

Abstract
Background & objectives:
Hairy cell leukaemia (HCL) is a B cell neoplasm which constitutes around 2 per cent of all the lymphoid leukaemias. It has a characteristic morphology and immunophenotypic profile. It is important to distinguish HCL from other B cell lymphoproliferative disorders due to availability of different chemotherapeutic agents. This study presents clinical, haematological and immunophenotypic profile of patients with HCL seen over a period of four years in a tertiary care hospital in north India.
Methods:
Twenty one cases of hairy cell leukaemia were analyzed for their clinical details, haemogram, bone marrow examination and immunophenotypic findings.
Results:
Age of the patients ranged from 28-76 yr with male predominance. Weakness and fever were commonest presentations. Splenomegaly, hepatomegaly, lymphadenopathy were seen in decreasing order of frequency. Anaemia was noted in all 21 patients, leukopenia in 15 and thrombocytopenia in 19 cases. Fourteen patients were pancytopenic. Bone marrow examination showed typical hairy cells in all cases. Immunophenotyping showed expression of CD19, CD20, CD103, CD25 and CD11c in all cases, while positivity was seen for CD79b in 93.7 per cent, kappa light chain restriction in 60 per cent and lambda in 40 per cent cases. Notably, 20 per cent showed CD10 and 12 per cent showed CD23 expression.
Interpretation & conclusions:
This study reveals some unusual findings in otherwise classical disease entity, like absence of palpable spleen, presence of lymphadenopathy, normal or elevated leukocyte counts, expression of CD10, which at times could be diagnostically challenging.
Keywords: Anaemia, hairy cell leukaemia, haematological profile, immunophenotyping
Hairy cell leukaemia (HCL) is a rare low grade B cell neoplasm, comprising 2 per cent of lymphoid leukaemias with a characteristic morphologic and immunophenotypic profile. Patients are predominantly middle age to elderly adults with a median age of
50 yr. HCL has a unique clinical course and a specific treatment protocol, and hence needs to be differentiated from other chronic lymphoproliferative disorders. Most common presenting symptoms include easy fatigability, weakness, fever, pain in left upper quadrant of abdomen and bleeding. Most patients present with splenomegaly, pancytopenia with a few circulating neoplastic cells. Monocytopenia is a characteristic finding. Other manifestations include hepatomegaly and recurrent opportunistic infections, while vasculitis, bleeding disorder, neurologic disorders are less common1. One third to about a half of the patients with HCL have been reported to be asymptomatic at the time of diagnosis2.
The neoplastic cells of HCL are small to medium in size, having abundant cytoplasm, exhibit hairy projections all over the cell surface, have oval to indented nucleus with spongy chromatin. HCL diffusely infiltrates the bone marrow and involves red pulp of the spleen. The peripheral blood usually has low total leukocyte count with only a few circulating hairy cells. The neoplastic cells typically show bright expression of B cell markers like CD19, CD20 and CD22 and are usually negative for CD5, CD23 and CD103. The surface expression of CD103, CD25, CD11c and CD123 is characteristic of HCL and is used as an important criterion for the diagnosis. However, in the past atypical immunophenotypes have been reported in otherwise morphologically classical HCL4.
The correct identification and diagnosis of HCL is clinically important due to availability of effective chemotherapeutic agents for the treatment. Single course of cladribine is known to induce high response rates5. The cell of origin of HCL is not clear. Phenotypically, hairy cells do not resemble any normal B cell subpopulations. However, an earlier study on immunoglobulin heavy chain variable (IgVH) genes revealed that the majority of HCL cases had somatic mutations, indicating that cells giving rise to HCL passed through the germinal center6. Molecular studies on this subset of cases may give insight to whether these cases represent a subset of HCL with possible germinal center origin. This retrospective study was planned to analyze the clinical presentation, haematological and immunophenotypic profile of patients of hairy cell leukaemia diagnosed over a period of four years in a north Indian tertiary care hospital.
