

Abstract
Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative disease. In humans, AD becomes symptomatic only after brain changes occur over years or decades. Three contiguous phases of AD have been proposed: (i) the AD pathophysiologic process, (ii) mild cognitive impairment due to AD, and (iii) AD dementia. Intensive research continues around the world on unique diagnostic markers and interventions associated with each phase of AD. In this review, we summarize the available evidence and new therapeutic approaches that target both amyloid and tau pathology in AD and discuss the biomarkers and pharmaceutical interventions available and in development for each AD phase.
Introduction
Alzheimer's disease (AD) is globally recognized as the most common form of dementia, with multiple studies projecting that by the year 2050, approximately 115 million people will be affected worldwide1. Due to the projected number of those affected, the economic, healthcare, and caregiver costs continue to have a place in public policy, but it is only recently that these issues are beginning to take a global center stage, especially in those regions of the world that are experiencing unprecedented increases in the life expectancy of their adult populations.
The burden of dementia is thought to disproportionately affect low-to-middle income countries. Fifty-eight per cent of all people with dementia worldwide live in these countries, and this number is expected to rise to 71 per cent by 2050. Estimates suggest that proportionate increases over the next twenty years in the number of people with dementia will be much steeper in low- and middle-income countries compared with high-income countries2,3. Data compiled from the World AD report of 2010 noted a predicted 40 per cent increase in persons with dementia in Europe, a 63 per cent increase in North America, a 77 per cent increase in southern Latin America, and an 89 per cent increase in the developed Asian Pacific countries4.
India, one of the most populous countries in the Asian Pacific region, is experiencing increased longevity among its adult population. According to the 2001 census, India was home to more than 76 million people aged 60 yr and older5. Although prevalence rates from distinct regional community-based studies of dementia in India have varied from 1.02 to 3.36 per cent in those above 60-65 yr of age6,7,8,9,10, a number lower than reported for other developing countries11, these rates are expected to increase dramatically as the Indian population ages. It is estimated that there are already approximately 1.5 million people affected by dementia in India, and this number is likely to increase by 300 per cent in the next four decades12. The challenges in India are similar to those of many low- to middle-income countries worldwide that are experiencing unprecedented increases in their populations that are ageing and developing dementia. Thus, to meet the needs of people with dementia worldwide, it remains of paramount importance to develop an infrastructure support system that supports health care workers in order to (i) incorporate the latest knowledge regarding diagnostic criteria for assessing cases of dementia and early detection of cognitive impairment, (ii) utilize a variety of different pharmacologic and treatment options while providing care, (iii) incorporate biomarker methods in clinical assessment when feasible and advisable, and (iv) implement evidence-based prevention strategies to entire groups or populations when available.
This review summarizes the latest biomarkers and pharmaceutical interventions available and in development for each AD phase, which ultimately may provide clues as to how to best utilize and integrate them in clinical practice.
Overview of the phases of Alzheimer's disease
Research activities regarding the prevalence of AD and its clinical and pathophysiological relationships have refined and altered the field's concept of AD, with longitudinal studies of age-related cognitive changes, using neuroimaging and neuropathology, confirming a marked temporal lag between the initiation of neuropathologic characteristics and the appearance of symptoms. In response to these findings, the National Institute on Aging (NIA) and the Alzheimer's Association (AA) convened working groups to gather expert opinions on the state of the field, which in 2011 culminated in the publication of a series of consensus reports on improved diagnostic strategies and full-spectrum disease characterization. Three contiguous phases, the AD pathophysiologic process, mild cognitive impairment (MCI) due to AD, and clinical AD dementia, were proposed13,14,15, not only to assist physicians in identifying diagnostic options, but also to provide a platform to develop primary prevention therapies (Figure)16.
Figure.
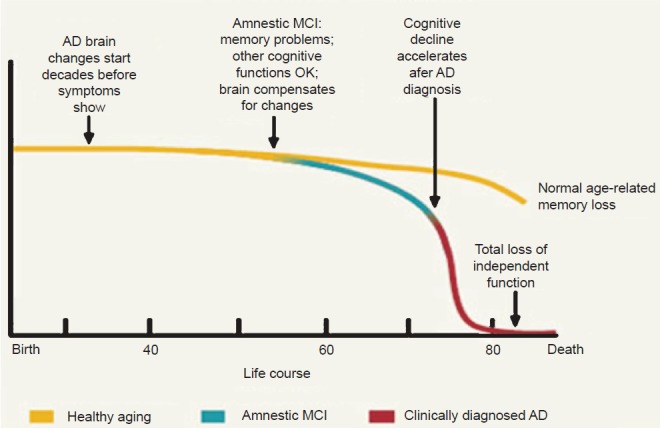
Cognitive trajectories over a lifetime and AD phases. National Institute on Aging's document titled “Alzheimer's Disease: Unraveling the Mystery”. Available from: https://www.nia.nih.gov/alzheimers/publication/alzheimers_disease-unraveling-mystery/preface, accessed on August 20, 2015. [Reproduced with permission from: www.nih.gov<http://www.nia.nih.gov/>].
Phases of Alzheimer's disease (Table I)13,14,15,16,17
Table I.
Summary of diagnostic biomarkers and pharmaceutical interventions for Alzheimer's disease (AD)
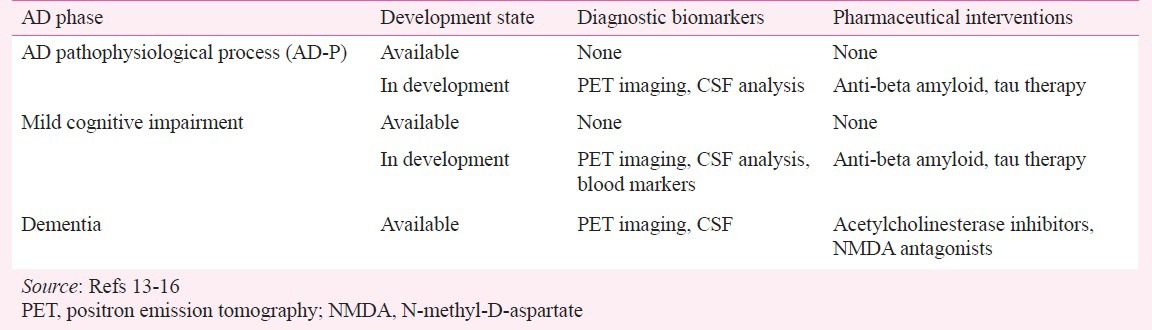
AD pathophysiological stage: The AD pathophysiological phase is asymptomatic, and distinguishing this condition from the cognitively normal elderly continues to be challenging18. Although the guidelines by the NIA-AA workgroups identified this phase as a distinct stage of AD, the proposed guidelines did not establish specific diagnostic criteria that could be used in typical clinical settings13,14. Rather, this stage is thought to reflect the period during which AD pathology may exist in persons who do not meet the criteria for either mild cognitive impairment or Alzheimer's disease, and who may or may not have clinical signs of subtle cognitive deficits19. The NIA-AA group's focus for this stage was to highlight the biomarker and developmental work that could help to track the onset and progression of mild cognitive impairment and AD, which is commonly hypothesized to proceed as follows: amyloidosis (Stage 1); synaptic dysfunction, stimulation of inflammatory processes, neuronal dysfunction, hyperphosphorylation of tau protein, accumulation of neurofibrillary tangles (NFTs) and neurodegeneration (Stage 2); and subtle cognitive changes (Stage 3)17.
