

Abstract
Fucosidosis is a rare lysosomal storage disease with clinical presentation of developmental retardation, coarse facial features, hepatosplenomegaly, dysostosis multiplex, and angiokeratomas. Here, a 7-year-old female patient with progressive dystonic movement disorder and loss of acquired motor skills is presented. Coarse facial feature and abnormal globuspallidus signaling in brain magnetic resonance imaging (MRI) led the patient to be investigated in terms of fucosidosis despite absence of hepatosplenomegaly, dysostosis multiplex, and angiokeratomas. Markedly decreased enzyme activity of alpha-fucosidosis led to the correct diagnosis.
Conclusion:
Various neurological findings have recently been reported in fucosidosis. However, neuroimaging findings have not been studied in detail except a few studies. It is critically important to discuss the wide neuroradiological spectrum of the disease and to highlight fucosidosis in differential diagnosis of bilateral pallidalhypointensity on T2-weighted images in brain MRI. In addition, description of atypical clinical findings of fucosidosis should avoid clinicians from diagnostic delay.
Keywords: Dystonia, fucosidosis, globus pallidus hypointensity, magnetic resonance imaging
Introduction
Fucosidosis (OMIM # 230000) is a rare lysosomal storage disorder, caused by α-L-fucosidase deficiency. α-L-fucosidase is responsible in degradation of fucose containing glycoprotein and glycolipids.[1] It was first described in 1968 by Durand and colleagues, in two brothers and enzyme was reported in the same year by Van Hoff and Hers.[2,3] Common clinical features include progressive cognitive and motor deterioration, coarse facial features, growth retardation, dysostosis multiplex, angiokeratomacorporis diffusum, and organomegaly.[4] Diagnosis is confirmed by demonstration of defective enzyme activity in plasma, leukocytes, and cultured fibroblasts. Genetic analysis can be performed to show the mutations in α-fucosidase structural gene (FUCA1) that is mapped on chromosome 1 and a second locus on chromosome 6 (FUCA2) that regulates the level of enzyme in plasma but not in leukocytes.[1]
There have been a few recent studies that mention cranial magnetic resonance imaging (MRI) findings in fucosidosis. Cerebral and cerebellar atrophy of various degrees depending on the progression of disease and white matter signal alterations in periventricular and subcortical areas were the first reported and well-known findings in brain MRI.[5] Bilateral pallidalhyperintense signaling on T1-weighted imaging and hypointense signaling on T2-weighted imaging are another typical but less reported MRI (magnetic resonance imaging), finding of fucosidosis.[6,7] Cerebral and cerebellar atrophy are common findings in lysosomal storage diseases; whereas, pallidal signaling alterations are characteristic for fucosidosis.[5,7]
Here, a 7-year-old female patient whose atypical clinical presentation leads a diagnostic delay of fucosidosis is reported. Evaluation of characteristic MRI (magnetic resonance imaging), findings in the light of clinical findings revealed the correct diagnosis.
Case Report
A 7-year-old female patient was admitted to our outpatient clinic with complaints of failure to thrive, loss of acquired motor skills, and generalized dystonia. She was the second child of nonconsanguineous parents and her prenatal, natal, and postnatal history was uneventful. First child of family was investigated because of severe spasticity and generalized dystonia. His cranial MRI (magnetic resonance imaging), demonstrated cerebellar atrophy and marked hypointense area on T2-weighted imaging in bilateral globuspallidus, indicating the presence of high levels of iron in echo-gradient MRI (magnetic resonance imaging), with similar pathological hypointense changes observed in bilateral substantianigra and tectum of the midbrain. According to his neuroradiological findings, diagnosis of neurodegeneration with brain iron accumulation disorders (NBIA) was suspected. He died at 14 years of age without a definite diagnosis.
