

Sir,
Chronic progressive external ophthalmoplegia (CPEO), phenotypically and genotypically heterogeneous disorders is characterised by progressive ptosis and ophthalmoparesis. Common neurologic findings include facial, bulbar and limb myopathies, peripheral neuropathy, ataxia, spasticity, gastrointestinal myopathy and neuropathy, deafness, vestibular dysfunction, dementia, episodic encephalopathy and calcification of the basal ganglia.
CPEO may be a nonspecific finding in patients with degenerative disorders of the nervous system.[1] CPEO can be part of well-defined mitochondrial syndromes. Molecular basis includes mitochondrial DNA depletion and/or mtDNA point mutations and deletions. CPEO can also manifest with oropharyngeal weakness, proximal muscle weakness, wasting and exercise intolerance and these are considered to lie on a spectrum of disease from pure CPEO to Kearns-Sayre syndrome.[2]
Pharyngeal symptoms have been noted in mitochondrial myopathy. However, only two prior case reports[3,4] have demonstrated findings of laryngeal involvement in mitochondrial Myopathy. Vocal palsy is rarely reported in CPEO and mitochondrial cytopathy. We report a case of CPEO with vocal cord palsy which to the best of our knowledge not described in India. The additional supportive diagnostic findings of ragged red, ragged blue fibers in muscle biopsy along with COX deficient fibers and electron microscopic evidence of abnormal mitochondria, videolaryngoscopic documentation of adductor palsy and genetic studies adds to the robust nature of our report. In large prospective studies available in Indian literature[5,6] in patients of CPEO with confirmed muscle biopsy and genetics, none had vocal cord palsy. None underwent electron microscopy.
A 23-year-old gentleman presented with gradually progressive drooping of both eyelids of 7 years duration and hoarseness of voice of 4-years duration. Tarsoraphy did not help his ptosis. There was no history of diplopia, vision loss, dysphagia, limb weakness or other systemic features. There was no family history. Examination revealed dysmorphism in the form of low set ears, high arched palate, and hallus valgus. He had bilateral ptosis with external ophthalmoplegia [Figure 1a]. Fundi and pupils were normal. He also had bifacial weakness with hoarseness of voice, vocal cord palsy [Figures 1b and c], normal palatal movements, mild neck flexor weakness and normal power.
Figure 1.
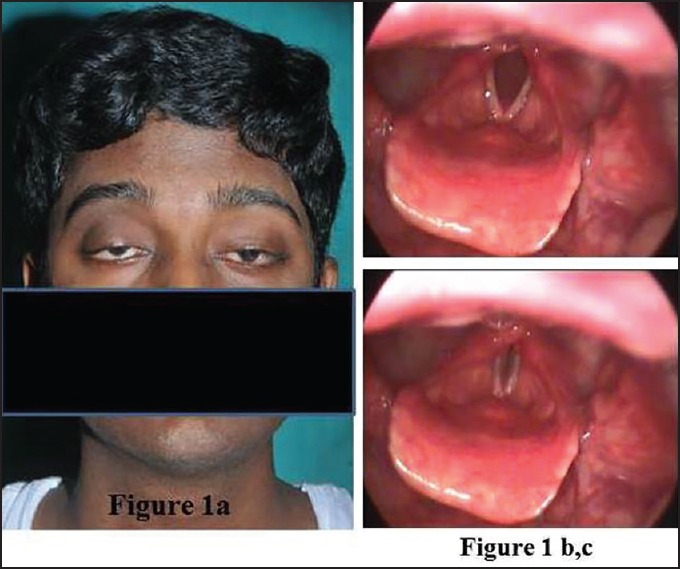
(a) Bilateral ptosis with restricted extraocular movements in all directions, (b, c) Video laryngoscopy depicting bilateral adductor palsy, (d) Video supplement-videolaryngoscopy depicting bilateral adductor palsy
On investigation, his blood sugars, creatinine phosphokinase, ammonia were normal. Lactate was 21.7 mg% (Normal 4.5-20). Cerebrospinal fluid (CSF) protein was normal. Cardiac evaluation, Magnetic resonance imaging (MRI) Brain and brainstem evoked responses were normal. Repetitive nerve stimulation was normal. Video laryngoscopy was suggestive of bilateral adductor palsy [Figure 1d, video supplement]. Left biceps muscle biopsy done was subjected to a battery of stains: Hematoxylin and Eosin (H&E), Modified Gomori Trichrome (MGT), Succinic dehydrogenase (SDH), Nicotinamide adenine dinucleotide tetrazolium reductase (NADHTR), Cytochrome C oxidase (COX), COX-SDH and Adenosine triphosphatase (ATPase) pH 9.4 and 4.6. Tiny pieces of muscle fixed in 2.5% gluteraldehyde were processed for electron microscopy. Moderate numbers of ragged blue fibres and ragged red fibers and COX deficient fibers were noted [Figure 2]. Electron microscopy revealed abnormal mitochondria with paracrystalline inclusions [Figure 3]. DNA isolated from muscle did not reveal mutations in the entire mitochondrial genome. There were no mutations found in Polymerase Gamma (POLG). Patient was given speech therapy and Co Q supplements with no significant improvement.
