by bold underline, predicted rho independent terminator with possible hairpin structure (WebGeSTer)
The novel ORF nog1 was C-terminally fused in frame to gfp to determine protein production. After induction, GFP fluorescence was increased compared to the uninduced control showing that the novel ORF nog1 is indeed translatable (Fig. 1b). However, we could not detect Nog1 induced from its natural promoter and fused to His and Myc tags (data not shown). Despite enrichment using Ni-columns and acetone precipitation, His-Myc-tagged Nog1 was not detectable in Western blots using Myc-tag antibodies. But acetone precipitation is prone to lose small proteins and Nog1 is predicted to be unstable and a membrane protein (see below), which may have rendered immunological detection of this short protein difficult. However, the above nog1::gfp fusion shows that the ribosomes pass the GTG start codon to translate the nog1 ORF.
Next, we predicted promoter, Shine-Dalgarno and terminator sequences for nog1, all of which were found (Fig. 1c). A possible transcription start site in the vicinity of the predicted promoter could be the read mapping farthest upstream in the strand-specific transcriptome sequencing (Fig. 1a). The read starts 40 bp upstream of the putative start codon. To verify this position, the +1 was determined using 5’-RACE. The latter result is in perfect agreement with the former since the indicated transcription start site of the new ORF was also found to be 40 bp upstream of the putative start codon GTG. Thus, this position is suggested to be the major +1 for nog1 (Fig. 1c, orange arrow).
Promoter activities of the genomic regions upstream of nog1 and citC
The promoter region immediately upstream of nog1 was fused to gfp and tested for inducibility. Unfortunately, cow dung was not usable for these experiments due to the opaqueness, autofluorescence, and the many other bacteria present in this substrate. Sterile filtration before and after dilution was attempted, but extremely difficult due to the high content in fine particulate matter. To ease analysis, we decided to test LB medium supplemented with a number of various inorganic and organic stressors in sub-inhibitory concentrations for promoter activity (see Methods). Using LB medium as a control, promoter Pnog1 activity was increased when cells were grown aerobically in the presence of 320 μM menadione (3-fold), 5 mM malonic acid (2.5-fold), and 150 mM magnesium chloride (1.5-fold; Fig. 2). The transcription of citC is only induced in the presence of citrate under anaerobic conditions [63], thus, these conditions were tested additionally (Fig. 2). PcitC becomes active about 2-fold when bacteria are grown anaerobically with the addition of 20 mM citrate. Interestingly, Pnog1 activity increased under anaerobiosis in plain LB, as well as in citrate supplemented LB (Fig. 2), and, in addition, when adding menadione (2-fold) or malonic acid (5-fold).
Fig. 2.
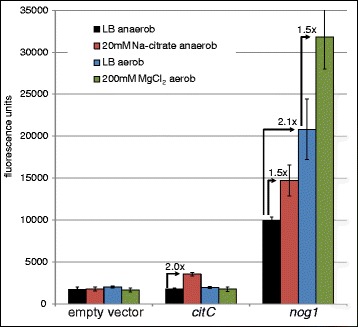
Fluorescence measurements of promoter::gfp fusions for empty vector control (left), PcitC (middle) and Pnog1 (right). The fluorescence has been normalized to OD600nm and the conditions are indicated
Metabolome of the translationally arrested mutants compared to the wild type
To uncover phenotypes related to the gene product Nog1, its translation was arrested by introducing a stop codon without changing the amino acid sequence of the mother reading frame citC (Δnog1, Fig. 3, left). Similarly, citC was arrested without changing Nog1 (Fig. 3, right). If nog1 were being a non-coding RNA, its function would not be disturbed by a single nucleotide exchange. In contrast, translation is arrested by the artificial stop codon and Nog1 production ceases. The metabolome of the mutant compared to wild type was analyzed by non-targeted profiling approaches using either gas chromatography coupled with mass spectrometry (GC-MS) or ion cyclotron resonance Fourier transform mass spectrometry (ICR-FT/MS).
Fig. 3.
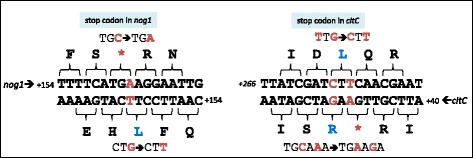
Mutations introduced to arrest the translation of nog1 (left) or citC (right) strand specifically. The numbers indicate the distance of the sequence shown from the start codons of nog1 and citC respectively. Note that the amino acid sequence of the reading frame in antisense remains unchanged in both cases. The positions of the mutations are shown in red
For GC-MS, six biological replicates of both wild type and Δnog1 were grown in plain LB and significantly different metabolites determined (p ≤ 0.05). Only a decrease of metabolites in the nog1-mutant was found. For instance, tryptophan had the highest fold decrease of about 4.7. Next were citric and isocitric acid, as well as succinate with a decrease of about 1.5 to 2-fold (further metabolites are in Additional file 6: Table S3). Hence, we believe there is a slight influence of the Δnog1 mutation on the primary metabolism, probably with a focus on the TCA cycle due to changes in citrate and succinate.