Material & Methods
A retrospective analysis of all chronic lymphoproliferative disorders (CLPD) diagnosed on immunophenotyping by flow cytometry in the department of Hematology, Postgraduate Institute of Medical Education and Research, Chandigarh, India, over a period of four years (November 2008 - November 2012) was carried out. A four colour (FITC, PE, APC and PerCP) panel of monoclonal antibodies including CD45, CD19, CD5, CD23, CD43, CD10, CD22, CD20, FMC7, CD79b, CD38, Kappa light chain, Lambda light chain, CD3, CD4, CD8, CD103, CD25, CD11c and CD123 (BD Pharmingen, USA) is routinely used in the department for evaluation of chronic lymphoproliferative disorders. All EDTA (ethylene diamine tetra acetic acid) anticoagulated bone marrow aspirates or peripheral blood samples were routinely processed using well standardized lyse-stain-wash procedure, acquired on BD FACS Canto II (BD Biosciences, USA) and analyzed by BD FACS Diva software (BD Biosciences, USA). All cases showing clonal B lymphocytes with surface expression of CD103, CD25, CD11c and CD123 were considered diagnostic for HCL and were selected for studying clinical presentation, haematological parameters and immunophenotyping profile. Cases that did not express classic immunophenotypic markers were specifically excluded. Cases of HCL variant who often lack CD25 were also excluded. Cases with incomplete information/work-up were excluded from the final analysis. In all, 21 cases had adequate clinical details, peripheral blood and bone marrow samples for review and complete immunophenotypic profile.
Results & Discussion
Twenty one patients with a confirmed diagnosis of HCL were selected for this study. HCL constituted approximately 5.3 per cent (21 out of 396 diagnosed cases of CLPD over 4 years) of all CLPD presenting at our institute. This figure was higher than that (2%) reported in western literature1. Age of our patients ranged from 28-76 yr with a median age of 55 yr, which was in concordance with western literature7. The youngest patient in the present study was 28 yr old, similar to a previous Indian study by Galani et al8 who reported a young individual of 26 yr with HCL. The male to female ratio was 6:1 which was similar to that reported in western literature8. All 21 patients in our study were symptomatic at presentation with weakness being the commonest presentation noted in 14 (66.6%). The second most common presenting feature was fever, seen in 11 (52.3%) patients followed by bleeding in three (14.3%) patients. Similar presenting features have been reported earlier9.
General physical examination showed splenomegaly, hepatomegaly and lymphadenopathy in 19 (90.5%), eight (38%) and six (28.6%) patients, respectively. Burke et al10 also reported splenomegaly in 90.5 per cent HCL patients, but only 19 per cent of their patients had hepatomegaly and none had lymphadenopathy at the time of presentation. Golomb et al11 reported splenomegaly, hepatomegaly and significant lymphadenopathy in 83, 19 and 10 per cent HCL cases, respectively. A higher frequency of lymphadenopathy was seen in our study, noted in around 28 per cent of patients and was similar to that published by Bouroncle12, reporting lymphadenopathy in 23 per cent of cases.
The haematological investigations revealed that all patients had anaemia with mean haemoglobin of 75 g/l (range 37-105 g/l), similar to other studies12,13. Fifteen patients (71.4%) were leukopenic, one had slightly higher total leukocyte counts (TLC) of 11.9×109/l, while five patients had normal TLC. The mean TLC was 3.24 x 109/l (range 0.3-11.9×109/l). One patient had mild leukocytosis (11.9x109/l), which is uncommon in classic HCL, although often seen in variant HCL. Monocytopenia was evident in all cases. The mean platelet count was 65.5 x 109/l (range 8-158×109/l), with 19 patients being thrombocytopenic. Pancytopenia was seen in 14 patients (66.6%). Anaemia (21/21) and thrombocytopenia (19/21) were more frequently seen at presentation than that reported in a previously published Indian study8. Bone marrow examination was done in all patients. A dry tap was observed in 15 (71.4%) and the remaining six patients showed occasional marrow particle, however, the smears were diluted with blood.