Our patient developed normal till 12 months of age. Unsupported ambulation that was acquired at 15 months became gradually unsteady and was lost soon after 5 years of age. Concurrently, her somatic growth slowed down and her length and weight fell less than −2 standard deviation score. Her physical examination revealed mild coarse facial features, Mongolian eye slant, arcuated eyebrows, high arched palate and exophthalmos in right eye. Her tongue was protruding due to macroglossia. There were red streaks on gingivae and blue-brown spots on tongue. She had no angiokeratomas in genital area and trunk. There was no hepatosplenomegaly. Her neurological examination revealed spastic tetraparesis dominated by dystonic posturing. Increased tendon reflexes and multilevel joint contractures were observed. She had a complete loss of voluntary movements including head control. Choreathetoid movements, especially marked on arms were observed during examination. Fundoscopic examination was normal and conventional radiographs did not reveal any signs of dysostosis multiplex.
Brain MRI (magnetic resonance imaging), showed mild hyperintense signal alterations in cerebral deep white matter and subcortical areas bilaterally. In addition, hyperintense signal alterations on T1-weighted imaging and hypointense signal alterations on T2-weighted imaging in bilateral globuspallidus, substantianigra, and nucleus ruber. Corpus callosum thinning and superior vermian atrophy was observed. MRI (magnetic resonance imaging), findings of our patient were found very similar to his brother [Figures 1 and 2].
Figure 1.
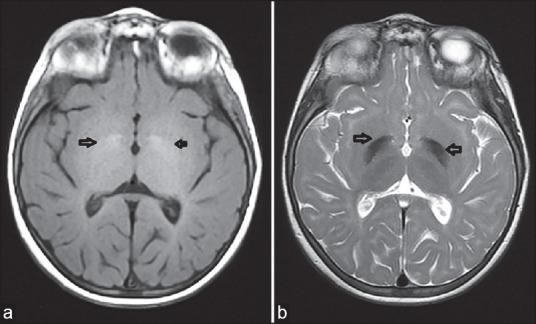
Cranial MRI (magnetic resonance imaging), findings of our patient: (a) Bilateral globus pallidus hyperintensity signaling on T1-weighted imaging; and (b) bilateral globus pallidus hypointensity signaling on T2-weighted imaging. MRI = Magnetic resonance imaging
Figure 2.
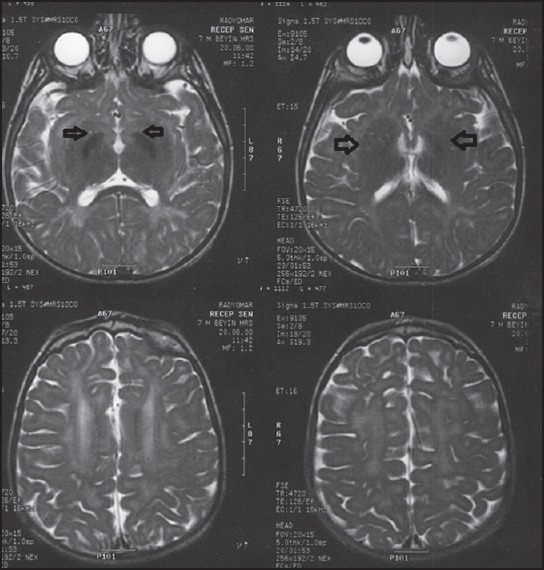
Cranial MRI findings of the patient's brother: Bilateral globus pallidus hypointensity signaling on T2-weighted imaging and hypomyelination
Based on clinical findings of coarse facial features, neuromotor deterioration, blue-brown spots on tongue, and abnormal MRI (magnetic resonance imaging), findings; fucosidosis was included in differential diagnosis. The enzymatic activity of α-fucosidase in plasma and lymphocytes was measured 0, 1 μmol/g.h. (normal ranges: 50-250 μmol/g.h.); that was consistent with fucosidosis.
Discussion
In this case, clinical and MRI (magnetic resonance imaging), findings of a 7-year-old female patient who is diagnosed as fucosidosis are presented. Diagnosis of disease is confirmed at the age of 7, in spite of the neurological findings that started at 12 months and the existence of a similar family history. In this case, lack of typical signs of fucosidosis as dysostosis multiplex, organomegaly, and angiokeratomacorporis diffusum in classical locations caused a diagnostic delay. Several aspects of this case should be instructive in evaluating the clinical and neuroradiological findings of fucosidosis.