Using the more sensitive and accurate ICR-FT/MS for profiling, several thousand features were detected (data not shown) and MassTRIX [34] annotated several hundred metabolites from different pathways. Significantly different molecules between wild type and mutant (p ≤ 0.05) included metabolites, e.g., of the glutathione metabolism (glutathione decreased 5.6-fold in Δnog1 compared to wild type). Furthermore, amino acid metabolism pathways were affected, since corresponding metabolites were found to be changed (see Additional file 7: Table S4). The latter fits with the observed change in tryptophan levels in the GC-MS experiment above. GC-MS and ICR-FT/MS complement each other. ICR-FT/MS is not able to detect small masses, e.g., of tricarboxylic acid cycle (TCA) intermediates, but glutathione metabolites were not identifiable in the GC-MS experiment. However, targeted investigations are needed to further support the above results since only a weak metabolic phenotype is found in both assays.
Fitness of translationally arrested Nog1 and CitC mutants
When wild type and Δnog1 mutant were grown separately under a variety of stress conditions, no differences in growth rates were observed (data not shown), except a slightly reduced growth of the Δnog1 mutant in medium supplemented with MgCl2 (Fig. 4a). In contrast, competitive growth assays comparing two strains (e.g., mutant versus wild type) in the same flask are highly sensitive tools to detect even small fitness differences between two strains [64, 65]. For competition experiments, the two strains were mixed in equivalent small cell numbers (50:50) and broth supplemented with MgCl2 at sub-inhibitory concentration was inoculated using the strain mixture. A change in relative fitness between the two strains becomes apparent by determining the fraction of one strain over the other. After 18 h of aerobic growth, the performance of each strain was measured (Fig. 4b). Interestingly, Δnog1 shows a clear loss of fitness compared to its parental strain when grown in plain LB. When using MgCl2-supplemented broth, the decrease in fitness of Δnog1 was even more pronounced, corroborating the finding of retarded growth in MgCl2-supplemented medium (compare to Fig. 4a). A mutant translationally arrested in citC, however, did not show any fitness differences when competitively grown against its parental strain (Fig. 4b). Accordingly, when Δnog1 was grown competitively against ΔcitC a similar decrease in fitness was observed comparable to the wild type strain, both in plain LB and medium supplemented with MgCl2.
Fig. 4.
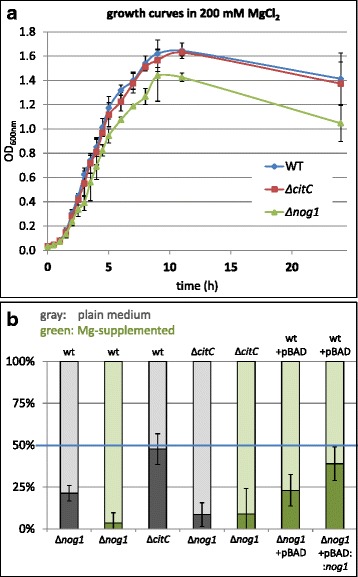
Growth phenotype of wild type, and ΔcitC and Δnog1. a Growth curves of wild type, and ΔcitC and Δnog1 strains grown separately in Schott bottles under shaking at 37 °C. The average OD values and the standard deviations from three replicates are given. b Percentage of strains after growth in competitive growth assays. Gray columns indicate plain LB medium, green columns LB supplemented with MgCl2. The experiments were repeated at least three times and standard deviations are shown. First column, growth of Δnog1 against wild type in LB; second column, Δnog1 against wild type in LB with MgCl2; third column, growth of ΔcitC against wild type in LB; fourth column, growth of Δnog1 against ΔcitC in LB; fifth column, growth of Δnog1 against ΔcitC in LB with MgCl2; sixth column, growth of Δnog1 + pBAD competing against wild type + pBAD in LB with MgCl2; seventh column, Δnog1 + pBAD::nog1 was grown together with wild type + pBAD in LB with MgCl2. The last two experiments were performed in the presence of ampicillin and arabinose
For complementation experiments, the nog1 ORF was cloned downstream of an arabinose inducible promoter in the plasmid pBAD. At first, the translationally arrested Δnog1 mutant and wild type, both transformed with an empty pBAD, were grown competitively in MgCl2-supplemented medium. Unexpectedly, the difference in competition, visible when using vector-less strains in this medium, diminished somewhat. Next, Δnog1 was transformed with pBAD::nog1 to compete against the wild type containing an empty pBAD. The Δnog1 strain containing pBAD::nog1 competed better against wild type containing the pBAD vector, although it could not be fully restored (Fig. 4b). We hypothesize that the indispensable addition of ampicillin and arabinose to the latter experiments disturbed the competition experiments to some extent.
Bioinformatics analysis of the Nog1 protein sequence
The new overlapping-encoded protein Nog1 is predicted to have 101 amino acids, a theoretical molecular weight of 11.15 kDa, a pI of 11.36, an aliphatic index of 117.72, and the GRAVY value is 0.564 [66]. No signal peptide or domain was predicted [38, 59]. The instability index was computed to be 42.05 which classifies the protein as being unstable [58]. Secondary structures and membrane domains were predicted, using a number of similar programs allowing comparison of the results for different algorithms. This assumes that secondary structures predicted by several programs are possibly more reliable. The Nog1 protein structure potentially consists of three short helices and several very short β-sheets. At least one membrane domain was predicted by all algorithms used (compare to Additional file 8: Figure S1). The protein may be localized in the inner membrane according to the software LocTree3 [60], most likely with the N-terminus outside and the C-terminus inside, as agreed on by two of three programs for transmembrane topology prediction [42, 43].