Thus, the biomarker research conducted on this stage has been directed at identifying subtle but measureable signs that reflect increases in deposition and synthesis of amyloid beta (Aβ) and/or tau20,21. Among the group of potentially useful biomarkers are those that measure changes in the cerebrospinal fluid (CSF) levels of a 42-amino acid form of Aβ, total tau protein, and phosphorylated tau at residue 181. Clinical research studies and assessment of these biomarkers have suggested that declining CSF Aβ42 levels can occur at least 20-25 yr prior to clinical dementia22. Because of the extensive prodromal phase of AD, other earlier-stage, less-invasive biomarker techniques, namely amyloid and tau neuroimaging, are positioned to aid and facilitate early diagnosis23,24.
Mild cognitive impairment (MCI): MCI due to AD, the second stage of AD, has become easier to identify over the years. The focus of the NIA-AA workgroup concerning this stage was to provide diagnostic criteria to enable practicing clinicians, many of whom may not have access to sophisticated imaging techniques or CSF analysis, to make a diagnosis of MCI by focusing their clinical skills and examinations on key areas of cognitive and social functioning. Specifically, the areas of focus included assessing whether a patient has had (i) a change in cognitive ability from a previous level, (ii) impairment in one or more cognitive domains (i.e., orientation, language, attention, executive function, memory), or (iii) complaints about or demonstration of mild problems in performing complex tasks once performed easily, without significant impact on social or occupational functioning14.
This clinical information, when coupled with the latest neuroimaging research scans conducted on persons with MCI25, is especially robust in providing a diagnostic target for future treatments. Patients with MCI have shown atrophied gray matter on MRI scans, especially in the hippocampus and entorhinal cortex26, whereas 18-fluorodeoxyglucose positron emission tomography (18F-FDG PET) scans show physiologic changes such as reduced metabolism in the temporoparietal cortex27 and decreased glucose metabolism in the posterior cingulate cortex28,29.
AD dementia: AD dementia, the third stage in the continuum, shows a distinct presentation from the two earlier stages. This is often the stage with which clinicians are most familiar, because in addition to the notable clinical changes in episodic memory and other cognitive functions such as executive function and language dysfunction patients are notably impaired in social or occupational functioning13. Further, MRI scans typically show pronounced atrophy of the hippocampus while volumetric data demonstrate pronounced atrophy in the middle temporal lobe associated with lower executive functioning and cognitive function30.
Early diagnosis and disease-modification therapies targeting specific disease mechanisms
The role of amyloid beta (Aβ) in AD research: Aβ appears to be involved in a variety of important functions in healthy subjects31. Among those functions are activation of enzyme kinases, regulation of cholesterol transport, mediation of synaptic plasticity, and pro-inflammatory activity32,33,34,35. In addition to its deposition in and around neural elements of the brain, Aβ is thought to maintain the integrity of cerebral vascular membranes; disruptions in this function may underlie cerebral amyloidal angiopathy36.
In the early stages of AD, one of the first physiologic abnormalities is increased Aβ deposition in several neocortical areas, such as the prefrontal, bilateral superior medial frontal, and lateral temporal cortex. Aβ deposition begins before any outward clinical symptoms are noted, and the increase in amyloid plaque density and distribution is often accompanied by a variety of other neuropathologic and morphologic changes, including increased accumulation and production of NFTs, and loss of total brain volume and hippocampal volume37,38,39. Similar changes are also found in persons who are either in the AD-P stage or the MCI-due-to-AD stage of the disease40,41,42, with memory deficits correlating to the accumulation of Aβ deposition and NFTs in the temporal neocortex43. Neuroimaging correlation with 18F FDG-PET imaging of persons with clinically-evident MCI has shown reduced perfusion in the temporoparietal cortex, while 18F amyloid PET shows increased Aβ deposition in the posterior cingulate cortex27,28. Lastly, the density and presence of Aβ aggregates have been noted in regions associated with executive function, processing speed, and verbal fluency44, and are pronounced in those with MCI that has progressed to clinical dementia45,46.
The role of genetics in AD onset: Several studies have attempted to better and more precisely define the genetic pathways directly associated with the evolution of AD. Mutations in three genes are now known to be associated with dominantly inherited AD. Trisomy 21 and the amyloid precursor protein (APP) gene, found in Down syndrome, result in Aβ and NFT accumulation and early-onset AD47. Cross-sectional findings from Dominantly Inherited Alzheimer Network (DIAN) investigations48 have demonstrated that in select groups of patients with autosomal-dominant genetic mutations in one of the three genes [APP or presenilin 1 or 2 (PSEN1 or PSEN2)], measurable changes in the brain can occur up to 20 years before the onset of clinical dementia48. In this select group, lower CSF levels of Aβ42 and accompanying higher tau protein were associated with dementia within three to four years49. DIAN also found that when increased Aβ42 in the blood was accompanied by decreased Aβ42 in CSF, amyloidal deposition in the brain and incidental AD were more likely48.
Numerous single nucleotide polymorphisms (SNPs) are also known to be associated with AD. Linkage studies performed approximately 20 years ago identified the first SNP50,51. Individuals with one copy of the apolipoprotein E ε4 (APOE4) gene have a 2-3-fold increase in lifetime risk of AD, and those who are homozygous may have up to a 10-fold increased risk. Whereas those possessing one ε2 allele (APOE2) is associated with a lower risk of AD52,53.
The precise mechanistic basis for this association remains obscure. It has been postulated that APOE-mediated cholesterol transport plays a role in prompting Aβ accumulation54. Ongoing research to explore these genetic associations are now being embedded in ongoing clinical trials. Strategies to use specific samples such as people with a high genetic risk, are being developed to evaluate new treatment modalities in cohorts of APOE positive homozygous persons and those with other genetic markers.
The era of genome-wide association studies (GWAS), with tens of thousands of cases and controls assembled from cohorts worldwide, has yielded nearly 30 additional SNPs55,56. The effects are quite small relative to the mutations and to APOE. However, these have yielded important new insights into the biology of AD. For example, a few of these SNPs are robustly related to pathologic indices of AD57. Rather, these appear in immune and inflammatory pathways. For example, CD33 is a singlet protein expressed on peripheral monocytes. SNPs have been associated with greater cell-surface expression of CD33 and reduced internalization of Aβ58. These monocytes appear in the human brain adjacent to amyloid deposits. Recently, genome-wide DNA methylation studies (MWAS) have been used to identify additional genes involved in AD59.
The role of biofluid biomarkers in AD research: Diagnostic biomarker analysis is also being investigated in the evaluation of cognitive disorders and AD. As mentioned previously, the early stages of AD may be detectable in the CSF. The relative titre of molecular biomarkers found in the CSF, such as Aβ42 and tau, can help trace the early process of AD60. Although CSF samples can be obtained via spinal taps, these procedures are invasive, so these are difficult to conduct routinely and are not regularly used in early diagnosis in academic- and community-clinic-based settings.
Recent developments that aim at analysis of routine blood samples has shown some promise in providing a platform for early AD detection and monitoring. For example, AD-linked alterations in ceramides and sphingomyelins have been postulated to play a role in amyloidogenesis and inflammatory stress related to neuronal apoptosis. Several clinical studies suggest that these biomarkers may have reproducibility in assessing early-stage AD61,62. Reduction of hypercholesterolaemia has been an area of interest in AD clinical trials and is another possible approach to AD detection and monitoring, as elevated cholesterol blood-level markers have been shown to be positively correlated to amyloid plaques in the brain63,64. Changes in the cholesterol metabolism-related gene expression in mononuclear cells from AD patients could have potential diagnostic implications, and blood samples stained with Oil Red O (ORO), a fat-soluble lysochrome dye used to identify neutral lipids, may help to distinguish healthy elderly patients from AD patients65.