Figure 2.
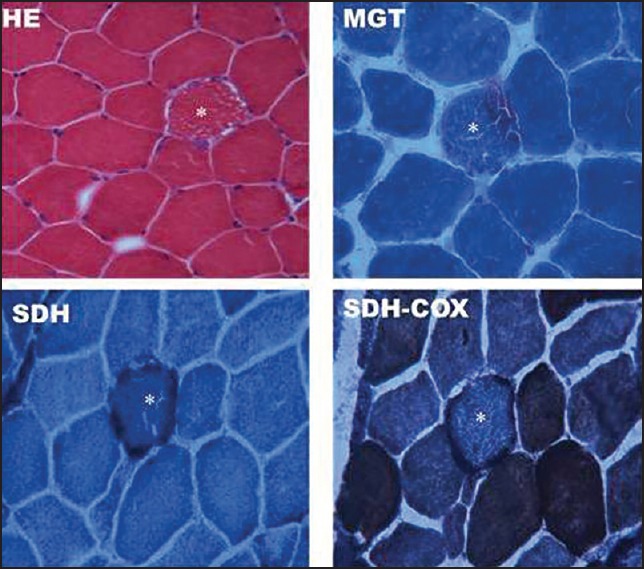
Transversely cut skeletal muscle tissue showing *ragged fibres (HE), ragged red fibers (MGT), ragged blue fibres (SDH) and COX deficient fibers (SDH-COX)
Figure 3.
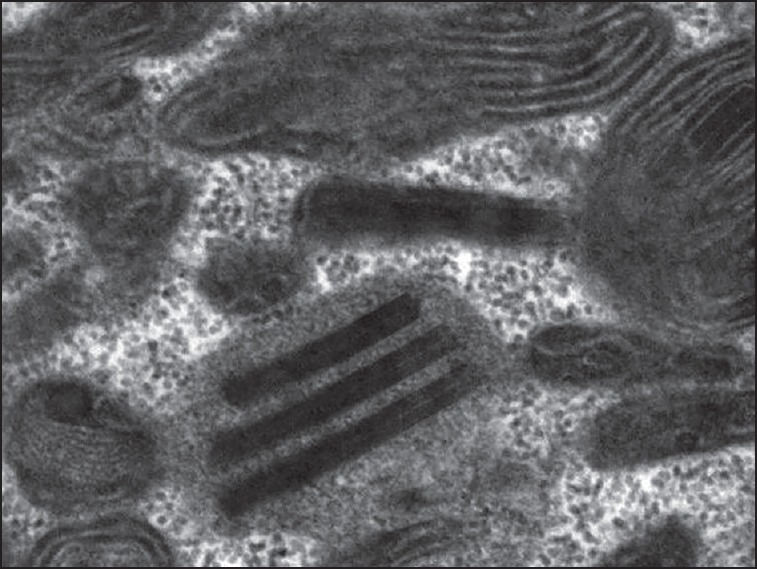
Electronmicrograph of skeletal muscle tissue showing abnormal mitochondria and paracrystalline inclusions × 49,000
Mendelian PEOs associated with multiple deletions of mtDNA may be caused by mutations in SLC25A4, encoding ANT1, TYMP encoding thymidine phosphorylase (autosomal recessive inheritance, CPEO, peripheral neuropathy and gastrointestinal dysmotility), POLG encoding subunit of mtDNA polymerase, C10 ORF2 encoding twinkle and OPA1 encoding a dynamin-related GTPase, causing autosomal dominant nonsyndromic optic atrophy and syndromic optic atrophy, CPEO, and deafness with variable signs. Mutations in the Twinkle gene, a cause of autosomal dominant progressive external ophthalmoplegia typically present with isolated extraocular muscle involvement. Proximal muscle weakness, sensory-axonal neuropathy, ataxia, dementia, Parkinsonism, bulbar symptoms including dysphagia, dysphonia, and dysarthria are also reported.[7] Patients with C10 ORF2-linked autosomal dominant PEO may have proximal muscle weakness, sensorineural hearing loss, ataxia, peripheral Neuropathy, cardiomyopathy, cataracts, depression, and endocrine abnormalities. (“CPEO+”) PEO caused by mutations in the POLG gene are associated with more complicated phenotypes.[8] POLG related disorders consist of a spectrum of overlapping phenotypes and it includes autosomal recessive progressive external ophthalmoplegia (arPEO) and autosomal dominant progressive external ophthalmoplegia (adPEO). Progressive PEO without systemic involvement is the hallmark of arPEO. Other features variably present are depression, premature ovarian failure, Parkinsonism, ataxia, and a phenotype Mitochondrial Neurogastrointestinal Encephalopathy Disease like phenotype.adPEO is characterised by ptosis and ophthalmoplegia. A generalized myopathy is present in most affected individuals, leading to early fatigue and exercise intolerance. Other features like Parkinsonism, premature ovarian failure, and depression are often referred to as “chronic progressive external ophthalmoplegia plus” (CPEO+).[9]
Hartley C et al., 1994 described the first case of laryngeal involvement in mitochondrial myopathy who had slowly progressive weakness of the voice, poor cough and poor glottic closure[3] Kelly EA et al., 2013[4] described the second case who had progressive dysphonia, dysphagia with positive family history of neuromuscular disease. His videostroboscopic laryngeal examination demonstrated marked early onset vocal fold atrophy. There is single documented case of Kearn Sayre syndrome with vocal cord palsy[10] and single documented case of Leigh disease presenting as progressive stridor and bilateral vocal cord paralysis.[11]