Flow cytometry remains the modality of choice in the diagnosis of HCL with the classic immunophenotypic profile consisting of bright expression of CD20 and CD22, presence of monotypic surface immunoglobulin, and co-expression of CD103, CD25, CD11c and CD123. Other markers, more often evaluated on immunohistochemistry include DBA.44, Annexin A1, T-bet and Cyclin D1. Immunophenotypic profile on multicolour flow cytometry of our patients has been summarized in the Table. In the present study CD19, CD20, CD103, CD25 and CD11c were expressed in all cases. Bright surface expression of CD22 was seen in 9/9 (100%) cases while CD79b was seen in 93.7 per cent patients. Kappa light chain restriction was seen in 60 per cent while lambda light chain restriction was seen in 40 per cent patients with HCL. Galani et al8 reported kappa restriction in 46 per cent and lambda light chain restriction in 50 per cent of their cases.
Table.
Immunophenotic profile of HCL patients
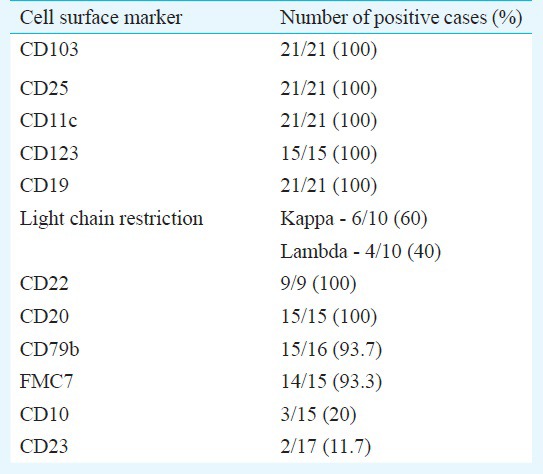
Most cases of HCL lack both CD10 and 5, although immunophenotypic variants are well known1. HCL variants usually express CD103 and CD11C but lack expression of CD2514. Annexin A1 is most specific marker since it is not expressed in any B cell lymphoma other than HCL. Expression of annexin A1 can be used to distinguish HCL from splenic marginal zone lymphoma and HCL variant which are both annexin A1 negative15. In our study, 20 per cent (3/15) cases were identified with expression of CD10. CD10 expression ranging from 5 to 26 per cent cases has been reported in some other studies3,4,16,17.
Of the 21 patients, treatment and follow up data were available for 15 patients only. Eight patients had received cladribine and six of them achieved complete remission at 12 months, one patient each in 30 and 60 months of follow up. Two patients underwent splenectomy and were asymptomatic during the follow up period of 18 and 48 months, respectively. One patient was treated with rituximab (600 mg for 4 days) in February 2011, but showed relapse and treated with cladribine in June 2012. This patient was in complete remission at the last follow up visit. Three patients died because of sepsis. One treatment naive patient remained asymptomatic at 14 months of follow up.
HCL is an uncommon haematological malignancy with unique clinical, haematological and immunophenotypic profile. However, a few patients with HCL may have an atypical presentation like absence of splenomegaly, enlarged lymph nodes, a normal or high total leukocyte count, which may be diagnostically challenging. Expression of CD103, CD25, CD11c and CD123 is considered unique for HCL, however, immunophenotypic variation like expression of CD10 and CD23 can also be seen.
Footnotes
Conflicts of Interest: None.