The role of imaging biomarker technologies in AD research: Gross neurodegeneration can easily be seen on structural MRI, and these changes correspond to reduced glucose uptake on 18-FDG PET. However, from a clinical perspective, crude visual inspection of anatomical changes noted on brain scans is not sufficient to establish a probable AD diagnosis, and more advanced analysis methods are needed. Although imaging with18 F-FDG PET is useful in highlighting typical hallmarks of AD (i.e. reduced regional metabolism)27, neuropathological findings demonstrate that AD pathology frequently coexists with other pathologies, making it difficult to attain diagnostic specificity with these structural and functional imaging methods66,67. Recent developments and refinements in 18F PET techniques, such as florbetapir PET imaging, have shown good agreement with autopsy-confirmed distribution and degree of Aβ pathology68. Although three amyloid tracers [florbetapir F 18 (AMYviD™, Lilly), florbetaben F 18 (Neuraceq™, Piramal Imaging), and flutemetamol F 18 (Vizamyl™, GE Healthcare)] have been approved by the US Food and Drug Administration (FDA) for use with PET imaging of the brain in adults being evaluated for AD and dementia, their clinical utility remains controversial. Furthermore, there have been no head-to-head comparisons that would inform the choice of one tracer over another. Although still in the early stages, PET tau imaging has now been developed and is currently under study. Other developing Aβ imaging technologies include (E)-5-styryl-1H-indole and (E)-6-styrylquinoline derivatives, which are specific to labelling plaques and could potentially be used with single-photon emission computed tomography (SPECT) agents69 and near-infrared fluorescence (NIRF) imaging probes, such as the intravenous agent THK-265, which also binds strongly to cortical Aβ70.
The role of pharmacologic strategies in AD research: Conventional therapies to treat AD, such as cholinesterase inhibitors (ChEIs) and NMDA receptor antagonists, have not been shown to have a disease-modifying effect or to impact the progression of AD over a prolonged period of time. As a result, novel treatments are being investigated and therapies are being designed to target or interfere with the amyloid cascade71,72, tau production and processing, or focus on neurotransmitter or neuroprotective agents in various stages of development (Table II)72,73,74,75,76.
Table II.
Disease-modifying approaches
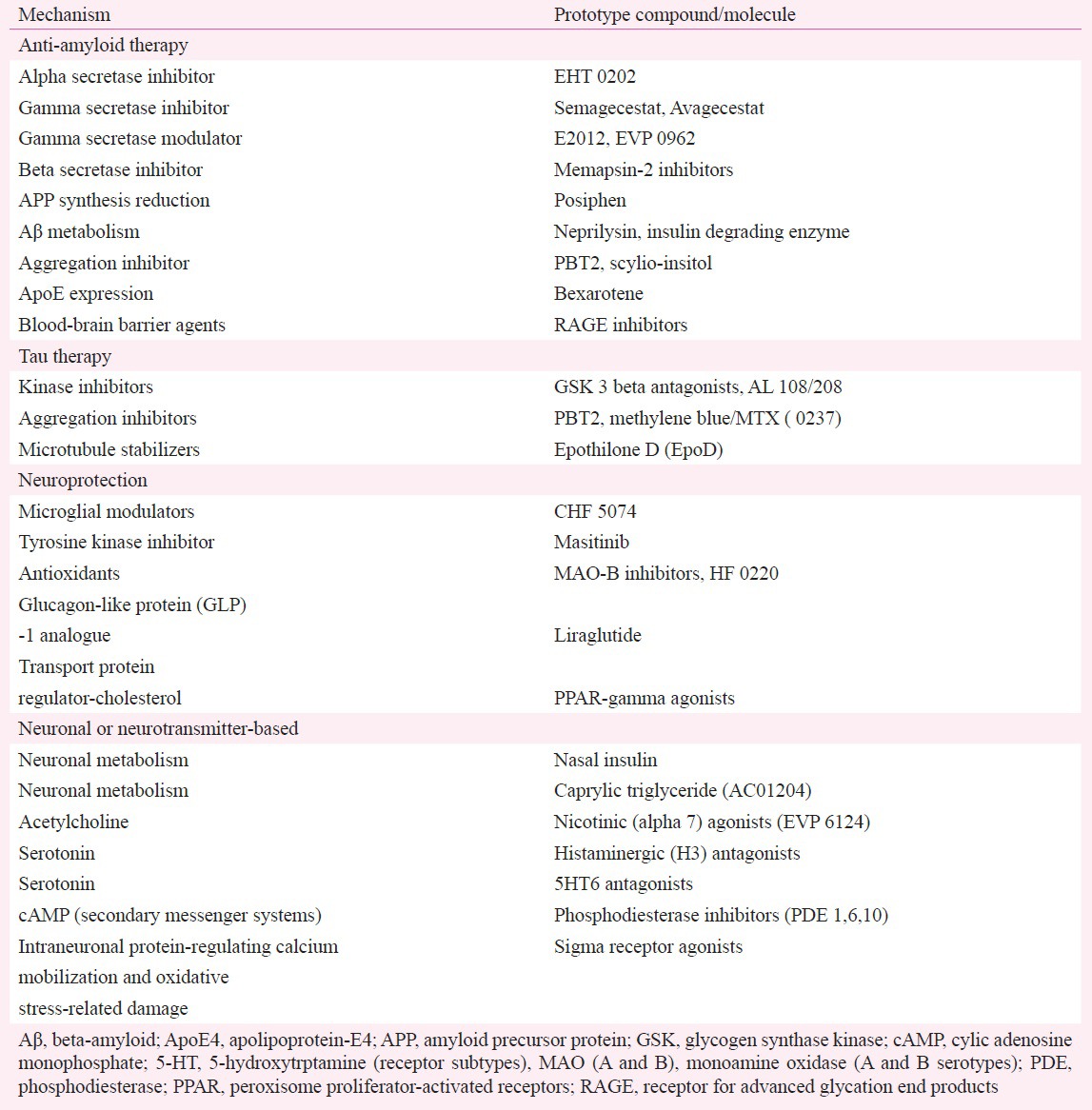
Amyloid-based treatments
The main mechanistic theory of AD is the amyloid hypothesis, according to which an imbalance in the production or clearance of the Aβ peptide results in accumulation of Aβ and initiation of a cascade of events leading to neurodegeneration and dementia77. This hypothesis has undergone an evolution, from one that was initially focused on the role of hard plaque in the development of disease and the removal of this plaque as the goal for disease-modifying drug development, to using specific soluble components of the plaques (oligomers and monomers) as potential drug targets. Thus, the latest anti-amyloid strategies comprise compounds with specific mechanisms of action, namely drugs that facilitate the clearance, inhibit the production, or prevent the aggregation of Aβ73.
The Aβ molecule is generated from the amyloid precursor protein and has multiple isoforms, comprising 36-45 amino acids with Aβ40 and Aβ42 isoforms, particularly tied to Aβ plaque deposition and AD74. Proteolytic cleavage of the Aβ precursor glycoprotein (AβPP) can be catalyzed by a series of secretase enzymes: (i) the alpha secretase cleaves AβPP within the intracellular region, preventing the formation and deposition of Aβ; (ii) cleavage of the amyloid precursor protein (APP) by β secretase produces a cell-membrane-bound fragment (C99); and (iii) cleavage of this fragment within its transmembrane domain by gamma secretase releases the intracellular segment of AβPP to produce Aβ73.