In classic infantile phenotype of disease, patients appear normal at birth and they develop progressive coarsening of features, retardation of linear growth, and cognitive development. Willems et al., reported that 79% of 77 patients who were diagnosed as fucosidosis had coarse features.[4] In another study, Ben et al., reviewed phenotypic spectrum of fucosidosis in Tunisia and reported that nine of 10 patients with coarse facies.[8] In our case mild dismorphic facial features were observed.
Dysostosis multiplex is the characteristic skeletal involvement of lysosomal storage diseases and is a common finding in fucosidosis. Ben et al., reported dysostosis multiplex in variable degrees in all of 10 cases; whereas, 58% of patients that were reported by Willems et al., presented dysostosis multiplex. Organomegaly was reported as 3 and 44%, respectively, by Ben et al., and Willems et al.[4,8] In our case, dysostosis multiplex and organomegaly were not observed.
In more indolent cases the first sign can be the development of angiokeratomas. They are marked especially over the buttocks and in genital area in childhood and may spread over the body later. Angiokeratomacorporis diffusum was reported in 52% of all cases by Willems et al., and four of 10 cases by Ben et al.[4,8] In our case, angiokeratomas were not observed in classical localization. They were first noted as red streaks on gingivae and then spread to tongue as angiokeratomas.
Fucosidosis is one of the neurodegenerative lysosomal storage diseases. Brain MRI (magnetic resonance imaging), findings of neurodegenerative diseases include delayed myelination, atrophy, and hyperintense signal alterations of corpus callosum, basal ganglia, and cerebral white matter on T2-weighted imaging. However, bilateral pallidalhyperintense and hypointense signal alterations on T1- and T2-weighted imaging, respectively, are not a common finding of the other neurodegenerative diseases.[9]
Common brain MRI (magnetic resonance imaging), findings in fucosidosis were described in several studies. Cerebral and cerebellar atrophy of various degrees depending on the progression of disease is well-documented.[5] Extensive and progressive white matter signal alterations in periventricular and subcortical areas, putamen, medullary lamina of thalamus, and internal and external capsules have been described because of diffuse demyelination.[5,10] Terepolsky et al., described bilateral pallidalhyperintense signaling on T1-weighted imaging and hypointense signaling on T2-weighted imaging as another typical but less reported MRI (magnetic resonance imaging), finding of fucosidosis. Soon after, Galluzzi et al., described these hyperintense spots that correspond to fiber tracts of the medial medullary laminae were described as “curvilinear streaks”.[10] Ben et al., reported low signal intensity of globuspallidum on T2-weighted imaging in two of 10 cases, but in these cases cerebral and cerebellar atrophy accompanied the abnormal pallidal image.[8] Cerebral and cerebellar atrophy is a common finding in lysosomal storage diseases; whereas, pallidal signaling alterations are characteristic for fucosidosis.[5,7] Our patient's brain MRI (magnetic resonance imaging), revealed bilateral mild hyperintense signal alterations in cerebral deep white matter and subcortical areas. In addition, bilateral pallidalhyposignaling with central hyperintensity on T2-weighted imaging was observed. Superior vermian atrophy was seen; whereas, no evidence of cerebellar atrophy was noted in spite of serious neurological deterioration at the time of imaging. MRI (magnetic resonance imaging), findings of our patient were found very similar to his brother who was misdiagnosed as NBIA. Considering the MR images as NBIA (neurodegeneration with brain iron accumulation disorders) led the misdiagnosis in the first sibling. NBIA (neurodegeneration with brain iron accumulation disorders) is one of the diseases that can present itself with same bilateral pallidalhypointensity image on T2; whereas, it does not cause hypomyelinization and atrophy like fucosidosis.[5,7]
In 2014, Gautschi et al. reported a 14-year-old female patient with mild morphological features, without the existence of dysostosis multiplex and organomegaly who was diagnosed as fucosidosis similar to our case. Her brain MRI (magnetic resonance imaging), revealed the same “eye of the tiger” sign. However, in comparison to our patient, she had wide, progressively spreading angiokeratomacorporisdiffusum in trunk that facilitates to lead the correct diagnosis of fucosidosis.[11]
Conclusion
This case mentions the wide clinical and radiological spectrum of fucosidosis. Lysosomal storage diseases should be in differential diagnosis of patients with progressive neurological disorders. Fucosidosis should be reminded even though common features such as dysostosis multiplex and organomegaly do not occur. Angiokeratoma corporisdiffusum should be carefully investigated and it should be reminded that in earlier ages it would present itself in atypical locations.