Phylogenetic analysis of nog1
In order to elucidate the phylogeny of nog1, a phylogenetic tree including EHEC was constructed of species harboring citC (only a representative subset was used omitting identical or very similar sequences) with Helicobacter as an out-group (Fig. 5). The phylogenetic tree calculation was independent of citC since it was based on a concatemer of 16S rDNA, atpD, adk, gyrB, purA and recA. All six genes belong to the core genome of the species included in the analysis. Homologs of citC were then analyzed for the presence of nog1 in their −2 antisense reading frame. The full reading frame nog1 was found in Escherichia or Shigella. In a few instances, its reading frame was destroyed, for example due to an insertion sequence or a frame shift which also destroyed citC (the only two of such species found are shown in Fig. 5). Several Salmonella species contain a nog1-like part in their citC sequence. Depending on the Salmonella strain, nog1 appears to be either elongated or fragmented, but whether nog1 is indeed functional in some Salmonella strains remains unknown. In all other genera, the nog1 reading frame is clearly fragmented (Fig. 5).
Fig. 5.

Phylogenetic tree of nog1-bearing E. coli and Shigella strains and species carrying nog1-like sequences. On the left, phylogenetic tree of representative bacterial strains containing a nog1 or nog1-like open reading frame within citC. The tree was inferred using the Maximum Likelihood method and is based on a concatemer of 16 s RNA, atpD, adk, gyrB, purA, and recA, thus, independent of citC and nog1 respectively. The percentage of trees which clustered together in 1000 bootstrap replicates is shown next to the branches. Helicobacter was used as out-group. On the right, an alignment of nog1 and nog1-like sequences within citC of the strains is shown. Black, sequence parts which can be aligned to nog1 of EHEC using BLAST2 and which might be translated; turquoise, N-terminal methionine; pink, translational stop; blue, frame shift mutation (which destroyed both, nog1 and citC); red, insertion of IS1 and duplicated amino acids ProAlaIle around the insertion site; this insertion also destroys nog1 and citC. Orange, regions likely not to be translated; green, insertion element in some Salmonella and Citrobacter strains, keeping the frames intact
Discussion
nog1 probably encodes the protein Nog1
In this paper we report that nog1 possesses an active promoter, activated in natural as well as lab conditions (Figs. 1a, and 2). A transcription start was determined, the gene is followed by a terminator sequence (Fig. 1c) and a protein can be expressed (Fig. 1b). In addition, we provided ample evidence that the ∆nog1 mutant shows a weak phenotype, especially in MgCl2-supplemented medium (Fig. 4; Additional file 6: Table S3; Additional file 7: Table S4). This data clearly shows that the gene is functional, although of apparently minor influence to the fitness of E. coli.
We were successful in demonstrating a stable gene product at the protein level when fusing Nog1 to GFP and detecting the latter by its fluorescence (Fig. 1b) but one could still hypothesize, despite this finding, that nog1 actually may encode a novel ncRNA rather than a protein in vivo. A number of relatively short RNAs are not translated and might function solely as ncRNAs at the RNA level [67, 68]. However, we suggest rejecting the “non-coding RNA hypothesis” of nog1 for the following five reasons.
First, a bioinformatics analysis of the open reading frame of nog1 (Fig. 1c, Additional file 8: Figure S1) shows clear characteristics of a protein encoding ORF, including a ribosome binding site in proper distance from the start codon. While it cannot be excluded that such sequence characteristics may occur just by chance, we consider this to be a remote option [69]. Second, if nog1 regulates citC via antisense silencing, nog1 should only be induced when citC mRNA is present [70]. However, nog1 is induced in cow dung where no transcript of citC could be detected (Fig. 1a). Promoter studies of this and other work show that citC is expressed only under anaerobic conditions, but nog1 was also detectable in aerobic conditions. Thus, it is quite unlikely that nog1 acts as regulatory RNA in an anti-sense fashion against citC since, for antisense-RNA regulation, the presence and base pairing of both sense and antisense transcripts is required [71]. Third, the introduction of a single base pair change only, to translationally arrest nog1, leads to a clear phenotype which would not be expected when nog1 is an antisense regulator. Also, a hypothetical ribozyme activity would require stable base pairing involving many nucleotides [72]. Single base pair changes, such as those used in this study to translationally arrest nog1 (Fig. 3), are unlikely to cause detectable differences in an RNA-RNA pairing event which involves many base pairs. Fourth, a translationally arrested ΔcitC mutant which introduced a nucleotide change synonymous in the overlapping nog1 did not, as was expected, display any fitness differences in competitive growth experiments (Fig. 4). Finally, supplementing the Δnog1 strain with a functional nog1 on a plasmid in trans means that Nog1 is produced, and the strain competes somewhat better against wild type in MgCl2-supplemented medium (Fig. 4b). However, if nog1 acts as ncRNA, this strain should compete even less against the wild type since the gene dosage of the plasmid-borne gene is higher and more ncRNA would be present to regulate its targets.
Potential function of Nog1
Transcription of nog1 is specifically induced in cow dung (Fig. 1a), and the promoter is activated by magnesium ions (Fig. 2), menadione and malonic acid. In addition, magnesium ions had an effect on growth of Δnog1 in growth and competitive growth assays (Fig. 4). However, the function of nog1 remains cryptic. It would have been interesting to elucidate the function of nog1 in cow dung further, but this is a difficult substrate to work with. For this reason we relied on lab media. Changes in metabolite concentrations of the Δnog1 mutant are small compared to wild type in plain LB, so nog1 is only slightly involved in any central metabolic reaction (e.g. TCA cycle, amino acid metabolism; Additional file 6: Table S3, Additional file 7: Table S4). Since the ∆nog1 mutant displayed a small growth disadvantage in MgCl2-supplemented medium (Fig. 4a), bacteria carrying nog1 may have an increased fitness in environments reflecting this or other unknown conditions. Whatever function this gene exerts, its modificatory action is at least strong enough to cause a phenotype in a competitive growth assay. Since, according to the bioinformatics analysis, Nog1 is membrane bound, it could either act as sensor or modificatory protein in the membrane. However, further experiments would have to support this hypothesis.