Secretase-targeted treatments
The goal of secretase-targeted therapies is fundamentally to either slow or arrest the enzyme-catalyzed steps that drive the formation of the pathogenic Aβ species75,76. One approach involves enhancing the alpha secretase processing of APP, which in turn generates a soluble extracellular N terminal fragment and a C terminal transmembrane fragment (C83), which precludes formation and deposition of the Aβ78,79,80. Gamma secretase is the enzyme responsible for the final step in Aβ generation, and determines the length of the Aβ peptide and the ratio of Aβ42 to Aβ40. Clinical trials with gamma secretases (i.e. tarenflurbil, semagacestat, and begacestate) have been performed, and although initial results looked promising, the trials failed to meet clinical endpoints and reported significant haematological, gastrointestinal, and skin related side effects. Modulation of gamma secretase activity by non-steroidal anti-inflammatory drug (NSAID)-like agents has been postulated to reduce brain Aβ42 concentrations and to prevent cognitive deficits in animal models of AD; however, several NSAID enantiomers, such as R-flurbiprofen, have failed to demonstrate clear efficacy in reducing levels of Aβ in clinical trials81. Beta-secretase, otherwise known as the beta-site amyloid protein precursor cleaving enzyme (BACE 1), is thought to initiate the amyloidogenic pathway by cleaving the amyloid precursor protein.
Metabolic approaches
Alzheimer's disease can also be considered a metabolic disease in which brain glucose utilization and energy production are impaired82,83,84,85,86. Metabolic abnormalities have been linked to brain insulin and insulin-like growth factor (IGF) resistance with disruption of signaling pathways that regulate neuronal survival, energy production, gene expression, and plasticity82. Abnormalities in insulin-signaling in the human brain have also been linked to AD87.
On a functional basis, insulin/IGF resistance causes downregulation of target genes that are needed for cholinergic homeostasis, and it compromises systems that mediate neuronal plasticity, memory, and cognition. Small clinical trials suggest that intranasal insulin improves both memory performance and metabolic integrity in the brains of patients suffering from AD dementia or MCI due to AD88,89. A larger clinical trial involving non-diabetic individuals is presently ongoing to determine whether or not insulin, when administered as a nasal spray, improves memory in adults with mild memory impairment or AD90.
Aβ aggregation inhibitors
The goal of this class of drugs is to inhibit Aβ aggregation, allowing for Aβ elimination or neutralizing the toxicity of the oligomers. One of the first aggregation inhibitors, tramiprosate (Alzhemed™), was reported to bind and maintain Aβ monomers in a non-fibrillary conformation to prevent the formation of neuritic plaques91. However, it did not show efficacy in a Phase III trial. Other trials examining scyllo-cyclohexanehexol (AZD 10, ELND 005) - a compound that competes with an intracellular messenger that stimulates Aβ aggregation, phosphatidylinositol - have also been performed. Initial findings showed that scyllo-cyclohexanehexol bound successfully to Aβ oligomers in a process that inhibited their aggregation and toxicity, and reduced plaque deposition and cognitive deficits92. However, this compound also failed to meet its endpoint in a Phase II trial.
APOE-dependent Aβ clearance
Another area of interest involves the role of APOE in Aβ clearance. Aβ clearance from the CNS is reduced by approximately 30 per cent in individuals with AD, and laboratory studies have suggested that APOE may facilitate clearance of Aβ peptides from the brain93.
Bexarotene, an antineoplastic skin cancer agent, was found to reverse AD and improve brain function in genetically engineered mice that exhibited AD-like symptoms. Investigators noted that after a single dose, 25 per cent of the toxic beta amyloid was removed from the mouse brains within 6 h94. Moreover, half of the plaques were removed 72 h later, and the mice began to show behavioural improvement. It is thought that bexarotene stimulated expression and higher levels of ApoE, which led to the intracellular clearance of beta amyloid. Human studies are planned for this drug to determine whether it crosses the blood-brain barrier and can, therefore, eliminate Aβ94.
Enzymatic clearance
Reduction of intracellular tissue plasminogen activation inhibitor 1 (PAI-1) increases plasmin-associated Aβ proteolysis, providing a basis for Aβ clearance via enzyme activation. Other compounds, such as the receptor for advanced glycation end products (RAGE) and low-density lipoprotein receptor-related protein 1 (LRP1), have also been shown to modulate Aβ transport at the blood-brain barrier95,96,97.
Aβ immunotherapy
Multiple immunotherapeutic agents are now undergoing multiple clinical trials. The Phase III testing of AN 1791, a vaccine with pre-aggregated Aβ, was designed to elicit a cell-mediated immune response to Aβ plaque pathology98. However, inappropriate immune responses in clinical trials, leading to meningoencephalitis, stopped further development of this vaccine. Another study showed that while immunotherapy could attenuate Aβ plaque deposition in AD models, it was ineffective if deposition was already under way99.
Reports that immunization with aggregated Aβ42 attenuated AD pathology in animal models100 led to the development of humanized serum immunologics, such as bapineuzumab, solanezumab, and intravenous immunoglobulin-G, specifically directed against Aβ101,102,103,104.
Although the results from these studies were not overwhelmingly convincing of a disease-modifying effect, a number of agents exhibited a slight beneficial effect on cognitive endpoints in the mild to moderate AD groups, and solanezumab is now being actively tested in large-scale population-based studies of cognitively unimpaired persons.
Emerging strategies
Clinical trials focusing on disease modification and progression in AD have shown modest results, which have led to the development of new trials that focus on the preclinical stages of AD, with aggressive treatment at those stages. The prevention trials mentioned in Table III use biomarker profiles, cognitive endpoints, or a combination of both in cognitively unimpaired persons at risk for developing Alzheimer's disease, to guide the design of the study and the drug intervention (Table III)105,106,107,108,109,110,111.
Table III.
Alzheimer's disease (AD) prevention trials
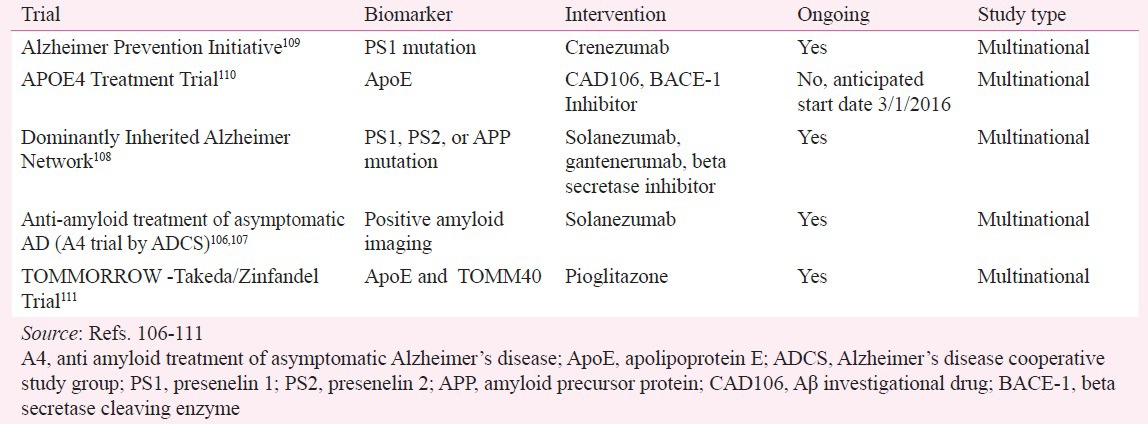
The recently launched A4 study is a clinical trial of 1,000 older individuals who have evidence of amyloid plaque buildup in their brains on PET amyloid imaging and who may be at risk for memory loss and cognitive decline due to AD. The A4 study will test the anti-amyloid investigational drug, solanezumab, in older individuals who do not yet show symptoms of AD cognitive impairment or dementia, with the aim of slowing memory and cognitive decline. The A4 study will also test whether anti-amyloid treatment can delay the progression of AD-related brain injury on imaging and other biomarkers106,107.