In this case, neuroradiological findings of brain MRI (magnetic resonance imaging), suggested differential diagnosis of fucosidosis. In our country prevalence of inherited metabolic diseases is frequent partially due to consanguineous marriages. As a result, having knowledge about cranial MRI (magnetic resonance imaging), changes due to metabolic disorders plays an important role to avoid diagnostic delays and misdiagnosis. Here, fucosidosis is highlighted in differential diagnosis of bilateral pallidalhypointensity on T2-weighted imaging in brain MRI (magnetic resonance imaging), even with or without white matter changes.
Diagnostic delay could be avoided with remembering the wide spectrum of clinical and radiological findings of fucosidosis with atypical presentations of disease.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.George HT. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Disorders of glycoprotein degradation: α-mannosidosis, β-mannosidosis, α-fucosidosis and sialidosis. Child B, Kinzler KW, Vogelstein B, assoc. 8th ed. The Metabolic and Molecular Basis of Inherited Metabolic Disease. 2001;3:3507–33. [Google Scholar]
- 2.Durand P, Borrone C, Della Cella G, Philippart M. Fucosidosis. Lancet. 1968;1:1198. [Google Scholar]
- 3.Van Hoff F, Hers HG. Mucopolysaccharidosis by absence of α-fucosidase. Lancet. 1968;1:1198. doi: 10.1016/s0140-6736(68)91895-3. [DOI] [PubMed] [Google Scholar]
- 4.Willems PJ, Gatti R, Darby JK, Romeo G, Durand P, Dumon JE, et al. Fucosidosis revisited: A review of 77 patients. Am J Med Genet. 1991;38:111–31. doi: 10.1002/ajmg.1320380125. [DOI] [PubMed] [Google Scholar]
- 5.Provenzale JM, Barboriak DQ, Sims K. Neuroradiological findings in fucosidosis, a rare lysosomal storage disease. AJNR Am J Neuroradiol. 1995;16:809–13. [PMC free article] [PubMed] [Google Scholar]
- 6.Jain P, Ramesh K, Mohamed A, Kumar A, Gulati S. Distinct neuroimaging features of fucosidosis. Neurology. 2012;78:e33. doi: 10.1212/WNL.0b013e3182452910. [DOI] [PubMed] [Google Scholar]
- 7.Terepolsky D, Clarke JT, Biaser SI. Evolution of the neuroimaging changes in fucosidosis type II. J Inher Metab Dis. 1996;19:775–81. doi: 10.1007/BF01799172. [DOI] [PubMed] [Google Scholar]
- 8.Ben Turkia H, Tebib N, Azzouz H, Abdelmoula MS, Bouguila J, Sanhaji H, et al. Phenotypic spectrum of fucosidosis in Tunisia. J Inherit Metabolic Dis. 2008;31:313–6. doi: 10.1007/s10545-008-0891-0. [DOI] [PubMed] [Google Scholar]
- 9.Cheon JE, Kim IO, Hwang YS, Kim KJ, Wang KC, Cho BK, et al. Leukodystrophy in children: A pictorial review of MR imaging features. Radiographics. 2002;22:461–76. doi: 10.1148/radiographics.22.3.g02ma01461. [DOI] [PubMed] [Google Scholar]
- 10.Galluzzi P, Rufa A, Balestri P, Cerase A, Federico A. MR brain imaging of fucosidosis type I. ANJR. 2001;22:777–80. [PMC free article] [PubMed] [Google Scholar]
- 11.Gautschi M, Merlini L, Calza AM, Hayflick S, Nuoffer JM, Fluss J. Late diagnosis of fucosidosis in a child with progressive fixed dystonia, bilateral pallidal lesions and red spots on the skin. Eur J Paediatr Neurol. 2014;18:516–9. doi: 10.1016/j.ejpn.2014.02.005. [DOI] [PubMed] [Google Scholar]