Recent origin of nog1 by overprinting
The citrate-lyase ligase gene citC, which is the mother gene of nog1, is taxonomically broadly distributed (mostly in γ-proteobacteria and firmicutes, but also in some other bacteria). In contrast, nog1 is found only in the Escherichia/Salmonella clade. Such a taxonomical distribution could be explained by an ancient overprinting event followed by many subsequent deletions of the nog1 reading frame in all clades except the Escherichia/Salmonella clade. However, for parsimony reasons we suggest a recent origin of nog1 after the separation of Escherichia/Salmonella from the rest of the enterobacteria (Fig. 5).
Based largely on research in eukaryotes, recently arisen genes are thought to be shorter and less important compared to genes with a longer evolutionary history [11, 73, 74]. After a short evolutionary history, novel genes should be less well-integrated in the cellular metabolism and, therefore, may provide only a limited fitness gain for the cell. Since young genes are expected to be not yet well-adapted, due to their short evolutionary history, they may be expressed at a low level only. New genes should be orphans [75] and overlapping genes have been proposed to be phylogenetically more restricted than the mother gene they overlap [17]. Finally, de novo formed genes may have a strain specific function only [76], which could be important for niche specific adaptations assumed for orphan genes [77].
We suggest that nog1, embedded completely in its mother frame citC, offers a number of characteristics which fit the hypothesis of nog1 being a young gene: (i) nog1 shows a restricted occurrence within the closely related genera of Escherichia/Salmonella, (ii) the gene is short, (iii) it is weakly expressed, (iv) it appears to be only marginally associated in E. coli’s central metabolism, and (v) the gene product provides only a small fitness advantage for its carrier. Conversely, the mother frame citC is widely distributed among many bacterial species since it is important to metabolize citrate (in the TCA) under anaerobic growth conditions [28]. Thus, we hypothesize that nog1 may have arisen by a recent overprinting event, probably after the split of the Escherichia/Shigella clade or after the divergence of Escherichia/Salmonella from the other γ-proteobacteria (Fig. 5).
Conclusions
According to Nekrutenko & He [78] “[…] genes with overlapping reading frames exemplify some of the most striking biological phenomena […]”. This is a statement with which we agree, especially since Johnson and Chisholm [14] proposed an information content constraint which should prevent non-trivially overlapping genes to form freely. Indeed, the initial sequence features of the citC mother reading frame, which would allow for a successful overprinting process producing a Nog1 protein with at least a weak initial function, are unknown and certainly require further investigation.
Bacteriological research has never been directed to discover overlapping genes. On the contrary, bacterial genome annotation programs systematically remove overlapping genes [73, 79–81], thereby efficiently preventing their discovery. Perhaps it is not surprising that with the exception of E. coli’s three overlapping genes, only a few further examples have been discovered rather accidentally in Streptomyces [82], Pseudomonas [83, 84] or Xanthomonas [85]. Functional analyses of overlapping genes in bacteria are even rarer.
Bacterial overlapping genes may be more common than expected
Four lines of argument lead us to suggest that overlapping genes in bacteria are more common than is currently assumed by microbiologists.
First, a systematic bioinformatics analysis of bacterial overlapping ORFs demonstrates that bacterial genomes contain a larger number of long overlapping ORFs than is expected based on a statistical analysis [86]. Random mutational drift would have eliminated this signal long ago if no selection pressures were stabilizing these ORFs. Indeed, Sabath, et al. [76] found that overlapping loci are under weak positive selection.
Second, during various comprehensive transcriptomic analyses of E. coli targeted to its differential gene expression in different habitats [27], we have noticed that many overlapping reading frames show a transcriptional signal which is clearly above background and is probably not due to ncRNA (data not shown). Such signals might be evidence for the existence of further overlapping genes.
Third, a bioinformatics analysis suggests that overlapping genes appear abundantly in some viruses [87, 88]. There is good evidence that all bacteria are parasitized by viruses and bacteriophage vastly outnumber bacterial cells on earth [89]. Furthermore, genomes of bacterial viruses are constantly being mixed, by various mechanisms, with the genomes of their hosts, which should result in an increase of overlapping genes in bacteria [90].
Fourth, for eukaryotes it has been suggested that completely new genes evolve frequently de novo from non-coding DNA and for a few cases even overprinting has been suggested [5, 10, 73]. Since prokaryotic genomes are densely packed with established genes [91], that source for novel genes is almost absent and we suggest overprinting as a more important alternative to gene duplication for these organisms acquisition of completely novel genes. This exploits a huge hypothetical hidden coding reserve potentially providing a “novelty pool“ for adaptation [92].
While in this paper we have presented specific experimental evidence for the existence of a fourth overlapping gene in E. coli EHEC, we suggest that this may be an initial finding only. For the origin of gene novelties in bacteria, such cases of new overlapping genes could lead to the establishment of overprinting as a potentially more significant alternative to gene duplication, once microbiologists and evolutionary biologists start to target experimental research along this research path.