The Longitudinal Evaluation of Amyloid Risk and Neurodegeneration study (LEARN), a substudy of the A4 study, will focus on causes of cognitive decline besides the buildup of Aβ in the brain112. In this substudy, participants will have an amyloid PET scan to determine the buildup of tau protein in the brain. The LEARN subcomponent of A4 will follow the screen failures for the A4 study (i.e. participants over time who do not have elevated amyloid) and will determine which biological changes are related to cognitive decline, including possible later amyloid buildup as well as increases in tau levels, helping to demonstrate a differential rate of clinical decline between amyloid-positive and amyloid-negative individuals on a standardized set of clinical outcomes.
Another study that has been enrolling individuals since 2012, the DIAN Trials Unit (DIAN-TU, NIA U01AG042791) study, targets persons who are either known to have a genetic mutation that causes autosomal-dominant Alzheimer's disease or who are unaware of their genetic status but have a parent or sibling with a known genetic mutation. Investigators will test two experimental drugs to assess their safety, side effects, and effect on imaging and biomarkers. Subtle, early changes in cognition will also be evaluated, though participants at this disease stage are unlikely to have more than minimal changes in cognitive measures during the study. Because many at-risk individuals decide not to know whether they have an AD-associated genetic mutation, some of the participants in this study will not have the disease-causing mutations. These “mutation-negative” individuals will be assigned to the placebo group. Mutation-positive individuals will receive one of two different therapies, gantenerumab or solanezumab, or a placebo108. Gantenerumab is a humanized monoclonal antibody that will be given subcutaneously every four weeks. Solanezumab, the other humanized monoclonal antibody used in this study (and in the A4 study), will be given as an intravenous infusion every four weeks.
Another study testing monoclonal antibody therapy in persons from a large extended family in Colombia, who carry the rare presenilin gene, is the crenezumab study by the Alzheimer Prevention Initiative (API)109. While brain swelling and tiny leaks of blood into the brain are seen with solanezumab (ARIA-E and ARIA-H), crenezumab, an injectable monoclonal antibody that targets the Aβ precursor protein in the brain, does not cause vasogenic oedema, so potentially larger doses of it can be given to patients. Study participants will be given regular injections of the drug or a placebo for at least five years. Utilization and measurement of traditional biomarkers employed in the two other large-scale studies, as previously discussed, will be employed in this study113.
The recently launched APOE4 Treatment Trial110 will test whether two anti-amyloid drugs - an active immunotherapy or an oral medication (BACE inhibitor) - can prevent or delay the emergence of AD symptoms in persons at high risk for developing the disease. Participants in this study will carry two copies of the apolipoprotein E (APOE4) gene, which is linked to late-onset AD, and will be randomized to either the treatment arm or a placebo.
Finally, a global Phase III, five-year, double-blind, placebo-controlled study called the TOMMORROW study will also examine genetic risk-factor influence on developing MCI due to AD. This study will use a genetic-based biomarker risk-screen assessment and evaluate whether a low dose of pioglitazone (AD-4833), a currently approved diabetes drug, can delay the onset of MCI due to AD in cognitively normal persons at high risk, as determined by the group risk-assignment algorithm111.
Clinical research and trials in India
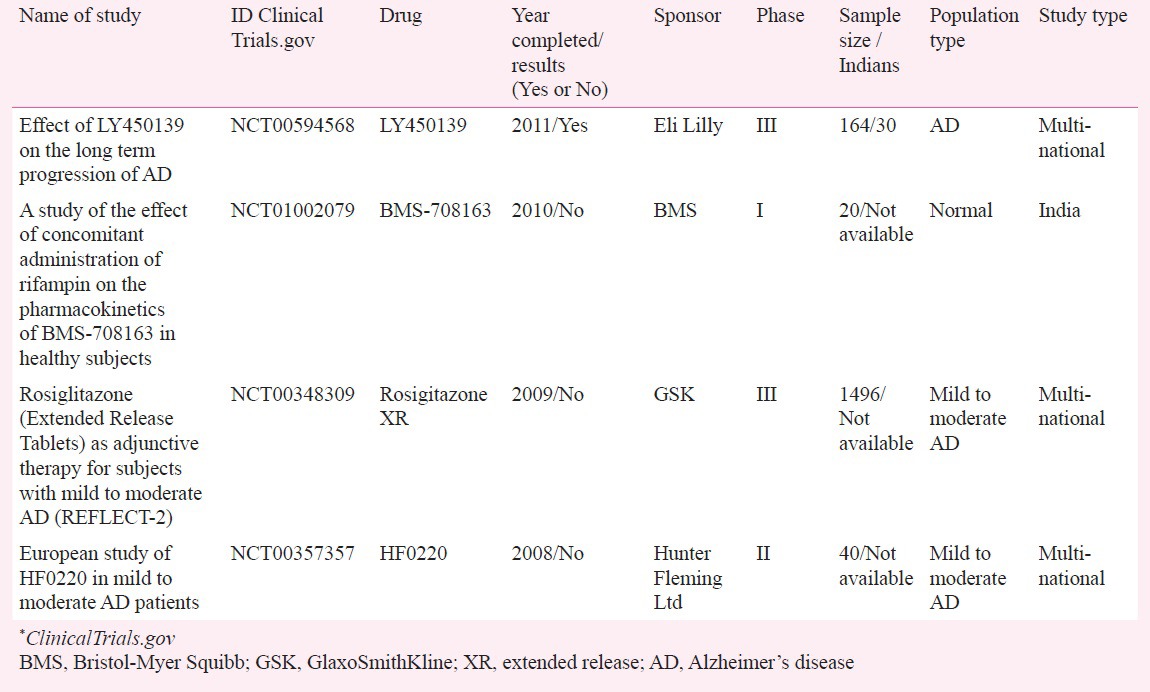
Despite various metrics and surveys suggesting that India's relatively low operational costs and increasingly ageing population (most of whom are relatively naïve regarding treatments for dementia) could be harnessed to fuel a more rapid programme of clinical trial development and activity, the most recent review of the Clinicaltrials.gov database notes only a handful of clinical trials being conducted in India, with most sponsored by international pharmaceutical companies (Table IV). Regulatory issues (i.e. prohibition of testing compounds developed in India on Indians with or without prior Phase I data)114, and difficulty in ensuring strict adherence to early-phase protocols and methodology, have been touted as hindrances in conducting early-phase clinical trials in India115.
Table IV.
Alzheimer's disease/dementia studies conducted in India (from https://clinicalTrials.gov)*
Conclusions
The advent of disease-modifying agents in AD has heightened the importance of developing blood, CSF, and imaging markers of progression. Such markers will increase our knowledge of this disease and help provide prognostic information to patients, and may also provide cost-effective ways to identify therapies that slow AD as opposed to providing only symptomatic benefit. These advances, however, do not come without some risks and limitations. Early diagnosis in persons with full insight can lead to catastrophic reactions upon disclosure of a risk for developing the disease. Access to new treatments will be restricted by the studies’ inclusion and exclusion criteria, availability of infusion services, and access to MRI and neurology resources for short-term complications. Several countries have already called for and put forth national plans to address the burden of dementia. With organizational pathways in place and additional funding to support these activities, there is a good chance that national and international strategies for AD diagnosis, treatment, and prevention may yield better tests and effective medications to prevent and treat this disease. Only through continual investment in research and cost-effective approaches to diagnosis, treatment, and care can future societal costs be anticipated and managed.