References
- 1.Foucar K, Falini B, Catovsky D, Stein H. Hairy cell leukaemia. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO classification of tumours of the haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008. pp. 188–90. [Google Scholar]
- 2.Malfuson JV, Gisserot O, Cremades S, Doghmi K, Fagot T, Souleau B, et al. Hairy-cell leukemia: 30 cases and a review of the literature. Ann Med Interne (Paris) 2003;154:435–40. [PubMed] [Google Scholar]
- 3.Chen YH, Tallman MS, Goolsby C, Peterson L. Immunophenotypic variations in hairy cell leukemia. Am J Clin Pathol. 2006;125:251–9. doi: 10.1309/PMQX-VY61-9Q8Y-43AR. [DOI] [PubMed] [Google Scholar]
- 4.Jasionowski TM, Hartung L, Greenwood JH, Perkins SL, Bahler DW. Analysis of CD10+ hairy cell leukemia. Am J Clin Pathol. 2003;120:228–35. doi: 10.1309/QVJD-31TE-G9UJ-18BQ. [DOI] [PubMed] [Google Scholar]
- 5.Goodman GR, Burian C, Koziol JA, Saven A. Extended follow-up of patients with hairy cell leukemia after treatment with cladribine. J Clin Oncol. 2003;21:891–6. doi: 10.1200/JCO.2003.05.093. [DOI] [PubMed] [Google Scholar]
- 6.Miranda RN, Cousar JB, Hammer RD, Collins RD, Vnencak-Jones CL. Somatic mutation analysis of IgH variable regions reveals that tumor cells of most parafollicular (monocytoid) B-cell lymphoma, splenic marginal zone B-cell lymphoma, and some hairy cell leukemia are composed of memory B lymphocytes. Hum Pathol. 1999;30:306–12. doi: 10.1016/s0046-8177(99)90010-2. [DOI] [PubMed] [Google Scholar]
- 7.Wanko SO, de Castro C. Hairy cell leukemia: an elusive but treatable disease. Oncologist. 2006;11:780–9. doi: 10.1634/theoncologist.11-7-780. [DOI] [PubMed] [Google Scholar]
- 8.Galani KS, Subramanian PG, Gadage VS, Rahman K, Ashok Kumar MS, Shinde S, et al. Clinico-pathological profile of hairy cell leukemia: critical insights gained at a tertiary care cancer hospital. Indian J Pathol Microbiol. 2012;55:61–5. doi: 10.4103/0377-4929.94858. [DOI] [PubMed] [Google Scholar]
- 9.Hoffman MA. Clinical presentations and complications of hairy cell leukemia. Hematol Oncol Clin North Am. 2006;20:1065–73. doi: 10.1016/j.hoc.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Burke JS, Byrne GE, Jr, Rappaport H. Hairy cell leukemia (leukemic reticuloendotheliosis). I. A clinical pathologic study of 21 patients. Cancer. 1974;33:1399–410. doi: 10.1002/1097-0142(197405)33:5<1399::aid-cncr2820330526>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 11.Golomb HM, Catovsky D, Golde DW. Hairy cell leukemia: a clinical review based on 71 cases. Ann Intern Med. 1978;89:677–83. doi: 10.7326/0003-4819-89-5-677. [DOI] [PubMed] [Google Scholar]
- 12.Bouroncle B. Leukemic reticuloendotheliosis (hairy cell leukemia) Blood. 1979;53:412–36. [PubMed] [Google Scholar]
- 13.Chatterjee T, Panigrahi I, Mahapatra M, Pati HP, Kumar R, Naithani R, et al. Hairy cell leukemia: clinical, pathological and ultrastructural findings in Asian-Indians. Indian J Cancer. 2008;45:41–4. doi: 10.4103/0019-509x.41768. [DOI] [PubMed] [Google Scholar]
- 14.Cessna MH, Hartung L, Tripp S, Perkins SL, Bahler DW. Hairy cell leukemia variant: fact or fiction. Am J Clin Pathol. 2005;123:132–8. doi: 10.1309/8qytyq1clqmhq9cl. [DOI] [PubMed] [Google Scholar]
- 15.Falini B, Tiacci E, Liso A, Basso K, Sabattini E, Pacini R, et al. Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1) Lancet. 2004;363:1869–70. doi: 10.1016/S0140-6736(04)16356-3. [DOI] [PubMed] [Google Scholar]
- 16.Robbins BA, Ellison DJ, Spinosa JC, Carey CA, Lukes RJ, Poppema s s0, et al. Diagnostic application of two-color flow cytometry in 161 cases of hairy cell leukemia. Blood. 1993;82:1277–87. [PubMed] [Google Scholar]
- 17.Juliusson G, Lenkei R, Liliemark J. Flow cytometry of blood and bone marrow cells from patients with hairy cell leukemia: phenotype of hairy cells and lymphocyte subsets after treatment with 2-chlorodeoxyadenosine. Blood. 1994;83:3672–81. [PubMed] [Google Scholar]