Availability of supporting data
All data and protocols supporting the results of this article are available in the repository labarchives.com (https://mynotebook.labarchives.com/) using the links https://doi.org/10.6070/H42N5083, https://doi.org/10.6070/H4XW4GSD, https://doi.org/10.6070/H4T43R3V, https://doi.org/10.6070/H4PG1PR0, https://doi.org/10.6070/H4JQ0Z24, https://doi.org/10.6070/H4DZ06BS, https://doi.org/10.6070/H4959FKC, and https://doi.org/10.6070/H45D8PVN. All the supporting data are included as additional files.
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft DFG (BO867/23-1,2,3; KE740/13-1,2,3, and SCHE316/3-1,2,3).
Abbreviations
- 5’-RACE
5’ rapid amplification of cDNA ends
- AP
Alkaline phosphatase
- BCIP
5-bromo-4-chloro-3-indolylphosphate
- blast
Basic local alignment search tool
- bp
Base pairs
- BSA
Bovine serum albumin
- EHEC
Enterohemorrhagic E. coli
- GC-MS
Gas chromatography coupled with mass spectrometry
- GFP
Green fluorescent protein
- GRAVY
Grand average of hydropathy
- ICR-FT/MS
Ion cyclotron resonance Fourier transform mass spectrometry
- IPTG
Isopropyl-β-D-thiogalactopyranosid
- LB
Luria-Bertani medium
- MCL
Maximum composite likelihood
- ML
Maximum likelihood
- MS Excel
Microsoft Excel
- Nal
Nalidic acid
- NBT
Nitroblue tetrazolium
- ncRNA
Non-coding RNA
- NEB
New England Biolabs
- nog1
Novel overlapping gene
- OD600 nm
Optical density at 600 nm wavelength
- ORF
Open reading frame
- PBS
Phosphate-buffered saline
- PCR
Polymerase chain reaction
- PVDF
Polyvinylidene difluoride
- Q3
Three-state description of α-helix, β-sheet and coil
- RPKM
Reads per kilobase per million mapped reads
- rpm
Revolutions per minute
- SDS
Sodium dodecyl sulfate
- TCA cycle
Tricarboxylic acid cycle
- Tris
2-amino-2-hydroxymethyl-propane-1,3-diol
- T‑test
Student’s test
Additional files
E. coli strains and plasmids. (PDF 116 kb)
Full description of the metabolomics measurements. (PDF 125 kb)
Accession numbers and genes used for the phylogenetic tree. (XLSX 154 kb)
Alignment of the concatenated sequences used for tree construction before gap deletion. (TXT 221 kb)
Alignment of the concatenated sequences with gaps deleted. (TXT 196 kb)
Full data set of the gas chromatography coupled with mass spectrometry (GC-MS). (XLSX 20 kb)
Full data set of the ion cyclotron resonance Fourier transform mass spectrometry (ICR-FT/MS). (XLSX 15 kb)
Overview of bioinformatics predictions for Nog1. (PDF 110 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LF conducted the experiments with the help of CP and wrote the manuscript. SSi helped in creating the mutants. CS conducted metabolomes with GC-MS, MW and PSK with ICR-FT/MS. KN was responsible for the bioinformatic and phylogenetic analyses. DAK, SiSch, StSch and KN conceived and supervised the study and helped draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Lea Fellner, Email: Lea.Fellner@dlr.de.
Svenja Simon, Email: simon@dbvis.inf.uni-konstanz.de.
Christian Scherling, Email: christian.scherling@klinkner.de.
Michael Witting, Email: michael.witting@helmholtz-muenchen.de.
Steffen Schober, Email: steffen.schober.de@ieee.org.
Christine Polte, Email: Christine.Polte@chemie.uni-hamburg.de.
Philippe Schmitt-Kopplin, Email: schmitt-kopplin@helmholtz-muenchen.de.
Daniel A. Keim, Email: keim@uni-konstanz.de
Siegfried Scherer, Email: siegfried.scherer@wzw.tum.de.
Klaus Neuhaus, Email: neuhaus@wzw.tum.de.