References
- 1.Qui C, De Ronchi D, Fratiglioni L. The epidemiology of the dementias: an update. Curr Opin Psychiatry. 2007;20:380–5. doi: 10.1097/YCO.0b013e32816ebc7b. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization and Alzheimer's Disease International. Dementia: a public health priority. [accessed on August 20, 2015]. Available from: http://www.who.int/mental_health/publications/dementia_report_2012/en .
- 3.Llibre Rodriguez JJ, Ferri CP, Acosta D, Guerra M, Huang Y, Jacobs KS, et al. Prevalence of dementia in Latin America, India, and China: a population-based cross-sectional survey. Lancet. 2008;372:464–74. doi: 10.1016/S0140-6736(08)61002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Alzheimer's Report 2010. [accessed on August 20, 2015]. Available from: http://www.alz.org/documents/national/Worldalzheimerreport2010.pdf .
- 5.Sathyanarayana Rao TS, Shaji KS. Demographic aging: implications for mental health. Indian J Psychiatry. 2007;49:78–80. doi: 10.4103/0019-5545.33251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaji S, Promodu K, Abraham T, Roy KJ, Verghese A. An epidemiological study of dementia in a rural community in Kerala, India. Br J Psychiatry. 1996;168:745–9. doi: 10.1192/bjp.168.6.745. [DOI] [PubMed] [Google Scholar]
- 7.Rajkumar S, Kumar S, Thara R. Prevalence of dementia in a rural setting: a report from India. Int J Geriatr Psychiatry. 1997;12:702–7. doi: 10.1002/(sici)1099-1166(199707)12:7<702::aid-gps489>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 8.Rajkumar S, Kumar S. Prevalence of dementia in the community: a rural-urban comparison from Madras, India. Australas J Aging. 1996;15:57–61. [Google Scholar]
- 9.Shaji S, Bose S, Verghese A. Prevalence of dementia in an urban population in Kerala, India. Br J Psychiatry. 2005;186:136–40. doi: 10.1192/bjp.186.2.136. [DOI] [PubMed] [Google Scholar]
- 10.Chandra V, Ganguli M, Pandav R, Johnston J, Belle S, DeKosky ST. Prevalence of Alzheimer's disease and other dementias in rural India: the Indo-US study. Neurology. 1998;51:1000–8. doi: 10.1212/wnl.51.4.1000. [DOI] [PubMed] [Google Scholar]
- 11.Prince MJ, de Rodriguez JL, Noriega L, Lopez A, Acosta D, Albanese E, et al. 10/66 Dementia research group. The 10/66 Dementia Research Group's fully operationalised DSM-IV dementia computerized diagnostic algorithm, compared with the 10/66 dementia algorithm and a clinician diagnosis: a population validation study. BMC Public Health. 2008;8:219. doi: 10.1186/1471-2458-8-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, et al. Alzheimer's Disease International. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–7. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.National Institute on Aging's document titled “Alzheimer's Disease: Unraveling the Mystery”. [accessed on September 11, 2015]. Available from: https://d2cauhfh6h4x0p.cloudfront.net/s3fs-public/alzheimers_disease_unraveling_the_mystery_0.pdf .
- 17.Jack CR, Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257–62. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bi X. Alzheimer disease: update on basic mechanisms. J Am Osteopath Assoc. 2010;110(9 Suppl 8):S3–9. [PubMed] [Google Scholar]
- 19.Bennett DA, Wilson RS, Boyle PA, Buchman AS, Schneider JA. Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol. 2012;72:599–609. doi: 10.1002/ana.23654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–88. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jack CR., Jr Alzheimer disease: new concepts on its neurobiology and the clinical role imaging will play. Radiology. 2012;263:344–61. doi: 10.1148/radiol.12110433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ringman JM, Coppola G, Elashoff D, Rodriguez-Agudelo Y, Medina LD, Gylys K, et al. Cerebrospinal fluid biomarkers and proximity to diagnosis in preclinical familial Alzheimer's disease. Dement Geriatr Cogn Disord. 2012;33:1–5. doi: 10.1159/000335729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging and the Alzheimer's Association. Alzheimers Dement. 2013;9:1–16. doi: 10.1016/j.jalz.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34:457–68. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 25.Aksu Y, Miller DJ, Kesidis G, Bigler DC, Yang QX. An MRI-derived definition of MCI-to-AD conversion for long-term, automatic prognosis of MCI patients. PLoS One. 2011;6:e25074. doi: 10.1371/journal.pone.0025074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitwell JL, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, et al. 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer's disease. Brain. 2007;130:1777–86. doi: 10.1093/brain/awm112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jagust W, Reed B, Mungas D, Ellis W, Decarli C. What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology. 2007;69:871–7. doi: 10.1212/01.wnl.0000269790.05105.16. [DOI] [PubMed] [Google Scholar]
- 28.Kikuchi M, Hirosawa T, Yokokura M, Yagi S, Mori N, Yoshikawa E, et al. Effects of brain amyloid deposition and reduced glucose metabolism on the default mode of brain function in normal aging. J Neurosci. 2011;31:11193–9. doi: 10.1523/JNEUROSCI.2535-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kantarci K, Senjem ML, Lowe VJ, Wiste HJ, Weigand SD, Kemp BJ, et al. Effects of age on the glucose metabolic changes in mild cognitive impairment. AJNR Am J Neuroradiol. 2010;31:1247–53. doi: 10.3174/ajnr.A2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oosterman JM, Oosterveld S, Rikkert MG, Claassen JA, Kessels RP. Medial temporal lobe atrophy relates to executive dysfunction in Alzheimer's disease. Int Psychogeriatr. 2012;24:1474–82. doi: 10.1017/S1041610212000506. [DOI] [PubMed] [Google Scholar]
- 31.Lahiri DK, Maloney B. Beyond the signaling effect role of amyloid-ß42 on the processing of APP, and its clinical implications. Exp Neurol. 2010;225:51–4. doi: 10.1016/j.expneurol.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tabaton M, Zhu X, Perry G, Smith MA, Giliberto L. Signaling effect of amyloid-beta(42) on the processing of AbetaPP. Exp Neurol. 2010;221:18–25. doi: 10.1016/j.expneurol.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baruch-Suchodolsky R, Fischer B. Abeta40, either soluble or aggregated, is a remarkably potent antioxidant in cell-free oxidative systems. Biochemistry. 2009;48:4354–70. doi: 10.1021/bi802361k. [DOI] [PubMed] [Google Scholar]
- 34.Igbavboa U, Sun GY, Weisman GA, He Y, Wood WG. Amyloid beta-protein stimulates trafficking of cholesterol and caveolin-1 from the plasma membrane to the Golgi complex in mouse primary astrocytes. Neuroscience. 2009;162:328–38. doi: 10.1016/j.neuroscience.2009.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamada M, Naiki H. Cerebral amyloid angiopathy. Prog Mol Biol Transl Sci. 2012;107:41–78. doi: 10.1016/B978-0-12-385883-2.00006-0. [DOI] [PubMed] [Google Scholar]
- 37.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 38.Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T, et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. J Neurosci. 2009;29:1860–73. doi: 10.1523/JNEUROSCI.5062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drzezga A, Becker JA, Van Dijk KR, Sreenivasan A, Talukdar T, Sullivan C, et al. Neuronal dysfunction and disconnection of cortical hubs in non-demented subjects with elevated amyloid burden. Brain. 2011;134:1635–46. doi: 10.1093/brain/awr066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–65. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin AL, Laird AR, Fox PT, Gao JH. Multimodal MRI neuroimaging biomarkers for cognitive normal adults, amnestic mild cognitive impairment, and Alzheimer's disease. Neurol Res Int 2012. 2012:907409. doi: 10.1155/2012/907409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schroeter ML, Stein T, Maslowski N, Neumann J. Neural correlates of Alzheimer's disease and mild cognitive impairment: a systematic and quantitative meta-analysis involving 1351 patients. Neuroimage. 2009;47:1196–206. doi: 10.1016/j.neuroimage.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chételat G, Villemagne VL, Bourgeat P, Pike KE, Jones G, Ames D, et al. Australian Imaging Biomarkers and Lifestyle Research Group. Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Ann Neurol. 2010;67:317–24. doi: 10.1002/ana.21955. [DOI] [PubMed] [Google Scholar]