Diagnosis of mtDNA deletion syndromes relies on presence of characteristic clinical finding, ragged- red fibers with the modified Gomori trichrome stain, hyperactive fibers with SDH, COX negative fibres, parking lot inclusions on transmission electron microscopy and decreased activity of respiratory chain complexes in muscle extracts. PCR and Southern blotting reveals common deletion.
Awareness of atypical presentations in CPEO helps in adequate management, avoidance of unnecessary surgery and in further mitochondrial research.
References
- 1.Fraser JA, Biousse V, Newman NJ. The Neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol. 2010;55:299–334. doi: 10.1016/j.survophthal.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol. 2012;47:64–74. doi: 10.3109/10409238.2011.632763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartley C, Ascott F. Laryngeal involvement in mitochondrial myopathy. J Laryngol Otol. 1994;108:685–7. doi: 10.1017/s0022215100127835. [DOI] [PubMed] [Google Scholar]
- 4.Kelly EA, Bock JM, Peltier AC, Oh SJ, Garrett CG. Mitochondrial Myopathy: A rare cause of early-onset vocal fold atrophy. Ann Otol Rhinol Laryngol. 2013;122:177–82. doi: 10.1177/000348941312200306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sundaram C, Meena AK, Uppin MS, Govindaraj P, Vanniarajan A, Thangaraj K, et al. Contribution of muscle biopsy and genetics to the diagnosis of chronic progressive external opthalmoplegia of mitochondrial origin. J Clin Neurosci. 2011;18:535–8. doi: 10.1016/j.jocn.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 6.Challa S, Kanikannan MA, Murthy JM, Bhoompally VR, Surath M. Diagnosis of mitochondrial diseases: Clinical and histological study of sixty patients with ragged red fibers. Neurol India. 2004;52:353–8. [PubMed] [Google Scholar]
- 7.Fratter C, Gorman GS, Stewart JD, Buddles M, Smith C, Evans J, et al. The clinical, histochemical, and molecular spectrum of PEO1 (Twinkle)-linked adPEO. Neurology. 2010;74:1619–26. doi: 10.1212/WNL.0b013e3181df099f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S, et al. Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann Neurol. 2002;52:211–9. doi: 10.1002/ana.10278. [DOI] [PubMed] [Google Scholar]
- 9.Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: Clinical and molecular genetic study. Lancet. 2004;364:875–82. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- 10.Diamantopoulou P, Ward VM, Harries ML. Kearns-Sayre syndrome: Presenting with vocal fold palsy. J Laryngol Otol. 2001;115:1021–2. doi: 10.1258/0022215011909675. [DOI] [PubMed] [Google Scholar]
- 11.Lin YC, Lee WT, Wang PJ, Shen YZ. Vocal cord paralysis and hypoventilation in a patient with suspected Leigh disease. Pediatr Neurol. 1999;20:223–5. doi: 10.1016/s0887-8994(98)00137-4. [DOI] [PubMed] [Google Scholar]