References
- 1.Ohno S. Evolution by gene duplication. New York: Springer Science & Business Media; 1970. [Google Scholar]
- 2.Espinosa-Cantu A, Ascencio D, Barona-Gomez F, DeLuna A. Gene duplication and the evolution of moonlighting proteins. Front Genet. 2015;6:227. doi: 10.3389/fgene.2015.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet. 2010;11(2):97–108. doi: 10.1038/nrg2689. [DOI] [PubMed] [Google Scholar]
- 4.Kondrashov FA. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proceedings Biological sciences/The Royal Society. 2012;279(1749):5048–5057. doi: 10.1098/rspb.2012.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neme R, Tautz D. Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genomics. 2013;14:117. doi: 10.1186/1471-2164-14-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruiz-Orera J, Messeguer X, Subirana JA, Alba MM. Long non-coding RNAs as a source of new peptides. eLife. 2014;3 doi: 10.7554/eLife.03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson BA, Masel J. Putatively noncoding transcripts show extensive association with ribosomes. Genome Biol Evol. 2011;3:1245–1252. doi: 10.1093/gbe/evr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andreatta ME, Levine JA, Foy SG, Guzman LD, Kosinski LJ, Cordes MH, et al. The recent de novo origin of protein C-termini. Genome Biol Evol. 2015;7(6):1686–1701. doi: 10.1093/gbe/evv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eaton KV, Anderson WJ, Dubrava MS, Kumirov VK, Dykstra EM, Cordes MH. Studying protein fold evolution with hybrids of differently folded homologs. Protein Eng Des Sel. 2015;28(8):241–250. doi: 10.1093/protein/gzv027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tautz D. The discovery of de novo gene evolution. Perspect Biol Med. 2014;57(1):149–161. doi: 10.1353/pbm.2014.0006. [DOI] [PubMed] [Google Scholar]
- 11.Carvunis AR, Rolland T, Wapinski I, Calderwood MA, Yildirim MA, Simonis N, et al. Proto-genes and de novo gene birth. Nature. 2012;487(7407):370–374. doi: 10.1038/nature11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grassé PP. Evolution of living organisms: evidence for a new theory of transformation. New York: Academic; 1977. [Google Scholar]
- 13.Ohno S. Birth of a unique enzyme from an alternative reading frame of the preexisted, internally repetitious coding sequence. Proc Natl Acad Sci U S A. 1984;81(8):2421–2425. doi: 10.1073/pnas.81.8.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson ZI, Chisholm SW. Properties of overlapping genes are conserved across microbial genomes. Genome Res. 2004;14(11):2268–2272. doi: 10.1101/gr.2433104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cock PJ, Whitworth DE. Evolution of gene overlaps: relative reading frame bias in prokaryotic two-component system genes. J Mol Evol. 2007;64(4):457–462. doi: 10.1007/s00239-006-0180-1. [DOI] [PubMed] [Google Scholar]
- 16.Sakharkar KR, Chow VT. Strategies for genome reduction in microbial genomes. Genome Inform. 2005;16(2):69–75. [PubMed] [Google Scholar]
- 17.Keese PK, Gibbs A. Origins of genes: "big bang" or continuous creation? Proc Natl Acad Sci U S A. 1992;89(20):9489–9493. doi: 10.1073/pnas.89.20.9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pavesi A, Magiorkinis G, Karlin DG. Viral proteins originated de novo by overprinting can be identified by codon usage: application to the "gene nursery" of Deltaretroviruses. PLoS Comput Biol. 2013;9(8) doi: 10.1371/journal.pcbi.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carter JJ, Daugherty MD, Qi X, Bheda-Malge A, Wipf GC, Robinson K, et al. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc Natl Acad Sci U S A. 2013;110(31):12744–12749. doi: 10.1073/pnas.1303526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon-Loriere E, Holmes EC, Pagan I. The effect of gene overlapping on the rate of RNA virus evolution. Mol Biol Evol. 2013;30(8):1916–1928. doi: 10.1093/molbev/mst094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chirico N, Vianelli A, Belshaw R. Why genes overlap in viruses. Proc Royal Soc B: Biol Sci. 2010;277(1701):3809–3817. doi: 10.1098/rspb.2010.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delaye L, Deluna A, Lazcano A, Becerra A. The origin of a novel gene through overprinting in Escherichia coli. BMC Evol Biol. 2008;8(1):31. doi: 10.1186/1471-2148-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fellner L, Bechtel N, Witting MA, Simon S, Schmitt-Kopplin P, Keim D, et al. Phenotype of htgA (mbiA), a recently evolved orphan gene of Escherichia coli and Shigella, completely overlapping in antisense to yaaW. FEMS Microbiol Lett. 2014;350(1):57–64. doi: 10.1111/1574-6968.12288. [DOI] [PubMed] [Google Scholar]
- 24.Kurata T, Katayama A, Hiramatsu M, Kiguchi Y, Takeuchi M, Watanabe T, et al. Identification of the set of genes, including nonannotated morA, under the direct control of ModE in Escherichia coli. J Bacteriol. 2013;195(19):4496–4505. doi: 10.1128/JB.00304-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McVeigh A, Fasano A, Scott DA, Jelacic S, Moseley SL, Robertson DC, et al. IS1414, an Escherichia coli insertion sequence with a heat-stable enterotoxin gene embedded in a transposase-like gene. Infect Immun. 2000;68(10):5710–5715. doi: 10.1128/IAI.68.10.5710-5715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sousa CP. East1 toxin and its presence in a changing microbial world. J Venomous Anim Toxins Trop Dis. 2003;9:4–52. doi: 10.1590/S1678-91992003000100002. [DOI] [Google Scholar]
- 27.Landstorfer R, Simon S, Schober S, Keim D, Scherer S, Neuhaus K. Comparison of strand-specific transcriptomes of enterohemorrhagic Escherichia coli O157:H7 EDL933 (EHEC) under eleven different environmental conditions including radish sprouts and cattle feces. BMC Genomics. 2014;15:353. doi: 10.1186/1471-2164-15-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bott M, Dimroth P. Klebsiella pneumoniae genes for citrate lyase and citrate lyase ligase: localization, sequencing, and expression. Mol Microbiol. 1994;14(2):347–356. doi: 10.1111/j.1365-2958.1994.tb01295.x. [DOI] [PubMed] [Google Scholar]