- 44.Rodrigue KM, Kennedy KM, Devous MD, Sr, Rieck JR, Hebrank AC, Diaz-Arrastia R, et al. 0β-Amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology. 2012;78:387–95. doi: 10.1212/WNL.0b013e318245d295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2008;29:1456–65. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 46.Okello A, Koivunen J, Edison P, Archer HA, Turkheimer FE, Någren K, et al. Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study. Neurology. 2009;73:754–60. doi: 10.1212/WNL.0b013e3181b23564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moncaster JA, Pineda R, Moir RD, Lu S, Burton MA, Ghosh JG, et al. Alzheimer's disease amyloid-beta links lens and brain pathology in Down syndrome. PLoS One. 2010;5:e10659. doi: 10.1371/journal.pone.0010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Dominantly inherited Alzheimer network. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Storandt M, Head D, Fagan AM, Holtzman DM, Morris JC. Toward a multifactorial model of Alzheimer disease. Neurobiol Aging. 2012;33:2262–71. doi: 10.1016/j.neurobiolaging.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roses AD. On the discovery of the genetic association of apolipoprotein E genotypes and common late-onset Alzheimer disease. J Alzheimers Dis. 2006;9(3 Suppl):361–6. doi: 10.3233/jad-2006-9s340. [DOI] [PubMed] [Google Scholar]
- 51.Riley JH, Allan CJ, Lai E, Roses A. The use of single nucleotide polymorphisms in the isolation of common disease genes. Pharmacogenomics. 2000;1:39–47. doi: 10.1517/14622416.1.1.39. [DOI] [PubMed] [Google Scholar]
- 52.Eisenstein M. Genetics: finding risk factors. Nature. 2011;475:S20–2. doi: 10.1038/475S20a. [DOI] [PubMed] [Google Scholar]
- 53.Cui Y, Liu B, Luo S, Zhen X, Fan M, Liu T, et al. Alzheimer's Disease Neuroimaging Initiative. Identification of conversion from mild cognitive impairment to Alzheimer's disease using multivariate predictors. PLoS One. 2011;6:e21896. doi: 10.1371/journal.pone.0021896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee CY, Tse W, Smith JD, Landreth GE. Apolipoprotein E promotes β-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012;287:2032–44. doi: 10.1074/jbc.M111.295451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452–8. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Escott-Price V, Bellenguez C, Wang LS, Choi SH, Harold D, Jones L, et al. Gene-wide analysis detects two new susceptibility genes for Alzheimer's disease. PLoS One. 2014;9:e94661. doi: 10.1371/journal.pone.0094661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shulman JM, Chen K, Keenan BT, Chibnik LB, Fleisher A, Thiyyagura P, et al. Genetic susceptibility for Alzheimer's disease neuritic plaque pathology. JAMA Neurol. 2013;70:1150–7. doi: 10.1001/jamaneurol.2013.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16:848–50. doi: 10.1038/nn.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, et al. Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2, and other loci. Nat Neurosci. 2014;17:1156–63. doi: 10.1038/nn.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shoji M. Molecular approaches to the treatment, prophylaxis, and diagnosis of Alzheimer's disease: clinical molecular and genetic studies on Alzheimer's disease. J Pharmacol Sci. 2012;118:345–9. doi: 10.1254/jphs.11r13fm. [DOI] [PubMed] [Google Scholar]
- 61.Han X, Rozen S, Boyle SH, Hellegers C, Cheng H, Burke JR, et al. Metabolomics in early Alzheimer's disease: identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS One. 2011;6:e21643. doi: 10.1371/journal.pone.0021643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mielke MM, Haughey NJ, Bandaru VV, Weinberg DD, Darby E, Zaidi N, et al. Plasma sphingomyelins are associated with cognitive progression in Alzheimer's disease. J Alzheimers Dis. 2011;27:259–69. doi: 10.3233/JAD-2011-110405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lesser GT, Haroutunian V, Purohit DP, Schnaider Beeri M, Schmeidler J, Honkanen L, et al. Serum lipids are related to Alzheimer's pathology in nursing home residents. Dement Geriatr Cogn Disord. 2009;27:42–9. doi: 10.1159/000189268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lesser GT, Beeri MS, Schmeidler J, Purohit DP, Haroutunian V. Cholesterol and LDL relate to neuritic plaques and to APOE4 presence but not to neurofibrillary tangles. Curr Alzheimer Res. 2011;8:303–12. doi: 10.2174/156720511795563755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mandas A, Abete C, Putzu PF, la Colla P, Dessì S, Pani A. Changes in cholesterol metabolism-related gene expression in peripheral blood mononuclear cells from Alzheimer patients. Lipids Health Dis. 2012;11:39. doi: 10.1186/1476-511X-11-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200–8. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dawe RJ, Bennett DA, Schneider JA, Arfanakis K. Neuropathologic correlates of hippocampal atrophy in the elderly: a clinical, pathologic, postmortem MRI study. PLoS One. 2011;6:e26286. doi: 10.1371/journal.pone.0026286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al. AV45-A07 Study Group. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011;305:275–83. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Y, Jia HM, Liu BL. (E)-5-styryl-1H-indole and (E)-6-styrylquinoline derivatives serve as probes for β-amyloid plaques. Molecules. 2012;17:4252–65. doi: 10.3390/molecules17044252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schmidt A, Pahnke J. Efficient near-infrared in vivo imaging of amyloid-β deposits in Alzheimer's disease mouse models. J Alzheimers Dis. 2012;30:651–64. doi: 10.3233/JAD-2012-112168. [DOI] [PubMed] [Google Scholar]
- 71.Boutajangout A, Sigurdsson EM, Krishnamurthy PK. Tau as a therapeutic target for Alzheimer's disease. Curr Alzheimer Res. 2011;8:666–77. doi: 10.2174/156720511796717195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Citron M. Alzheimer's disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–98. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 73.Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–84. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 74.Frisardi V, Solfrizzi V, Imbimbo PB, Capurso C, D’Introno A, Colacicco AM, et al. Towards disease-modifying treatment of Alzheimer's disease: drugs targeting beta-amyloid. Curr Alzheimer Res. 2010;7:40–55. doi: 10.2174/156720510790274400. [DOI] [PubMed] [Google Scholar]
- 75.Hampel H. Current insights into the pathophysiology of Alzheimer's disease: selecting targets for early therapeutic intervention. Int Psychogeriatr. 2012;24(Suppl 1):S10–782. doi: 10.1017/S1041610212000579. [DOI] [PubMed] [Google Scholar]
- 76.Galimberti D, Scarpini E. Disease-modifying treatments for Alzheimer's disease. Ther Adv Neurol Disord. 2011;4:203–16. doi: 10.1177/1756285611404470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110:1129–34. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- 78.Parvathy S, Ehrlich M, Pedrini S, Diaz N, Refolo L, Buxbaum JD, et al. Atorvastatin-induced activation of Alzheimer's alpha secretase is resistant to standard inhibitors of protein phosphorylation-regulated ectodomain shedding. J Neurochem. 2004;90:1005–10. doi: 10.1111/j.1471-4159.2004.02521.x. [DOI] [PubMed] [Google Scholar]
- 79.Sato N, Shinohara M, Rakugi H, Morishita R. Dual effects of statins on Aβ metabolism: upregulation of the degradation of APP-CTF and Aβ clearance. Neurodegener Dis. 2012;10:305–8. doi: 10.1159/000334534. [DOI] [PubMed] [Google Scholar]
- 80.Vincent B, Cisse MA, Sunyach C, Guillot-Sestier MV, Checler F. Regulation of beta APP and PrPc cleavage by alpha-secretase: mechanistic and therapeutic perspectives. Curr Alzheimer Res. 2008;5:202–11. doi: 10.2174/156720508783954749. [DOI] [PubMed] [Google Scholar]
- 81.Mancuso C, Siciliano R, Barone E, Butterfield DA, Preziosi P. Pharmacologists and Alzheimer disease therapy: to boldly go where no scientist has gone before. Expert Opin Investig Drugs. 2011;20:1243–61. doi: 10.1517/13543784.2011.601740. [DOI] [PubMed] [Google Scholar]
- 82.Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm. 1998;105:423–38. doi: 10.1007/s007020050068. [DOI] [PubMed] [Google Scholar]
- 83.Hoyer S. The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: an update. J Neural Transm. 2002;109:341–60. doi: 10.1007/s007020200028. [DOI] [PubMed] [Google Scholar]
- 84.Hoyer S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur J Pharmacol. 2004;490:115–25. doi: 10.1016/j.ejphar.2004.02.049. [DOI] [PubMed] [Google Scholar]
- 85.Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8:247–68. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- 86.Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease--is this type 3 diabetes? J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- 87.Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1, resistant, IRS-1 dysregulation and cognitive decline. J Clin Invest. 2012;122:1316–38. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Craft S, Baker LD, Montine TJ, Minoshima S, Watson S, Claxton A, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2008;70:440–8. doi: 10.1212/01.WNL.0000265401.62434.36. [DOI] [PubMed] [Google Scholar]
- 90.US National Institutes of Health. Study of nasal insulin to fight forgetfulness – short-acting insulin aspart (SNIFFQuick) Unique ID: NCT02462161. [accessed on August 20, 2015]. Available from: https://clinicaltrials.gov .
- 91.Aisen PS, Gauthier S, Ferris SH, Saumier D, Haine D, Garceau D, et al. Tramiprosate in mild-to-moderate Alzheimer's disease - a randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study) Arch Med Sci. 2011;7:102–11. doi: 10.5114/aoms.2011.20612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Salloway S, Sperling R, Keren R, Porsteinsson AP, van Dyck CH, Tariot PN, et al. ELND005-AD201 Investigators. A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology. 2011;77:1253–62. doi: 10.1212/WNL.0b013e3182309fa5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12:856–61. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, et al. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–6. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Soeda S, Koyanagi S, Kuramoto Y, Kimura M, Oda M, Kozako T, et al. Anti-apoptotic roles of plasminogen activator inhibitor-1 as a neurotrophic factor in the central nervous system. Thromb Haemost. 2008;100:1014–20. doi: 10.1160/th08-04-0259. [DOI] [PubMed] [Google Scholar]
- 96.Erickson MA, Niehoff ML, Farr SA, Morley JE, Dillman LA, Lynch KM, et al. Peripheral administration of antisense oligonucleotides targeting the amyloid-β protein precursor reverses AβPP and LRP-1 overexpression in the aged SAMP8 mouse brain. J Alzheimers Dis. 2012;28:951–60. doi: 10.3233/JAD-2011-111517. [DOI] [PubMed] [Google Scholar]
- 97.Perrone L, Sbai O, Nawroth PP, Bierhaus A. The complexity of sporadic Alzheimer's disease pathogenesis: the role of RAGE as therapeutic target to promote neuroprotection by inhibiting neurovascular dysfunction. Int J Alzheimers Dis 2012. 2012:734956. doi: 10.1155/2012/734956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wisniewski T, Boutajangout A. Vaccination as a therapeutic approach to Alzheimer's disease. Mt Sinai J Med. 2010;77:17–31. doi: 10.1002/msj.20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta 42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 100.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 101.Panza F, Frisardi V, Solfrizzi V, Imbimbo BP, Logroscino G, Santamato A, et al. Immunotherapy for Alzheimer's disease: from anti-β-amyloid to tau-based immunization strategies. Immunotherapy. 2012;4:213–38. doi: 10.2217/imt.11.170. [DOI] [PubMed] [Google Scholar]
- 102.Blennow K, Zetterberg H, Rinne JO, Salloway S, Wei J, Black R, et al. AAB-001 201/202 Investigators. Effect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer disease. Arch Neurol. 2012;69:1002–10. doi: 10.1001/archneurol.2012.90. [DOI] [PubMed] [Google Scholar]
- 103.Samadi H, Sultzer D. Solanezumab for Alzheimer's disease. Expert Opin Biol Ther. 2011;11:787–98. doi: 10.1517/14712598.2011.578573. [DOI] [PubMed] [Google Scholar]
- 104.Dodel R, Balakrishnan K, Keyvani K, Deuster O, Neff F, Andrei-Selmer LC, et al. Naturally occurring autoantibodies against beta-amyloid: investigating their role in transgenic animal and in vitro models of Alzheimer's disease. J Neurosci. 2011;31:5847–54. doi: 10.1523/JNEUROSCI.4401-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Strobel G. Collaborative umbrella CAPS three prevention trial initiatives. Alzheimer's Association International Conference. 2012. [accessed on August 9, 2015]. Available from: http://www.alzforum.org/new/detailaspid=3232 .
- 106.Strobel G, Zaraib BD. Solanuzemab selected for Alzheimer's A4 prevention trial. Human Amyloid Imaging. 2013. [accessed on August 1, 2015]. Available from: http://www.alzforum.org/new/detailasp?id=3379 .
- 107.Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6:228fs13. doi: 10.1126/scitranslmed.3007941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.National Institute on Aging. Dominant Inherited Alzheimer Network trial: an opportunity to prevent dementia (DIAN TU) c2013. Available from: https://www.nia.nih.gov/alzheimers/clinical-trials/dominantly-inherited-alzheimer-network-trialopportunity-prevent-dementia .
- 109.Reiman EM, Langbaum JB, Fleisher AS, Caselli RJ, Chen K, Ayutyanont N, et al. Alzheimer's Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis. 2011;26(Suppl 3):321–9. doi: 10.3233/JAD-2011-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.APOE treatment study. [accessed on August 20, 2015]. Available from: http://banneralz.org/research-plus-discovery/alzheimers-prevention-initiative.aspx .
- 111.US National Institutes of Health. Biomarker qualification for risk of mild cognitive impairment (MCI) due to Alzheimer's disease (AD) and safety and efficacy evaluation of pioglitazone in delaying its onset (TOMMORROW) [accessed on August 20, 2015]. Available from: https:// clinicaltrials.gov .
- 112.US National Institutes of Health. Longitudinal evaluation of amyloid risk and neurodegeneration - the LEARN study. [accessed on August 20, 2015]. Available from: https://clinicaltrials.gov .
- 113.US National Institutes of Health. A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomaldominant Alzheimer disease, including a placebo-treated non-carrier cohor. [accessed on August 20, 2015]. Available from: https:// clinicaltrials.gov .
- 114.Drugs and Cosmetics Rules 1945. India: 2003. Government of India. Schedule Y: Requirements and guidelines for permission to import and/or manufacture of new drugs for sale or to undertake clinical trials. [Google Scholar]
- 115.Srinivasan S. Patient protection in clinical trials in India: some concerns. Perspect Cln Res. 2010;1:101–3. [PMC free article] [PubMed] [Google Scholar]