- 29.Perna NT, Plunkett G, 3rd, Burland V, Mau B, Glasner JD, Rose DJ, et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409(6819):529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 30.Miller WG, Leveau JH, Lindow SE. Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol Plant Microbe Interact. 2000;13(11):1243–1250. doi: 10.1094/MPMI.2000.13.11.1243. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J, Russell DW. Molecular cloning. A laboratory manual. 3. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 32.Sarker MR, Cornelis GR. An improved version of suicide vector pKNG101 for gene replacement in Gram-negative bacteria. Mol Microbiol. 1997;23(2):410–411. doi: 10.1046/j.1365-2958.1997.t01-1-00190.x. [DOI] [PubMed] [Google Scholar]
- 33.Scherling C, Roscher C, Giavalisco P, Schulze ED, Weckwerth W. Metabolomics unravel contrasting effects of biodiversity on the performance of individual plant species. PLoS One. 2010;5(9) doi: 10.1371/journal.pone.0012569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wägele B, Witting M, Schmitt-Kopplin P, Suhre K. MassTRIX reloaded: combined analysis and visualization of transcriptome and metabolome data. PLoS One. 2012;7(7) doi: 10.1371/journal.pone.0039860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS. MetaboAnalyst 2.0--a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012;40(Web Server issue):W127-133. [DOI] [PMC free article] [PubMed]
- 36.Saeed A, Sharov V, White J, Li J, Liang W, Bhagabati N, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34(2):374. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 37.Mitra A, Kesarwani AK, Pal D, Nagaraja V. WebGeSTer DB--a transcription terminator database. Nucleic Acids Res. 2011;39(Database issue):D129–135. doi: 10.1093/nar/gkq971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, et al. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013;41(Database issue):D348–352. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linding R, Russell RB, Neduva V, Gibson TJ. GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003;31(13):3701–3708. doi: 10.1093/nar/gkg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlessinger A, Punta M, Yachdav G, Kajan L, Rost B. Improved disorder prediction by combination of orthogonal approaches. PLoS One. 2009;4(2) doi: 10.1371/journal.pone.0004433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morgulis A, Gertz EM, Schäffer AA, Agarwala R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J Comput Biol. 2006;13(5):1028–1040. doi: 10.1089/cmb.2006.13.1028. [DOI] [PubMed] [Google Scholar]
- 42.Tusnády GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17(9):849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 43.Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 44.Rost B, Fariselli P, Casadio R. Topology prediction for helical transmembrane proteins at 86 % accuracy–Topology prediction at 86 % accuracy. Protein Sci. 1996;5(8):1704–1718. doi: 10.1002/pro.5560050824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leman JK, Mueller R, Karakas M, Woetzel N, Meiler J. Simultaneous prediction of protein secondary structure and transmembrane spans. Proteins. 2013;81(7):1127–1140. doi: 10.1002/prot.24258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petersen B, Lundegaard C, Petersen TN. NetTurnP--neural network prediction of beta-turns by use of evolutionary information and predicted protein sequence features. PLoS One. 2010;5(11) doi: 10.1371/journal.pone.0015079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geourjon C, Deleage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci. 1995;11(6):681–684. doi: 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- 48.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 49.Chandonia JM, Karplus M. Neural networks for secondary structure and structural class predictions. Protein Sci. 1995;4(2):275–285. doi: 10.1002/pro.5560040214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garnier J, Gibrat J, Robson B, Doolittle R. GOR secondary structure prediction method version IV. Methods Enzymol. 1996;266:540–553. doi: 10.1016/S0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 51.Combet C, Blanchet C, Geourjon C, Deleage G. NPS@: network protein sequence analysis. Trends Biochem Sci. 2000;25(3):147–150. doi: 10.1016/S0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- 52.Bigelow HR, Petrey DS, Liu J, Przybylski D, Rost B. Predicting transmembrane beta‐barrels in proteomes. Nucleic Acids Res. 2004;32(8):2566–2577. doi: 10.1093/nar/gkh580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32(suppl 2):W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pollastri G, McLysaght A. Porter: a new, accurate server for protein secondary structure prediction. Bioinformatics. 2005;21(8):1719–1720. doi: 10.1093/bioinformatics/bti203. [DOI] [PubMed] [Google Scholar]
- 55.Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013;41(W1):W349–W357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rost B, Sander C. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins: Structure, Function, and Bioinformatics. 1994;19(1):55–72. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- 57.Ofran Y, Rost B. ISIS: interaction sites identified from sequence. Bioinformatics. 2007;23(2):e13–16. doi: 10.1093/bioinformatics/btl303. [DOI] [PubMed] [Google Scholar]
- 58.Walker JM. The proteomics protocols handbook. New York: Springer; 2005. [Google Scholar]
- 59.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 60.Goldberg T, Hecht M, Hamp T, Karl T, Yachdav G, Ahmed N, Altermann U, Angerer P, Ansorge S, Balasz K et al. LocTree3 prediction of localization. Nucleic Acids Res. 2014;42(Web Server issue):W350-355. [DOI] [PMC free article] [PubMed]
- 61.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nei M, Kumar S. Molecular evolution and phylogenetics. New York: Oxford University Press; 2000. [Google Scholar]
- 63.Scheu PD, Witan J, Rauschmeier M, Graf S, Liao YF, Ebert-Jung A, et al. CitA/CitB two-component system regulating citrate fermentation in Escherichia coli and its relation to the DcuS/DcuR system in vivo. J Bacteriol. 2012;194(3):636–645. doi: 10.1128/JB.06345-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deutschbauer A, Price MN, Wetmore KM, Shao W, Baumohl JK, Xu Z, et al. Evidence-based annotation of gene function in Shewanella oneidensis MR-1 using genome-wide fitness profiling across 121 conditions. PLoS Genet. 2011;7(11) doi: 10.1371/journal.pgen.1002385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Macho AP, Beuzon CR. Insights into plant immunity signalling: the bacterial competitive index angle. Plant Signal Behav. 2010;5(12):1590–1593. doi: 10.4161/psb.5.12.13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 67.Thomason MK, Storz G. Bacterial antisense RNAs: how many are there, and what are they doing? Annu Rev Genet. 2010;44:167–188. doi: 10.1146/annurev-genet-102209-163523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gottesman S, Storz G. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol. 2011;3(12): doi:10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed]
- 69.Dill KA, MacCallum JL. The protein-folding problem, 50 years on. Science. 2012;338(6110):1042–1046. doi: 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]
- 70.Lasa I, Villanueva M. Overlapping transcription and bacterial RNA removal. Proc Natl Acad Sci U S A. 2014;111(8):2868–2869. doi: 10.1073/pnas.1324236111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lasa I, Toledo-Arana A, Gingeras TR. An effort to make sense of antisense transcription in bacteria. RNA Biol. 2012;9(8):1039–1044. doi: 10.4161/rna.21167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cech TR. Ribozymes, the first 20 years. Biochem Soc Trans. 2002;30(Pt 6):1162–1166. doi: 10.1042/bst0301162. [DOI] [PubMed] [Google Scholar]
- 73.Tautz D, Domazet-Loso T. The evolutionary origin of orphan genes. Nat Rev Genet. 2011;12(10):692–702. doi: 10.1038/nrg3053. [DOI] [PubMed] [Google Scholar]
- 74.Zhao L, Saelao P, Jones CD, Begun DJ. Origin and spread of de novo genes in Drosophila melanogaster populations. Science. 2014;343(6172):769–772. doi: 10.1126/science.1248286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van Passel MW, Marri PR, Ochman H. The emergence and fate of horizontally acquired genes in Escherichia coli. PLoS Comput Biol. 2008;4(4) doi: 10.1371/journal.pcbi.1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sabath N, Wagner A, Karlin D. Evolution of viral proteins originated de novo by overprinting. Mol Biol Evol. 2012;29(12):3767–3780. doi: 10.1093/molbev/mss179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilson GA, Bertrand N, Patel Y, Hughes JB, Feil EJ, Field D. Orphans as taxonomically restricted and ecologically important genes. Microbiology. 2005;151(Pt 8):2499–2501. doi: 10.1099/mic.0.28146-0. [DOI] [PubMed] [Google Scholar]
- 78.Nekrutenko A, He J. Functionality of unspliced XBP1 is required to explain evolution of overlapping reading frames. Trends Genet. 2006;22(12):645–648. doi: 10.1016/j.tig.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 79.Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, et al. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol. 2007;5(3) doi: 10.1371/journal.pbio.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wood DE, Lin H, Levy-Moonshine A, Swaminathan R, Chang YC, Anton BP, et al. Thousands of missed genes found in bacterial genomes and their analysis with COMBREX. Biol Direct. 2012;7:37. doi: 10.1186/1745-6150-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23(6):673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tunca S, Barreiro C, Coque JJ, Martin JF. Two overlapping antiparallel genes encoding the iron regulator DmdR1 and the Adm proteins control siderophore and antibiotic biosynthesis in Streptomyces coelicolor A3(2) FEBS J. 2009;276(17):4814–4827. doi: 10.1111/j.1742-4658.2009.07182.x. [DOI] [PubMed] [Google Scholar]
- 83.Kim W, Silby MW, Purvine SO, Nicoll JS, Hixson KK, Monroe M, et al. Proteomic detection of non-annotated protein-coding genes in Pseudomonas fluorescens p f0–1. PLoS One. 2009;4(12) doi: 10.1371/journal.pone.0008455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Silby MW, Levy SB. Overlapping protein-encoding genes in Pseudomonas fluorescens p f0–1. PLoS Genet. 2008;4(6) doi: 10.1371/journal.pgen.1000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Balabanov VP, Kotova VY, Kholodii GY, Mindlin SZ, Zavilgelsky GB. A novel gene, ardD, determines antirestriction activity of the non-conjugative transposon Tn5053 and is located antisense within the tniA gene. FEMS Microbiol Lett. 2012;337(1):55–60. doi: 10.1111/1574-6968.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mir K, Neuhaus K, Scherer S, Bossert M, Schober S. Predicting statistical properties of open reading frames in bacterial genomes. PLoS One. 2012;7(9) doi: 10.1371/journal.pone.0045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neuhaus K, Oelke D, Fürst D, Scherer S, Keim D. Towards automatic detecting of overlapping genes - Clustered BLAST analysis of viral genomes. Lect Notes Comput Sci. 2010;6023:228–239. doi: 10.1007/978-3-642-12211-8_20. [DOI] [Google Scholar]
- 88.Rancurel C, Khosravi M, Dunker AK, Romero PR, Karlin D. Overlapping genes produce proteins with unusual sequence properties and offer insight into de novo protein creation. J Virol. 2009;83(20):10719–10736. doi: 10.1128/JVI.00595-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Clokie MR, Millard AD, Letarov AV, Heaphy S. Phages in nature. Bacteriophage. 2011;1(1):31–45. doi: 10.4161/bact.1.1.14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chibani-Chennoufi S, Bruttin A, Dillmann ML, Brussow H. Phage-host interaction: an ecological perspective. J Bacteriol. 2004;186(12):3677–3686. doi: 10.1128/JB.186.12.3677-3686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ochman H, Davalos LM. The nature and dynamics of bacterial genomes. Science. 2006;311(5768):1730–1733. doi: 10.1126/science.1119966. [DOI] [PubMed] [Google Scholar]
- 92.Okamura K, Feuk L, Marques-Bonet T, Navarro A, Scherer SW. Frequent appearance of novel protein-coding sequences by frameshift translation. Genomics. 2006;88(6):690–697. doi: 10.1016/j.ygeno.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 93.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]